

Abstract
Recent studies highlight the importance of translational control in determining protein abundance, underscoring the value of measuring gene expression at the level of translation. We present a protocol for genome-wide, quantitative analysis of in vivo translation by deep sequencing. This ribosome profiling approach maps the exact positions of ribosomes on transcripts by nuclease footprinting. The nuclease-protected mRNA fragments are converted into a DNA library suitable for deep sequencing using a strategy that minimizes bias. The abundance of different footprint fragments in deep sequencing data reports on the amount of translation of a gene. Additionally, footprints reveal the exact regions of the transcriptome that are translated. To better define translated reading frames, we describe an adaptation that reveals the sites of translation initiation by pre-treating cells with harringtonine to immobilize initiating ribosomes. The protocol we describe requires 5 – 7 days to generate a completed ribosome profiling sequencing library. Sequencing and data analysis requires a further 4 – 5 days.
This unit describes the ribosome profiling approach for comprehensive and quantitative analysis of in vivo translation. The Basic Protocol describes profiling in cultured mammalian cells harvested with rapid detergent lysis. Accurate ribosome profiling requires cell lysates that reflect the true status of in vivo translation, and the lysis described in the basic protocol does not typically result in dramatic ribosome accumulation or run-off depletion at gene termini that would indicate perturbation of ribosomes. Brief treatment with elongation inhibitors such as cycloheximide or emetine (Alternate Protocol 1) can stabilize the in vivo state of elongating ribosomes but may induce ribosome accumulation at sites of translation initiation. Initiation inhibitors such as harringtonine or lactimidomycin (Alternate Protocol 2) further enrich ribosomes at start sites, allowing genome-wide profiling of translation initiation. Lysates from any of these procedures can be nuclease-treated to footprint ribosomes and the footprints can be converted into a sequencing library according to the method described in the basic protocol. After deep sequencing, data from this library can be analyzed using the approach described in Support Protocol 1.
NOTE: Experiments involving RNA require careful technique to prevent RNA degradation; see Chapter 4, Section I introduction.
BASIC PROTOCOL: RIBOSOME PROFILING IN CULTURED MAMMALIAN CELLS
Ribosome profiling characterizes translation in vivo by deep sequencing of ribosome-protected mRNA fragments. The basic profiling protocol comprises cell lysis, nuclease footprinting, ribosome purification, and sequencing library generation and validation. Cells are rapidly lysed with detergent and the lysates are digested with E. coli RNase I in order to degrade mRNA not protected by the ribosome. Ribosomes, containing protected mRNA footprints, are purified by sedimentation through a sucrose cushion during ultracentrifugation. RNA is recovered from the ribosomal pellet and used to generate a sequencing library using an approach optimized for low sequence bias. This approach for sequencing short RNA fragments combines the preadenylylated linker ligation originally developed for microRNA cloning with an intramolecular circularization of first-strand cDNA to avoid a second intermolecular ligation. The final library can be sequenced directly on the Illumina platform.
Materials
Cultures of adherent mammalian cells such as mouse embryonic stem cells (mESCs), HEK293 cells (Ingolia 2012), HeLa cells, mouse neutrophils (Guo 2009), or PC3 cells (Hsieh 2012).
Phosphate-buffered saline, pH 7.2, ice-cold (PBS, see APPENDIX 2)
Lysis buffer, ice-cold (see recipe)
100 U / μl RNase I
20 U / μl SUPERase*In (Life Technologies)
1 M sucrose cushion, ice-cold (see recipe)
miRNeasy RNA isolation kit (Qiagen)
Chloroform
15 mg / ml GlycoBlue
10 mM Tris, pH 8 (see APPENDIX 2)
- 10 μM upper and lower size marker oligoribonucleotides:
- 5'-AUGUACACGGAGUCGAGCUCAACCCGCAACGCGA-(Phos)
- 5'-AUGUACACGGAGUCGACCCAACGCGA-(Phos)
- The designation (Phos) indicates 3' phosphorylation. Note that all residues are ribonucleotides.
10,000x SYBR Gold (Life Technologies)
10× T4 polynucleotide kinase buffer
10 U / μl T4 polynucleotide kinase
10× T4 RNA ligase 2 buffer
50% w/v PEG 8000
200 U / μl T4 RNA ligase 2, truncated
- 0.5 μg / μl preadenylylated 3' linker:
- 5'-rAppCTGTAGGCACCATCAAT-(X)
- The designation rApp indicates 5' adenylylation and (X) indicates a blocked 3' terminus.
- 1.25 μM reverse transcription primer:
- 5'-(Phos)-AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC-(SpC18)-CACTCA-(SpC18)-TTCAGACGTGTGCTCTTCCGATCTATTGATGGTGCCTACAG.
- The designation (Phos) indicates 5'-phosphorylation and–(SpC18)-indicates a hexa-ethyleneglycol spacer.
5x First-strand buffer
10 mM each dNTP mix
0.1 M dithiothreitol
200 U / μl M-MuLV reverse transcriptase, RNase H−
1 N NaOH
10x CircLigase buffer
1 mM ATP
50 mM MnCl2
100 U / μl CircLigase I (Epicentre)
Subtraction oligo mix (see recipe)
20x SSC (see APPENDIX 2)
MyOne C1 Streptavidin DynaBeads (Life Technologies)
2x bind/wash buffer (see recipe)
1x bind/wash buffer (see recipe)
5x Phusion HF buffer
- 100 μM forward library PCR primer:
- 5'- AATGATACGGCGACCACCGAGATCTACAC
- 100 μM indexed reverse library PCR primers:
- 5'-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCG.
- The underlined NNNNNN indicates the reverse complement of the index sequence captured during Illumina sequencing.
2 U / μl Phusion polymerase
QuantiFluor dsDNA System (Promega)
BioAnalyzer High Sensitivity DNA kit (Agilent)
TruSeq SBS v3 kit, 50 cycles (Illumina)
TruSeq SR Cluster Kit v3, cBot, HiSeq (Illumina)
Cell lifter
Non-stick RNase-free microfuge tubes
26-gauge needle
1ml syringe
13 × 51 mm polycarbonate ultracentrifuge tube
Optima TLX ultracentrifuge (Beckman Coulter)
TLA 100.3 rotor (Beckman Coulter)
Blue light transilluminator
DynaMag-2 magnetic separation rack (Life Technologies)
Fluorimeter
2100 BioAnalyzer (Agilent)
GAII or HiSeq deep sequencer (Illumina)
Additional reagents and equipment for purification and concentration of DNA and RNA (UNIT 2.1A), non-denaturing polyacrylamide gel electrophoresis and subsequent recovery of DNA (UNIT 2.7), and denaturing polyacrylamide gel electrophoresis and subsequent recovery of DNA and RNA (UNIT 2.12).
Cell lysis
-
1
Aspirate media from one 10 cm dish of adherent cells. Place the dish on ice, gently wash with 10. ml of ice-cold PBS, and aspirate the PBS thoroughly.
-
2
Drip 400 μl of ice-cold lysis buffer onto cells, taking care to cover the entire surface of the dish. Tip the dish and scrape cells down the slope into the lysis buffer pooled in the lower corner. Pipette lysis buffer from this pool back towards the top of the dish and scrape again down the slope of the dish.
-
3
Pipette cells in lysis buffer and withdraw the entire contents of the dish to a microfuge tube on ice. Pipette several times to disperse cell clumps and incubate 10 minutes on ice. Triturate cells ten times through a 26 gauge needle.
-
4
Clarify the lysate by centrifugation for 10 minutes at 20,000 × g, 4°C and recover the supernatant.
Nuclease footprinting and ribosome recovery
-
5
Take 300 μl of clarified lysate from step 4 and add 7.5 μl of 100 U / μl RNase I. Incubate 45 min at room temperature with gentle mixing, e.g., on a nutator.
-
6
Add 10.0 μl of 20 U / μl SUPERase*In to stop nuclease digestion.
-
7
Transfer digestion to a 13 mm × 51 mm polycarbonate ultracentrifuge tube and underlay 0.90 ml of 1 M sucrose cushion by carefully positioning a pipette tip (or a cannula or similar tool) at the very bottom of the tube and slowly dispensing the sucrose solution. The lysate should float on top of the sucrose, leaving a visible interface between the layers.
-
8
Pellet ribosomes by centrifugation in a TLA100.3 rotor at 70,000 rpm, 4°C for 4 hr.
-
9
Mark the outside edge of the ultracentrifuge tube, where the ribosome pellet will be found, before removing the tube from the rotor. Gently pipette the supernatant out of the tube. The ribosomal pellet is glassy and translucent, and may not be visible until the supernatant is removed.
-
10
Resuspend the ribosomal pellet directly in 700μl of Qiazol reagent from the miRNeasy kit.
-
11
Purify RNA from the resuspended ribosomal pellet using the miRNeasy kit according to the manufacturer's instructions for purifying total RNA including small RNA. Collect the eluate in a non-stick RNase-free tube.
-
12
Precipitate RNA from the elution by ethanol or isopropanol precipitation (see UNIT 2.1A) using 1.5 μl of 15 mg / ml GlycoBlue as a carrier. Resuspend the RNA pellet in 5.0 μl of 10 mM Tris pH 8.
Footprint fragment purification
-
13
Separate RNA fragments by denaturing polyacrylamide gel electrophoresis (PAGE) through at 15% polyacrylamide gel (see UNIT 2.12). Run a sample of upper and lower size marker oligonucleotides, 10 pmol each, on a separate lane of the same gel.
-
14
Stain nucleic acids by incubating the gel in 1x SYBR Gold in 1x TBE for 3 minutes, then rinsing briefly in 1x TBE.
-
15Visualize the gel on a blue light transilluminator (see Figure 1a). Excise a gel slice for each RNA sample, using the size range delineated by the marker oligonucleotides, as well as a gel slice containing the size markers themselves. Extract RNA from these gel slices (see UNIT 2.12).Process size marker oligos in parallel with other samples, to provide an internal control for library generation as well as a size standard for later PAGE purification steps.Use a blue light transilluminator, as even brief exposure to ultraviolet light induces photodamage that interferes with reverse transcription.
-
16
Resuspend RNA from gel extraction and precipitation in 10.0 μl of 10 mM Tris pH 8 and transfer to a clean non-stick RNase-free microfuge tube.
-
17
Denature 90 seconds at 80°C, and equilibrate to 37°C.
-
18For each reaction, combine and mix the following components to the denatured RNA (total volume should be 50 μl):
- 33. μl of nuclease-free water
- 5.0 μl of 10x T4 polynucleotide kinase buffer
- 1.0 μl of 20 U / μl SUPERase*In
- 1.0 μl of 10 U / μl T4 polynucleotide kinase
If multiple samples are being processed in parallel, prepare a master mix containing these components for all samples together. -
19
Incubate 1 hour at 37°C. Heat-inactivate the enzyme for 10 min at 70°C.
-
20
Precipitate RNA from the dephosphorylation reaction by ethanol or isopropanol precipitation (see UNIT 2.1A) using 1.0 μl of 15 mg / ml GlycoBlue as a carrier.
Figure 1.

Representative gels from intermediate product purification. (a) Size selection of ribosome footprint fragments. The footprinting samples are derived from HeLa lysates with 5 – 15 μg input RNA. The blue bracket indicates the gel region that should be excised. (b) Purification of ligation products. Two marker samples are shown, one of which contains only the lower and upper marker oligonucleotides, the other of which was produced by carrying forward the markers from the size selection gel through dephosphorylation and ligation. The blue bracket indicates the gel region that should be excised. The blue arrowhead indicates the unreacted linker. (c) Purification of reverse transcription products. The blue bracket indicates the gel band that should be excised. The blue arrowhead indicates the unextended RT primer, which should be avoided. (d) Purification of PCR products. The blue bracket indicates the ~175 nt product band that should be purified. The blue arrowhead indicates the ~145 nt background band derived from unextended RT primer that should be avoided. The blue asterisk indicates the partial duplexes resulting from re-annealing as the PCR amplification approaches saturation. (e) BioAnalyzer profile of a high-quality sequencing library. A single 176 nt peak is present. (f) BioAnalyzer profile of a sequencing library with significant background from unextended RT primer. The background manifests as smaller DNA fragments that comprise 5–10% of the total DNA present in the sample; completely unextended RT primer yields a 144 bp PCR product. The DNA in this peak will produce sequencing data, but the sequence will consist of the linker sequence with no footprint.
Linker ligation
-
21
Resuspend dephosphorylated RNA from step 20 in 8.5 μl of 10 mM Tris pH 8 and transfer to a clean non-stick RNase-free microfuge tube.
-
22
Add 1.5 μl of 0.5 μg / μl preadenylated 3' linker to the dephosphorylated RNA and denature 90 seconds at 80°C, then cool to room temperature.
-
23Add the following components to the denatured RNA samples (total volume should be 20. μl):
- 2.0 μl of 10x T4 Rnl2 buffer
- 6.0 μl of 50% w/v PEG 8000
- 1.0 μl of 20 U / μl SUPERase*In
- 1.0 μl of 200 U / μl T4 RNA ligase 2 (truncated)
If multiple samples are being processed in parallel, prepare a master mix containing these components for all samples together. High-concentration solutions of polyethylene glycol are quite viscous--pipette carefully. -
24
Incubate 2.5 hours at room temperature.
-
25Add 338 μl water to each reaction and precipitate RNA from the linker ligation reaction by ethanol or isopropanol precipitation (see UNIT 2.1A) using 2.0 μl of 15 mg / ml GlycoBlue as a carrier.Diluting the reaction with water before precipitation reduces the concentration of polyethylene glycol in the precipitation reaction, which can otherwise interfere with the formation of a tight nucleic acid pellet.
-
26
Resuspend purified ligation products in 5.0 μl of 10 mM Tris pH 8.
-
27
Separate RNA by denaturing PAGE through at 15% polyacrylamide gel (see UNIT 2.12).
-
28
Stain nucleic acids by incubating the gel in 1x SYBR Gold in 1x TBE for 3 minutes, then rinsing briefly in 1x TBE.
-
29
Visualize the gel on a blue light transilluminator (see Figure 1b). Excise a gel slice for each RNA sample, based on the size range delineated by the marker oligonucleotide ligation products, as well as a gel slice containing the ligated size markers themselves. Extract RNA from these gel slices (see UNIT 2.12).
Reverse transcription
-
30
Resuspend the gel-purified ligation products from step 29 in 10.0 μl of 10 mM Tris pH 8 and transfer to a clean PCR tube.
-
31
Add 2.0 μl of 1.25 μM reverse transcription primer to the ligation products. Denature 2 min at 80°C in a thermal cycler and then place on ice. Cool the thermal cycler to 48°C.
-
32To the denatured mix of ligation products and primer, add the following (total volume should be 20.0 μl):
- 4.0 μl of 5x first-strand buffer
- 1.0 μl of 10 mM each dNTP mix
- 1.0 μl of 0.1 M dithiothreitol
- 1.0 μl of 20 U / μl SUPERase*In
- 1.0 μl of 200 U / μl M-MuLV Reverse Transcriptase RNase H-
If multiple samples are being processed in parallel, prepare a master mix containing these components for all samples together. -
33
Incubate 30 min at 48°C.
-
34
Hydrolyze RNA by adding 2.2 μl of 1 N NaOH to each reaction and incubate 20 min at 98°C.
-
35
Add 57. μl of nuclease-free water to each reverse transcription reaction and precipitate DNA from the reverse transcription reaction by ethanol or isopropanol precipitation (see UNIT 2.1A). Resuspend DNA in 5.0 μl of 10 mM Tris pH 8.
-
36
Separate the reverse transcription products from the unextended primer by denaturing PAGE (see UNIT 2.12). Run a control sample containing 2.5 pmol reverse transcription primer on one lane of this gel.
-
37
Stain nucleic acids by incubating the gel in 1x SYBR Gold in 1x TBE for 3 minutes, then rinsing briefly in 1x TBE.
-
38
Visualize the gel on a blue light transilluminator (see Figure 1c). Excise the extended reverse transcription product for each sample and extract DNA from these gel slices (see UNIT 2.12).
Circularization
-
39
Resuspend reverse transcription products from step 38 in 15.0 μl of 10 mM Tris pH 8 and transfer to a PCR tube.
-
40Add the following to the reverse transcription products (total volume should be 20.0 μl):
- 2.0 μl of 10x CircLigase buffer
- 1.0 μl of 1 mM ATP
- 1.0 μl of 50 mM MnCl2
- 1.0 μl of 100 U / μl CircLigase I
-
41
Incubate for 1 hr at 60°C and then heat-inactivate for 10 min at 80°C.
rRNA depletion
-
42Combine the following in a PCR tube (total volume should be 10.0 μl):
- 5.0 μl of circularization reaction from step 41
- 1.0 μl of subtraction oligo mix
- 1.0 μl of 20x SSC
- 3.0 μl of nuclease-free water
-
43
Place PCR tubes in a thermal cycler and denature 90 sec at 100°C, and then anneal at 0.1°C / second to 37°C. Incubate 15 min at 37°C.
-
44
Vortex MyOne C1 Streptavidin DynaBeads. Transfer 250 μg of beads to a clean non-stick microfuge tube and place the tube on a magnetic rack for 1 minute to isolate beads. Gently withdraw all liquid from the tube, remove the tube from the rack, and resuspend in 1 volume 1x bind/wash buffer. Repeat this procedure twice more.
-
45
Place the beads on a magnetic rack for 1 min to isolate beads, withdraw the final wash, and resuspend in 10.0 μl of 2x bind/wash buffer. Equilibrate beads at 37°C.
-
46
Transfer 10.0 μl of subtraction reaction from step 43 directly from the PCR tube in the thermal cycler to washed magnetic beads. Incubate 15 min at 37°C with mixing.
-
47
Transfer tubes directly to a magnetic rack and isolate beads for one minute. Recover 17.5 μl eluate from the depletion and transfer it to a new non-stick microfuge tube.
-
48
Precipitate RNA from the dephosphorylation reaction by ethanol or isopropanol precipitation (see UNIT 2.1A) using 1.0 μl of 15 mg / ml GlycoBlue as a carrier.
-
49
Resuspend depleted DNA in 5.0 μl of 10 mM Tris pH 8.
PCR amplification and barcode addition
-
50Combine the following on ice (total volume should be 100 μl):
- 5.0 μl of subtracted DNA from step 49
- 20.0 μl of 5x Phusion HF buffer
- 2.0 μl of 10 mM each dNTP mix
- 0.5 μl of 100 μM forward library primer
- 0.5 μl of 100 μM indexed reverse library primer
- 71. μl of nuclease-free water
- 1.0 μl of 2 U / μl Phusion polymerase
Select a different indexed primer for each sample that will be pooled together for sequencing. -
51
Set up 5 PCR tube strips and transfer a 16.7 μl aliquot of the PCR mixture into one tube in each strip.
-
52
Perform semi-quantitative PCR amplification with varying numbers of cycles by placing all strip tubes in the thermal cycler and starting a program with the conditions below. Remove strips successively at the very end of the extension step after 6, 8, 10, and 12 extension cycles, leaving the last strip in the thermal cycler until the end of cycle 14.

-
53
Separate the library products from the adapter-only background by nondenaturing polyacrylamide gel electrophoresis (see UNIT 2.7).
-
54
Stain nucleic acids by incubating the gel in 1x SYBR Gold in 1x TBE for 3 minutes, then rinsing briefly in 1x TBE.
-
55
Visualize the gel on a blue light transilluminator (see Figure 1d). Excise the sequencing library PCR product for each sample and extract DNA from these gel slices (see UNIT 2.7).
-
56
Resuspend library DNA in 15.0 μl of 10 mM Tris pH 8.
-
57
Quantify DNA in 1.0 μl of the extracted sample using the QuantiFluor assay for dsDNA according to the manufacturers instructions.
-
58
Quantify and characterize the library by preparing a dilution, with a DNA concentration no greater than 500 ng / μl, and using the High Sensitivity DNA chip on the Agilent BioAnalyzer according to the manufacturer's protocol (see Figure 1, e and f).
ALTERNATE PROTOCOL 1: PRE-TREATMENT OF CULTURED CELLS WITH ELONGATION INHIBITORS
Cells are treated briefly with elongation inhibitors before proceeding to lysis as described in the basic protocol.
Materials
Adherent cultured cells as described in the Basic Protocol
100 mg / ml cycloheximide in DMSO
100 mg / ml emetine in DMSO
Elongation inhibitor treatment
-
1
Add cycloheximide to culture media at a final concentration of 100 μg / ml, or emetine to a final concentration of 20 μg / ml, mix quickly, and return to incubator for 1 min.
-
2
Proceed with ribosome profiling starting at Basic Protocol, step 1.
ALTERNATE PROTOCOL 2: PRE-TREATMENT OF CULTURED CELLS WITH INITIATION INHIBITORS
Cells are treated with inhibitors of translation initiation before proceeding to perform ribosome profiling according to the basic protocol.
Materials
Adherent cultured cells as described in the Basic Protocol
2 mg / ml harringtonine in DMSO
50 mM lactimidomycin in DMSO
Initiation inhibitor treatment
-
1
Add harringtoine to culture media at a final concentration of 2 μg / ml, mix quickly, and return to the incubator for 3 minutes. Alternately, add lactimidomycin to a final concentration of 50 μM and return to the incubator for 30 minutes.
-
2
Proceed with ribosome profiling starting at Basic Protocol, step 1.
SUPPORT PROTOCOL 1: PRIMARY ANALYSIS OF RIBOSOME PROFILING DATA
Genome sequence and transcript structures are downloaded and indexed only once. Sequence data from each sample is pre-processed to remove adapter sequences, aligned against rRNA sequence to remove contaminating ribosomal reads, and then aligned to the indexed genome with a splicing-aware alignment tool.
Materials
64-bit computer running Linux, with at least 4 GB of RAM
FastX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html)
Bowtie software (http://bowtie-bio.sourceforge.net/index.shtml)
TopHat software (http://tophat.cbcb.umd.edu/)
SAMtools software (http://samtools.sourceforge.net/)
Downloading and indexing reference sequences
-
1
Obtain species-specific rRNA sequences (e.g. NR_003285.2, NR_003286.1, NR_003287.1, and NR_023363.1 for human cells) and place them in a single Fasta-format sequence file.
-
2
Build a bowtie index for the rRNA sequences:

-
3
Download the genome reference as a Fasta file. The current human genome reference, GRCh37, is available at ftp://ftp.ncbi.nlm.nih.gov/genbank/genomes/Eukaryotes/vertebrates_mammals/Homo_sapiens/GRCh37/Primary_Assembly/assembled_chromosomes/FASTA/ with a single compressed Fasta-format file per chromosome. The download can be automated using the following command:

-
4
Index the genomic reference Fasta files using bowtie-build. The GRCh37 genome downloaded as a collection of Fasta files, described above, can be indexed with the following command:

-
5
Obtain a GTF-format annotation for the exact genome reference sequence downloaded and indexed above. For the GRCh37 human genome sequence, the UCSC genome browser annotations can be downloaded from their table browser (http://genome.ucsc.edu/cgi-bin/hgTables?org=Human&db=hg19). Select the group “Genes and Gene Prediction Tracks”, track “UCSC Genes”, table “knownGene”, output format “GTF”, and use “get output” to retrieve the file. Rename it as hg19.gtf.

Pre-processing and aligning ribosome profiling data
-
6
Uncompress the raw sequence data, clip the 3' adapter sequence from the end, and collect the pre-processed results. The standard CASAVA 1.8 output is a collection of gzip-compressed FastQ files in a directory named Project_YYY/Sample_XXX. To perform all preprocessing steps in series, use the following command:

-
7
Align trimmed sequencing reads to an rRNA reference using the Bowtie short-read alignment program, discard the rRNA alignments, and collect unaligned reads.

-
Align non-rRNA sequencing reads to a genomic reference using the TopHat splicing-aware short read alignment program.
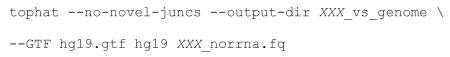
-
Extract perfect-match alignments from TopHat output.
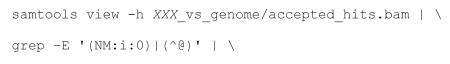
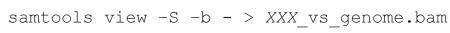
-
REAGENTS AND SOLUTIONS
Use sterile, RNase-free water, reagents, and techniques in all recipes and protocol steps. For common stock solutions, see APPENDIX 2.
Polysome buffer
20 mM Tris, pH 7.4
150 mM NaCl
5 mM MgCl2
1 mM DTT
100 μg / ml cycloheximide
Prepare immediately prior to use
Lysis buffer
Polysome buffer
1% v/v Triton X-100
25 U / ml Turbo DNase I
Prepare immediately prior to use and store on ice
Sucrose cushion
Polysome buffer
1 M sucrose
20 U / ml SUPERase*In
Prepare immediately prior to use and store on ice
Subtraction oligo mix
Combine up to twenty biotinylated subtraction oligonucleotides, each at 10 μM. Design oligonucleotide sequences to target species- and sample-specific high abundance rRNA sequences, identified empirically. Use forward-strand rRNA sequence for oligos, incorporate a 5'-biotin-TEG, and purify oligos by HPLC.
Bind/wash buffer, 2x
2 M NaCl
1 mM EDTA
5 mM Tris pH 7.4
0.2% v/v Triton X-100
Store up to 1 year at room temperature
Bind/wash buffer, 1x
Dilute from 2x bind/wash buffer and store up to 1 year at room temperature
COMMENTARY
Background Information
Gene expression profiling is a central tool for understanding cellular physiology and regulation. Historically, studies of gene expression have typically measured mRNA abundances rather than rates of protein synthesis, in large part because such data are much easier to obtain. The focus on overall mRNA levels increased with the emergence of microarrays (Brown and Botstein, 1999) and more recently RNA-Seq (Mortazavi et al., 2008; Nagalakshmi et al., 2008) as comprehensive and quantitative expression profiling techniques. These measurement techniques have revolutionized our ability to monitor the internal state of cells, but they have naturally led to a focus on transcriptional regulatory networks. However, mRNA and protein levels are imperfectly correlated in yeast and in mammalian cells (Ghaemmaghami et al., 2003; Lu et al., 2007; Schwanhausser et al., 2011; Vogel, 2011), and translational control can play a critical role in modulating gene expression (Sonenberg and Hinnebusch, 2009; Vogel, 2011).
Ribosome profiling monitors translation directly by deep sequencing of ribosome-protected mRNA fragments. The protein being synthesized by a ribosome is, of course, determined by the mRNA sequence it is decoding. A translating ribosome encloses a ~30 nt portion of this mRNA template and protects it from nuclease digestion. These ribosome-protected mRNA fragments have previously been used to map the positions of ribosomes in homogeneous in vitro translation reactions (Steitz, 1969; Wolin and Walter, 1988). Dramatic advances in sequencing technology (Bentley et al., 2008) now make it possible to characterize the complex pool of fragments produced by nuclease footprinting of ribosomes from living cells. Each ribosome produces a footprint fragment whose sequence indicates which mRNA it was translating, as well as its precise position on the transcript. Deep sequencing of ribosome footprints thus provides information about ribosome positions, which is inaccessible to existing polysome profiling approaches for measuring translation, as well as measuring expression quantitatively.
Applications of ribosome profiling
Ribosome footprint sequences indicate which portions of the genome are actually being translated into protein. These translated sequences include conventional protein-coding genes, but also reading frames that encode short peptides. A few short ORFs have been identified genetically (Kondo et al., 2010), and ribosome profiling data has revealed many more (Brar et al., 2011; Ingolia, 2010; Ingolia et al., 2011). Thus, it is likely that the number of small peptides is much larger than currently known. In order to be translated, a sequence must first be transcribed. Recent studies have revealed great diversity in the mammalian transcriptome (Trapnell et al., 2010b), though many of these transcripts lack long open reading frames. Short ORFs on traditional and noncanonical messages can be difficult to identify reliably by computational approaches (Dinger et al., 2008), but ribosome profiling has proven to be a highly useful tool for exploring the peptide coding potential of these RNAs (Ingolia et al., 2011).
In addition to discovering these novel ORFs, ribosome profiling data leads to revisions in the annotation of known genes. In many cases, footprinting data indicates the translation of extended or truncated protein isoforms. These alternate protein products can have functions that are distinct from or antagonistic to those of the annotated variant. Alternate protein isoforms can arise from the translation of different mRNA isoforms or from the use of alternative initiation sites on the same transcript. When multiple isoforms are co-expressed, translation from upstream initiation sites can obscure the presence of downstream initiation. These internal initiation sites are revealed by ribosome profiling after treatment with harringtonine or lactimidomycin, drugs that immobilize ribosomes immediately after translation initiation and results in footprint accumulation at all initiation sites (Brar et al., 2012; Fresno et al., 1977; Robert et al., 2009; Schneider-Poetsch et al., 2010). Thus, the presence or absence of ribosome footprints at downstream AUG codons in harringtonine-treated samples marks sites of potential internal initiation that lead to shorter protein isoforms. More generally, initiation site profiling with harringtonine can be combined with ribosome profiling to detect elongating ribosomes over the entire reading frame to produce an experimentally-based annotation of the translated products from a genome (Ingolia et al., 2011).
Still, the broadest application of ribosome profiling may be measurements of gene expression at the level of actual protein synthesis. Each ribosome footprint corresponds to a translating ribosome, so the number of footprints produced from a transcript should correspond to the number of ribosomes engaged in synthesizing the encoded protein. This is proportional to the amount of the protein being produced and to the time required to produce it. We have shown that the speed of protein synthesis is broadly consistent across different groups of genes (Ingolia et al., 2011). Thus under a given condition, the translation time of an ORF is simply proportional to its length. One can therefore determine the rate at which a protein is being produced by measuring the density of ribosome footprints on its transcript. Ribosome footprint density thus can be used in place of mRNA abundance measurements to quantify gene expression. It can also be combined with mRNA abundance measurements to identify translational regulation as changes in protein expression that cannot be explained by transcript levels (Brar et al., 2011; Ingolia et al., 2009). RNA-Seq measurements of transcript abundance measurements can be made by analysis of randomly fragmented complete mRNA in parallel with ribosome footprints (Ingolia, 2010; Ingolia et al., 2009) or by other standard approaches (Levin et al., 2010).
Finally, ribosome profiling provides an approach for studying the mechanics of translation and of co-translational processes in vivo. Just as the overall number of ribosome footprints on a gene indicates how many ribosomes are typically translating it, the number of footprints centered on a codon should reflect how often a ribosome is found at that particular spot. If ribosomes stall at a specific point when translating a gene, then ribosomes will spend more time there than elsewhere, producing a corresponding excess of footprints. We have used this excess of ribosome footprints to detect peptide-mediated translational stalling in mammalian cells (Ingolia et al., 2011) and RNA-mediated stalling in bacteria (Li et al., 2012). Ribosome footprint density has also been applied to determine codon-specific elongation rates in bacteria (Li et al., 2012) and yeast (Qian et al., 2012), as well as C. elegans and human cultured cells (Stadler and Fire, 2011). It has also been applied to monitor co-translational processes in protein biogenesis, including chaperone association and protein secretion (Oh et al., 2011; Reid and Nicchitta, 2012).
Convergence of expression profiling techniques
Ribosome profiling bridges the gap between global measurements of steady-state mRNA and protein levels. As such, it will be particularly valuable to compare ribosome profiling and mass spectrometry measurements of protein expression levels. At present, sequencing technologies provide deeper measurement than mass spectrometry measurements in most circumstances. However, steady-state measurements by mass spectrometry are sensitive to protein degradation as well as synthesis. In fact, high-quality ribosome profiling and proteomic measurements may offer a new approach to determine the turnover rate of native proteins in unperturbed cells. Similar interpolation between RNA-Seq and mass spectrometry measurements recently quantified the large contribution of translation to steady-state protein levels (Schwanhausser et al., 2011).
Until now, the translational status of mRNAs typically has been assessed by separating intact ribosome-mRNA complexes based on the total number of ribosomes bound to a transcript. A genomic adaptation of this assay, called polysome profiling, measures the mRNA constituents of different ribosome number fractions using microarrays (Arava et al., 2003; Sampath et al., 2008). Ribosome profiling has technical advantages over polysome profiling for making routine expression measurements, but polysome profiling can complement ribosome footprinting experiments, particularly for performing mechanistic studies of translational control. Ribosome profiling provides more precise expression measurement, because it avoids the difficulty in resolving the exact number of ribosomes bound to highly ribosome-loaded transcripts. Failure to separate these transcripts can obscure changes in the exact number of ribosomes bound to them and thereby compress the dynamic range of polysome profiling experiments. Ribosome profiling also avoids certain technical hurdles that arise in polysome profiling. While many skilled investigators reliably obtain high-quality, intact polysomes, RNA degradation remains a challenge. Ribosome profiling requires only the nuclease footprint from single ribosomes, so it is less sensitive to compromised RNA integrity. Finally, ribosome profiling can distinguish between ribosomes translating protein-coding genes and those translating regulatory upstream open reading frames.
Polysome profiling monitors the translational status of entire transcripts, which provides data that cannot be determined from footprint sequencing measurements that focus on the activities of individual ribosomes. Thus, polysome profiling can distinguish between a uniform decrease in the number of ribosomes on all copies of a transcript and a complete repression of a sub-population of mRNAs, a phenomenon that was revealed by polysome profiling of mouse ES cells (Sampath et al., 2008). By contrast, ribosome profiling would simply detect a quantitative decrease in the ensemble-averaged rate of protein synthesis in either case. Similarly, polysome profiling may have a greater ability to measure differences in the translation of alternate transcript isoforms, particularly when they differ in their 5' or 3' untranslated regions. These measurements could complement ribosome profiling data to provide insight into the molecular mechanism of translational regulation.
Critical Parameters
Cell lysis
Ribosome profiling begins with the preparation of cell lysates where ribosome-mRNA complexes accurately reflect in vivo translation. The best approach for lysate preparation will vary based on the sample being analyzed. Traditionally, polysomes have been “stabilized” by treating cells with translation elongation inhibitors before cell lysis. We found that in mammalian cells, brief treatment with such drugs causes an accumulation of ribosomes in the first five to ten codons of all genes (Ingolia et al., 2011). This may well reflect an artifactual accumulation of ribosomes that initiate during drug treatment and stall translation shortly thereafter. We therefore favor the in situ detergent lysis of adherent, cultured cells because it seems to produce the least opportunity for perturbation between normal growth and ribosome extraction. However, this approach is not suitable for all samples. In Saccharomyces cerevisiae, we found that elongation inhibitors suppressed changes in translation that occurred during cell harvesting (Ingolia et al., 2009), and similar polysome stabilization may be necessary in other situations as well. Drug pre-treatment may provide other valuable information. For instance, brief pre-treatment of cells with harringtonine or lactimidomycin enriches ribosomes specifically on initiation sites, enhancing the detection and annotation of translated sequences (Fritsch et al., 2012; Ingolia et al., 2011; Lee et al., 2012).
We also found that cryogenic pulverization of frozen yeast produced effective lysis and homogenization under conditions that blocked biological responses (Ingolia, 2010). This technique is also applicable in mammalian cells and tissues that require physical disruption for ribosome extraction. Indeed, flash freezing of tissues followed by cryogenic pulverization and thawing in the presence of translation inhibitors provides a particularly robust and simple approach for the analysis of animal-derived samples (del Prete et al., 2007).
Nuclease footprinting
Nuclease footprinting converts ribosome positions into RNA sequence tags that can be analyzed by deep sequencing. We have found that similar digestion conditions can be used to footprint ribosomes in yeast, HeLa, and mouse ES cell lysates, suggesting that extensive optimization is not needed for profiling in various eukaryotic systems. However, we have identified an effect of lysis buffer conditions on the size and reading frame precision of ribosome footprints (Ingolia et al., 2012). Lower salt and magnesium improve the reading frame resolution ribosome footprints without substantially altering overall measurements of gene expression.
Ribosome recovery
Following nuclease digestion, we separate intact ribosome-footprint complexes from cell lysates prior to RNA extraction. We originally performed sucrose density gradient purification of 80S ribosome particles. However, sucrose density gradient fractionation is challenging, and in fact represents a substantial barrier to analyzing translation through traditional polysome approaches. More recently, we purified ribosomes by sedimenting them through a sucrose cushion, which provides a more accessible density-based separation. We did not observe increased contamination with untranslated but protein-bound RNA sequences, such as 3'UTRs, in samples purified by sucrose cushion, though it is in principle less specific than sucrose gradient purification and it may be more important to verify that RNA fragments show the characteristic size and reading frame distribution of true ribosome footprints. Other approaches are possible as well— ribosomes can be purified by gel filtration (Jelenc, 1980), and in certain systems genetic manipulation can be used to add epitope tags to ribosomes, enabling affinity purification (Heiman et al., 2008; Sanz et al., 2009).
Linker ligation
Deep sequencing analysis typically requires libraries containing specific linker sequences; in the case of the Illumina sequencer employed here, the library is double-stranded DNA with defined sequences flanking the target fragment. In our previous work, we identified and worked to minimize significant biases in the conversion of RNA footprints into a sequencing library. These biases are present in all RNA-Seq libraries, but cause particular difficulties in analyzing ribosome footprinting data. While our approach achieved notably good uniformity (Levin et al., 2010), it involved the addition of a poly-(A) tail to each sequence. This degenerate sequence complicated bioinformatic analyses. We have subsequently shown that an optimized RNA ligation of a preadenylated linker (Lau et al., 2001) can achieve comparable results, and both the genetically modified RNA ligase and the chemically modified linker required for this approach are now commercially available. We have also altered the sequences of reverse transcription and PCR primers used in the protocol to allow sequencing with the standard Illumina primers. This includes the option of adding a 6 nt index that can be read in the same manner as the indices added to standard Illumina libraries.
Ribosomal RNA depletion
Ribosomal RNA contamination substantially decreases the amount of informative sequence data obtained in a ribosome profiling experiment. This is unsurprising, as there are several kilobases of rRNA in each ribosome-footprint complex, but only ~28 bases of footprint mRNA. We observed that a few specific rRNA fragments represented a large fraction of the overall contamination present in the 26 – 34 nt size window that we purified, presumably because nuclease digestion of intact ribosomes results in reproducible cleavage at a limited number of positions. We therefore depleted first-strand cDNAs derived from these high-abundance contaminants by hybridization to biotinylated sense strand oligonucleotides followed by removal of the duplexes through streptavidin affinity.
Analysis and interpretation
Ribosome footprint sequencing data can be preprocessed and then aligned to the genome using tools available for RNA-Seq analysis (Langmead et al., 2009; Trapnell et al., 2010b). Most of the bioinformatics challenges, such as the alignment of reads across splice junctions and the possibility of multiple distinct genomic alignments, are similar in these data. Considerable specific rRNA contamination can remain even after depletion by subtractive hybridization. Thus, we also implement a bioinformatics filter to remove these sequences first. Examination of the positive alignments from this filter points to specific contaminating rRNA sequences that can be targeted with additional biotinylated subtraction oligos. We also identify some contaminating sequences derived from other abundant non-coding RNAs (ncRNAs), such as tRNAs and snRNAs. These contaminants typically derive from a single specific position, whereas ribosome footprints will cover many positions along a reading frame, and ncRNA fragments will show an atypical length distribution. The extent of rRNA and ncRNA contamination can vary, particularly when global changes in protein synthesis alter the fraction of active ribosomes, and thus the number of ribosome-protected footprints relative to other RNAs.
Many applications of ribosome profiling, including expression measurements, depend on comparing the numbers of aligned sequencing reads between genes, samples, or specific codons. Quantitative analysis of ribosome profiling data, like RNA-Seq data (Mortazavi et al., 2008), is powerful, but this analysis must account for limitations that arise in this sort of data. Two major concerns that have been studied in RNA-Seq data are systematic sequence-dependent biases and stochastic sampling errors (Anders and Huber, 2010; Bullard et al., 2010; Hansen et al., 2010; Levin et al., 2010; Li et al., 2010; Roberts et al., 2011; Trapnell et al., 2010a). RNAs will be captured during library generation with differing efficiencies, perhaps due to sequence or structural preferences of the enzymes used in library generation (Hafner et al., 2011; Linsen et al., 2009; Zhuang et al., 2012). While we strove to minimize these biases, they are present in all sequencing samples (Levin et al., 2010) and it is important to avoid confusing library generation biases with differences in the underlying abundance of different footprints. These sequence biases are minimized in expression measurements due to averaging across the entire sequence of the mRNA; comparison of the same gene across different samples, one of the most frequent uses of profiling data, provides further protection from these effects. Stochastic error also arises, and is most serious when comparing small absolute numbers of reads. Several statistical approaches have been developed to estimate and model this error in RNA-Seq data, which can exceed the expectation derived from Poisson statistics (Anders and Huber, 2010; Bullard et al., 2010; Garber et al., 2011; Trapnell et al., 2010a). Many of these techniques and tools should be directly applicable to ribosome profiling expression measurements.
Troubleshooting
Low yield in linker ligation will result in more unligated RNA at ~30 nt and less ligated RNA product at ~50 nt (see Figure 1b). One common cause is failure of the dephosphorylation reaction, which is sensitive to residual salt from precipitated RNA as well as to other contaminants. Note that the lower and upper size marker RNAs are chemically phosphorylated and serve as an internal control for both dephosphorylation and subsequent linker ligation. Take care to remove all liquid from the RNA pellet, dry it thoroughly, and resuspend the pellet in a small volume while avoiding residual salt on other parts of the precipitation tube before transferring it to a new, clean tube.
A lower ~145 nt band during non-denaturing PAGE of library PCR products represents background derived from an unextended RT primer (see Figure 1d). When the amount of RT product is unusually low, this background will comprise a greater fraction of the total DNA. To decrease this background, excise the RT product band precisely and avoid the background haze. In some cases, reducing the amount of RT primer may help as well.
A broad, slower-migrating smear during PAGE of the library PCR prodcuts indicates excessive amplification (see Figure 1d). When the PCR amplification consumes a large fraction of the total oligonucleotides present in the reaction mixture, re-annealing of library strands becomes kinetically competitive with primer annealing. Re-annealed library duplexes have long complementary sequences on each end, but typically contain non-complementary inserts, causing slow and heterogeneous migration relative to the fully complementary library duplex. Use product bands from reactions with fewer PCR cycles.
Anticipated Results
The protocol typically produces 550 – 600 μl lysate. RNA extraction from an aliquot of this lysate indicates a yield of 25 – 50 μg total RNA from one 10 cm dish of 50% – 80% confluent HEK293 cells. The RNA yield from the footprinting pellet is typically 40% – 50% of the total RNA input, resulting in 6 μg – 15 μg of RNA from 300 μl lysate. The lost RNA includes non-coding RNAs that do not enter the sucrose cushion, such as tRNAs, as well as mRNA and rRNA that is degraded during the footprinting digest. We have successfully prepared libraries from as little as 2 μg of ribosomal pellet RNA.
Gel electrophoresis of the footprinting RNA will reveal a broad array of specific and fairly reproducible bands (Figure 1a), most presumably derived from the rRNA. The marker oligos will guide the excision of the gel region that contains the footprint fragments, which may not be visible as a discrete band (Figure 1a). They also provide a positive control through subsequent PAGE purification steps. The ligated control oligos indicate a specific region that should be excised in the linker ligation reaction (Figure 1b). While the marker reverse transcription products do still produce a discernible doublet (Figure 1c), the RT product in general forms a much tighter band because the relative length variation is lower, and it is not necessary to excise a broad region. The PCR products should produce a discrete band that is ~175 nt long (Figure 1d).
Time Considerations
Library generation, described in the Basic Protocol, typically requires one week in total. Cell lysate can be flash-frozen in liquid nitrogen and stored at −80°C. Nuclease digestion, ribosome purification, and RNA extraction should be performed without interruption, over the course of ~6 hours. After RNA extraction, each of the four gel extraction steps can be performed overnight, setting the overall time-table for the library generation experiment. Intermediate nucleic acid samples can be stored at −20°C or at −80°C at different stages, either as a part of isopropanol precipitation or after resuspension in nuclease-free water. Completed DNA libraries can be stored indefinitely at −20°C. Sequencing requires 2 – 3 days, and primary analysis described in the Support Protocol.
LITERATURE CITED
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O'Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. High-Resolution View of the Yeast Meiotic Program Revealed by Ribosome Profiling. Science. 2011 doi: 10.1126/science.1215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335:552–557. doi: 10.1126/science.1215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nat Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Prete MJ, Vernal R, Dolznig H, Mullner EW, Garcia-Sanz JA. Isolation of polysome-bound mRNA from solid tissues amenable for RT-PCR and profiling experiments. Rna. 2007;13:414–421. doi: 10.1261/rna.79407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;4:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno M, Jimenez A, Vazquez D. Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem. 1977;72:323–330. doi: 10.1111/j.1432-1033.1977.tb11256.x. [DOI] [PubMed] [Google Scholar]
- Fritsch C, Herrmann A, Nothnagel M, Szafranski K, Huse K, Schumann F, Schreiber S, Platzer M, Krawczak M, Hampe J, Brosch M. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 2012 doi: 10.1101/gr.139568.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nature methods. 2011;8:469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Hafner M, Renwick N, Brown M, Mihailovic A, Holoch D, Lin C, Pena JT, Nusbaum JD, Morozov P, Ludwig J, Ojo T, Luo S, Schroth G, Tuschl T. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. Rna. 2011;17:1697–1712. doi: 10.1261/rna.2799511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Brenner SE, Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010;38:e131. doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 2010;470:119–142. doi: 10.1016/S0076-6879(10)70006-9. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelenc PC. Rapid purification of highly active ribosomes from Escherichia coli. Anal Biochem. 1980;105:369–374. doi: 10.1016/0003-2697(80)90472-8. [DOI] [PubMed] [Google Scholar]
- Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, Kobayashi S, Payre F, Kageyama Y. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;329:336–339. doi: 10.1126/science.1188158. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee S, Liu B, Huang SX, Shen B, Qian SB. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2424–2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nature methods. 2010;7:709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GW, Oh E, Weissman JS. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012 doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jiang H, Wong WH. Modeling non-uniformity in short-read rates in RNA-Seq data. Genome Biol. 2010;11:R50. doi: 10.1186/gb-2010-11-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsen SE, de Wit E, Janssens G, Heater S, Chapman L, Parkin RK, Fritz B, Wyman SK, de Bruijn E, Voest EE, Kuersten S, Tewari M, Cuppen E. Limitations and possibilities of small RNA digital gene expression profiling. Nature methods. 2009;6:474–476. doi: 10.1038/nmeth0709-474. [DOI] [PubMed] [Google Scholar]
- Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Becker AH, Sandikci A, Huber D, Chaba R, Gloge F, Nichols RJ, Typas A, Gross CA, Kramer G, Weissman JS, Bukau B. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell. 2011;147:1295–1308. doi: 10.1016/j.cell.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Yang J-R, Pearson NM, Maclean C, Zhang J. Balanced Codon Usage Optimizes Eukaryotic Translational Efficiency. PLoS Genet. 2012;8:e1002603. doi: 10.1371/journal.pgen.1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid DW, Nicchitta CV. Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. J Biol Chem. 2012;287:5518–5527. doi: 10.1074/jbc.M111.312280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert F, Carrier M, Rawe S, Chen S, Lowe S, Pelletier J. Altering chemosensitivity by modulating translation elongation. PLoS One. 2009;4:e5428. doi: 10.1371/journal.pone.0005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath P, Pritchard DK, Pabon L, Reinecke H, Schwartz SM, Morris DR, Murry CE. A hierarchical network controls protein translation during murine embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2008;2:448–460. doi: 10.1016/j.stem.2008.03.013. [DOI] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6:209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Fire A. Wobble base-pairing slows in vivo translation elongation in metazoans. Rna. 2011;17:2063–2073. doi: 10.1261/rna.02890211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz JA. Polypeptide chain initiation: nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature. 1969;224:957–964. doi: 10.1038/224957a0. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010a;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010b;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C. Translation's coming of age. Mol Syst Biol. 2011;7:498. doi: 10.1038/msb.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin SL, Walter P. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 1988;7:3559–3569. doi: 10.1002/j.1460-2075.1988.tb03233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang F, Fuchs RT, Sun Z, Zheng Y, Robb GB. Structural bias in T4 RNA ligase-mediated 3'-adapter ligation. Nucleic Acids Res. 2012 doi: 10.1093/nar/gkr1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
KEY REFERENCES
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the ribosome profiling technique and its application in budding yeast.
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the application of ribosome profiling and initiation site mapping in mammalian cells.
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]; Original publication of the ribosome profiling protocol presented here.