

Abstract
Linear polymers have been considered the best molecular structures for the formation of efficient protein conjugates due to their biological advantages, synthetic convenience and ease of functionalization. In recent years, much attention has been dedicated to develop synthetic strategies that produce the most control over protein conjugation utilizing linear polymers as scaffolds. As a result, different conjugate models, such as semitelechelic, homotelechelic, heterotelechelic and branched or star polymer conjugates, have been obtained that take advantage of these well-controlled synthetic strategies. Development of protein conjugates using nanostructures and the formation of said nanostructures from protein-polymer bioconjugates are other areas in the protein bioconjugation field. Although several polymer-protein technologies have been developed from these discoveries, few review articles have focused on the design and function of these polymers and nanostructures. This review will highlight some recent advances in protein-linear polymer technologies that employ protein covalent conjugation and successful protein-nanostructure bioconjugates (covalent conjugation as well) that have shown great potential for biological applications.
Keywords: protein, polymer, telechelic, nanostructures, covalent conjugation
1. Introduction
Polymer science is currently in the process of reinventing itself. Polymers are now not only being developed for the creation of common materials; in recent years, polymers have become an important tool for biological sciences. Synthetic polymers and polymeric assemblies have been designed to interact with biological moieties, such as proteins and nucleic acids, through recognition or hybridization for innovative approaches of interest to biomedicine. In general, research in this area has been focused on creating novel materials to improve the efficiency of therapeutics delivery [1–4], diagnostics and biological assays [5–14].
Although drug development has mainly focused on small molecules that act as activators or inhibitors of proteins, it is also well recognized that there is great potential for proteins themselves to be utilized for therapeutic purposes [15–16]. Therefore, efforts towards protein engineering and discovery of new therapeutic proteins have increased in recent years. Unfortunately, administration of these proteins suffers from a number of limitations including: inability to cross biological barriers, degradation in biological systems, toxicity problems, poor solubility and biodistribution [17]. For these and many other reasons, the conjugation of biocompatible polymeric materials to proteins has garnered interest. Bioconjugation, the covalent (or non-covalent) modification of biomolecules with synthetic molecules or macromolecules [18], is the most common method used to obtain polymer-biomolecule hybrids. The improvement in therapeutic performance of proteins when associated to these materials is the source of substantial growth in this area of research [19].
Many synthetic routes facilitate access to polymers and polymeric macro- and nanostructures to incorporate biological moieties, such as proteins. A critical feature of successful protein bioconjugation is that the protein should retain their native properties upon conjugation to the polymer. Therefore, successful conjugation depends on the availability of reactive moieties on protein surfaces, reaction conditions at which the conjugation is performed, and solubility of reagents [20–21]. Amino acid residues such as lysine, glutamate and cysteine are commonly used for protein conjugation with polymeric materials containing complementary functional groups. However, these amino acids can be inaccessible for direct attachment to the artificial polymer scaffolds due to variations in protein structure and conformation. Methods have been developed to circumvent this issue. For instance, if a protein does not contain any accessible amino acids, it can be genetically engineered to conveniently place one within its amino acid sequence [22–24]. Alternatively, artificial amino acids can be incorporated during protein synthesis by modifying cellular molecular machinery [25–26]. These modifications have shown great success in protein structure retention and stabilization. A wide variety of core materials have been conjugated to proteins, such as gold nanoparticles [27], organic small molecules [28] and polymers [29]. Although not as extensively explored, protein conjugation to polymeric nanostructures and formation of amphiphiles from conjugates have also been achieved (vide infra).
There has been a significant surge in research activity concerning bioconjugates between proteins and polymers. This has inspired many reviews and notes on the synthesis and application of protein-polymer conjugates [29–32]. However, to the best of our knowledge, little attention has been paid to the conjugation of proteins to polymeric nanoassemblies. In this review, therefore, we will not dwell on the synthetic techniques utilized for bioconjugation. We will focus on a select few examples of protein conjugate technologies developed so far and highlight two main areas of study within bioconjugation research: (1) protein conjugation with linear polymers (most common) and (2) protein conjugation with polymeric nanostructures or macromolecular assembly formation from conjugates (growing research area). We specifically restrict our analysis to covalent conjugation of polymeric materials, since stabilization of proteins within a complex biological environment is one of the greater advantages of covalent conjugation compared to non-covalent conjugation methods.
2. Protein Conjugation with Polymers
It is noteworthy that reverse addition fragmentation chain-transfer (RAFT) and atom transfer radical polymerization (ATRP) are understandably the most common synthetic strategies utilized for the preparation of polymers for protein conjugation [29]. These polymeric scaffolds can be covalently attached to proteins through three well-known conjugation strategies: (i) attachment of a preformed polymer to an unmodified or linker-modified protein, also called “grafting to” method; (ii) in situ bioconjugation by previously modifying a protein with initiation sites in which a polymer is then “grafted from” the protein microinitiator; and (iii) anchoring of proteins on the pendant side chains of a polymer; the “grafting through” method. For successful bioconjugation, it is essential that the polymeric scaffolds bring certain features to the protein conjugate (e.g. stability to mechanical and proteolytic degradation) without affecting the intrinsic property of the protein. For this reason, it is imperative that we target well-defined polymeric structures and utilize the right conjugation method.
The location at which the protein is conjugated to a polymeric material, conjugation of the protein to a precise single site or to different areas of the protein for instance, can greatly affect the pharmacological behavior of the therapeutic protein. Earlier efforts toward protein conjugation focused on the attachment of poly(ethyleneglycol) based linear polymers to the protein; “PEGylation”, as it is known. PEGylation represents the most common and successful conjugation strategy [3, 33–37]. PEGylated proteins have demonstrated good bioavailability, thermal stability, enhanced proteolytic resistance and even therapeutic potency [19, 30, 38]. On the other hand, PEGylation chemistry presents some challenges such as side reactions, poor selectivity in substitutions and lack of homogeneity of the conjugates [32, 38]. Efforts have been put into refining conjugation strategies to overcome these issues. Newer technologies, such as “smart” linear polymer-protein conjugates, have been demonstrated to circumvent some of the problems encountered in the PEGylation method. “Smart” polymers are linear polymers synthesized with chemo-specific functional groups having the capability of targeting specific protein sites and are responsive to external stimuli changes, such as pH, light, temperature, enzymatic cleavage, etc. Many of these polymers are telechelic. Telechelic polymers are those that contain end-functional groups capable of further polymerization [39–42] and are capable of binding to a specific number of proteins (Figure 1). Methods utilized for achieving this class of linear polymer-protein conjugates will be further discussed in the following sections.
Figure 1.

Representation of different types of polymer-protein conjugates. (2 column fitting image)
2.1 Monomeric Conjugation
2.1.1 Semitelechelic Polymer-protein Conjugates
Monomeric conjugates are arguably the most common type of conjugates; in this case, the protein is anchored by a single linear polymer (Structure I in Figure 1). Protein conjugates can be achieved by the covalent attachment of semitelechelic polymers through specific amino acid residues. As mentioned earlier, lysine and cysteine are the preferred amino acid residues for conjugation with polymers, mainly because of the relatively large number of complementary functional groups that are available for reaction with amines and thiols. Although cysteine residues are common in proteins, these thiol moieties are often tasked with stabilizing protein secondary and tertiary structures through the formation of disulfides. Therefore, free cysteine thiols are relatively scarce in proteins. This scarcity however, also provides an opportunity; one can attach a controlled number of polymers to a protein, depending on the number of free thiols available. Indeed, a versatile bioconjugation strategy has been developed for the in situ preparation of protein-“smart” polymer conjugates starting with a bovine serum albumin (BSA) macroinitiator (Scheme 1) [43]. BSA, a protein that contains a free cysteine at amino acid 34, was chosen as a model protein due to its availability, robustness, controlled number of reactive functional groups and stability throughout various purification procedures. After a protein reduction process, the number of thiol groups was maximized to achieve initiator conjugation. The initiator, based on a thiol-reactive pyridyl disulfide molecule, was conjugated to the modified BSA protein under mild conditions through a disulfide bond. The conjugation was first confirmed by electrospray ionization mass spectroscopy (ESI-MS). Subsequent in situ polymerization of BSA macroinitiator with N-isopropylacrylamide (NIPAAm) leads to the “smart” polymer conjugate. After demonstrating the concept with a BSA macroinitiator, this method was applied to a mutant form of lysozyme, a small protein that provides protection from bacteria by breaking down polysaccharide walls [43,44]. The enzymatic activity of the pNIPAAm conjugate was assessed and determined to be retained, when compared to the unmodified mutant lysozyme.
Scheme 1.

Polymerization from BSA or Lysozyme macroinitiator. (2 columns fitting image)
More recently, a new class of therapeutic conjugates based on dextrin, semitelechelic poly[N-(2-hydroxypropyl)methacrylamide] (ST-HMPA) and trypsin was targeted (Scheme 2) [45]. Dextrin I (8,000 g/mol), dextrin II (61,000 g/mol) and ST-HPMA-trypsin conjugates were synthesized utilizing 1:1 molar ratio of polymer:protein, anticipating that dextrin conjugates would have more than one polymer attachment site per trypsin. Conjugates were characterized by analyzing their molecular weights using gel permeation chromatography (GPC) and free protein content was determined by gel electrophoresis (SDS-PAGE). These analyses showed the conjugates with trypsin content of 54, 17 and 3 wt% can be prepared, while the reaction often returned less than 5% unreacted protein. The aim of this work was to study the effect of polymer on protein activity, autolysis and thermal stability. After conjugation, trypsin showed a decrease in activity of about 20%, a behavior observed in all three conjugate types. Moreover, conjugates showed both better resistance to autolysis and increased thermal stability compared to native trypsin. Interestingly, ST-HPMA-trypsin conjugates displayed lower t1/2 for autolysis and lower thermal stability than dextrin conjugates despite the fact that they are of similar molecular weights. This study underlined the potential of higher molecular weight dextrin II conjugates demonstrating improved stability and protein masking, followed by reactivation.
Scheme 2.

Bioconjugation of succinoylated dextrin and ST-HPMA (2 column fitting)
In 2011, a well-defined semitelechelic biodegradable poly N-(2-hydroxypropyl) methacrylamide-lysozyme conjugate (polyHPMA-lysozyme) was reported [46]. PolyHPMA has been widely targeted for biomedical applications due to its potential pharmacological advantages. A biodegradable protein reactive group, thiazolidine-2-thione, was synthesized and employed as a functional group for RAFT polymerization. This functional chain transfer agent is capable of reacting with amine residues present on the protein surface to form amide covalent conjugation between the functional polyHPMA chain and the model protein, lysozyme. Two different polymers were synthesized by RAFT polymerization: a) polymer 1, a hydroxyl-terminated polyHPMA with Mn 12,000 and b) polymer 2, a thiazolidine-2-thione-terminated polyHPMA (polymer 2a, Mn: 12,700 and polymer 2b, Mn: 16,200) (Scheme 3). Bioactivities of the protein-polymer conjugates were tested using MI cells as substrates. It was demonstrated that polymers’ molecular weights had an impact on the suppression of conjugate bioactivity. On the other hand, after cleavage of the polymer chains and protein release, recovery of activity was observed. Cytotoxicity studies confirmed that conjugates were non-toxic; in vivo tests, carried out through subcutaneous injection of wild protein and conjugates into nude mice, indicated that pHPMA effectively increased proteolytic activity of the targeting protein.
Scheme 3.

Chemical structures of polymer 1, hydroxyl-terminated polyHPMA and polymer 2, thiazolidine-2-thione-terminated polyHPMA and conjugation with lysozyme. (2 column fitting image)
2.2 Multimeric Conjugation
2.2.1 Homotelechelic Polymer-protein Conjugates
Most of the protein-polymer conjugates made are associated to only one protein. The reality is that in nature, the majority of proteins exist in their multimer form, many of which exist as homodimers [47]. Consequently, it is important to be able to mimic these super-structures to potentially retain protein function in the conjugate state. Thus, tethering more than one protein to a polymeric scaffold would provide a technology with superior performance (Structure II in Figure 1).
To date, polymers with the same end group have been synthesized, introducing the possibility for several applications [48, 39]. Early reports on the preparation of conjugates from homotelechelic polymers showed the use of oligo(ethylene glycol) having azido and amino groups as starting materials [49, 50]. However, to the best of our knowledge, controlled radical polymerization was first employed to synthesize homodimeric protein conjugates in 2009 [51]. Here, a difunctional pNIPAAM polymer was synthesized by RAFT polymerization using a bistrithiocarbonate chain transfer agent (CTA) and a small percentage of nitrobenz-2-oxa-1,3-diazole (NBD) monomer (fluorescent marker), as shown in Scheme 4. It is important to achieve control over the polymer molecular weight as the polymeric spacer length can drastically affect the activity of the protein dimers [51, 52]. After extensive purification by dialysis, the conjugate Mn was found to be 8,990 to 11,500 Da, as determined by GPC, ESI and MALDI-TOF techniques, suggesting that the purified polymer had the expected symmetric structure. Dimeric structures were also verified by carrying out aminolysis of the polymer with butylamine. Once the desired dimeric polymer was obtained, radical addition of a protein reactive azo-initiator was introduced to the polymer chain ends. This initiator contained maleimide functional groups. Under mild acidic conditions, this group can react with cysteines faster than with amines, thus making this a convenient functional group for selective cysteine conjugation [53]. V131C mutant T4 lysozyme (T4L) was selected as the model protein to investigate conjugation efficiency, given that this protein contains one free cysteine. The bioconjugation reaction was carried out at 4 °C and the resulting conjugates were analyzed by SDS-PAGE, which showed new bands at molecular weight ~26 kDa (single protein-polymer conjugate, 79%) and 48 kDa (dimer protein-polymer conjugate, 21%) compared to unmodified lysozyme (18 kDa). Removal of the monomer adduct was performed with a standard cation exchange resin.
Scheme 4.

Schematic representation of homotelechelic protein conjugate formation. (Single column fitting image)
2.2.2 Heterotelechelic Polymer-protein Conjugates
When dual protein conjugation is possible, homotelechelic polymers (Structure II in Figure 1) have certain functional advantages over semi-telechelic polymers. However, chemoselective end groups can be appropriate for available functional groups in a specific protein, but not necessarily for two different proteins. To address this need, heterotelechelic polymers containing two different chemospecific chain end groups have been developed [54–56]. These polymers have the ability to incorporate two different proteins that are not necessarily attached through the same amino acid functional group (Structure III in Figure 1). This method allows for the immobilization of proteins that have different pharmacokinetic/therapeutic or catalytic functions. An example of this versatile multimeric technology involved the synthesis of a RAFT agent bearing an azide end chain fragment and a dithiopyridine fragment, which was used for the polymerization of N-isopropylacrylamide (NIPAAm), styrene, and oligo(ethyle glycol) acrylate (Scheme 5). This polymer was further functionalized with biotin/avidin, glutathione, and bovine serum albumin (BSA) biomolecules via click and thiol-disulfide exchange chemistries [59]. Similarly, synthesis of a heterotelechelic biotin-maleimide pNIPAAm polymer was achieved for the formation of streptavidin (SAv)-BSA polymer conjugates [60].
Scheme 5.

Heterotelechelic polymer conjugation with Biotin and BSA or Glutathione.
More recently, this same technology was optimized through incorporation of a disulfide linkage between the biotin functional group and the maleimide-terminated polymer [57] (Scheme 6). This functional group was integrated into the polymer in order to achieve reversible modification of surfaces with proteins, demonstrating tolerance to further reactions. Post-polymerization, maleimide was installed via radical coupling and removal of a trithiocarbonate moiety. The ability of the functional end groups to be conjugated to proteins was confirmed using a mutant V131C T4 lysozyme (T4L). Conjugation of this protein to the maleimide group without alteration to the disulfide bond was confirmed by SDS-PAGE. The crude conjugate was then incubated with SAv protein for conjugation through the biotin functionality. SDS-PAGE analysis indicated an increase in molecular weight from 37 kDa (T4L conjugate) to 125 kDa (with SAv conjugated). Reversible protein immobilization on SAv anchored surface was tested, demonstrating conjugate release after addition of a reducing agent, tris(2-carboxyethyl)phosphine hydrochloride (TCEP).
Scheme 6.

Schematic representation of polymer-protein conjugate formation and immobilization with a second protein via biotin-SAv interaction. (2 column fitting image)
Also, a recent report showed a modified bifunctional RAFT agent, S,S-bis[α ′-dimethyl- ″-(2-pyridyldisulfide)ethyl acetate] trithiocarbonate (BDPET), which resulted in a macro-RAFT agent modified with two pyridyl disulfide groups [58]. BSA was incubated with the polymer resulting in 50% conjugation. After obtaining the protein-polymer conjugate, polymerization with water soluble oligo(ethylene glycol) acrylate (OEG-A) was performed. This conjugate was successfully further functionalized with thiocholesterol and rhodamine B using the available pyridyl disulfide group.
2.2.3 Branched and Star Polymer-protein Conjugates
Protein conjugation with branched polymers (Structure IV in Figure 1) seems to provide a special masking of the protein through the formation of an umbrella-like structure, which allows the protein to potentially have a longer circulation time and consequently, a better therapeutic performance [61]. Several proteins have been conjugated with these so-called “second generation” PEGylation agents, which have been shown to exhibit improvements in protein stability and performance [62–65]. An interesting variation of these structures involves a polymer with the conjugating moiety located at the center of the polymer chain. Here, pyridyldisulfide is placed at the middle of poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA) for conjugation to BSA through cysteine [61]. The branched polymer structure was confirmed by inducing polymer hydrolysis by refluxing with hexylamine. The resultant polymer was analyzed by GPC through comparison with the original polymer. The conjugation reaction was carried out at pH 7.4 with excess polymer for 20 hours and the product was analyzed using SDS-PAGE. Conjugate formation was confirmed by observing an increase in the molecular weight of the polymer after BSA addition. Nonetheless, the incomplete consumption of the free thiols was ascribed to steric hindrance, since the functional group is in the middle of the polymer. After dissociation of the conjugate by cleavage of the disulfide linkages using mercaptoethanol, the protein band was again observed on the gel, indicating protein release.
Another method of improving protein performance is by the formation of star conjugates (Structure V in Figure 1). In 2009, a multimeric star polymer based on poly(NIPAAm) was successfully conjugated to lysozyme [66]. Maleimide functional groups were introduced in the star polymer arms, allowing site-specific conjugation of the lysozyme to the polymer, thus generating a star (multimeric) polymer-protein conjugate (Scheme 7). These conjugates were characterized using electrospray ionization gas-phase electrophoretic mobility macromolecule analysis (ESI-GAMAA), MALDI-TOF, SDS-PAGE and liquid chromatography tandem mass spectrometry (LC-MS/MS) of trypsin-digested conjugates, confirming successful covalent attachment of the protein to the star polymer.
Scheme 7.
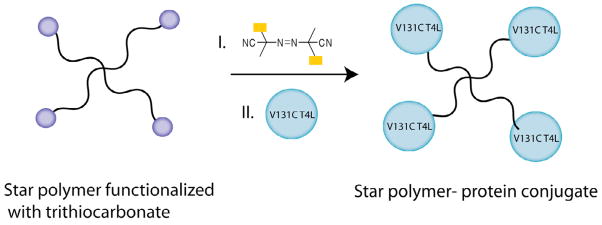
Schematic representation of star protein-polymer conjugate formation. (Single column fitting)
3. Protein Conjugation with Polymeric Nanostructures or Nanostructure Assembly from Polymer-protein Conjugates
Advances in nanotechnology have made nanoparticles an important part of the construction of therapeutic delivery systems. For instance, nanoparticles can be functionalized with receptor ligands to increase specificity. These provide a promising platform for advantageous use of the enhanced permeation and retention (EPR) effect when delivering therapeutics to cancer cells [67]. Several polymeric nanostructures have been developed for protein conjugation, including dendrimers and crosslinked nanogels (Structures III and IV in Figure 2). Although literature in this topic is more limited than polymer-protein conjugates, likely because it is in its early stages, it is worth discussing advances in this area and the potential polymeric nanostructures have as novel conjugate systems. Even though systems that conjugate proteins by physical adsorption exist [68–73], we will limit our discussion to covalent conjugation.
Figure 2.
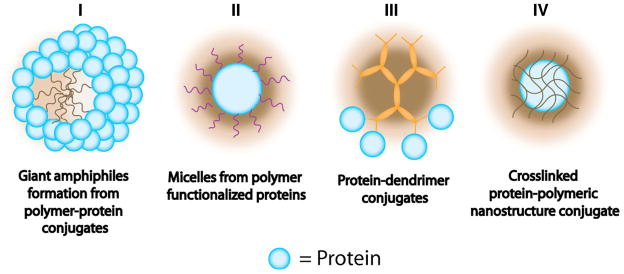
Representation of different types of polymeric nanostructure-protein conjugates. (2 column fitting image)
3.1 Formation of Protein Conjugate Amphiphilic Nanoassemblies
As far as the construction of well-ordered nanoscale amphiphiles from protein conjugates is concerned, giant amphiphile chimeras should be highlighted (Structure I in Figure 2). The so-called “giant amphiphiles” are amphiphilic chimeras formed from polymer-protein conjugates, in which its amphiphilic properties are provided by a diblock copolymer where one of the blocks is the protein [74]. As in many conjugation techniques, the strategy for the synthesis of giant amphiphiles also requires specific reaction steps and conditions to obtain well-defined amphiphilic conjugates [74].
It has been shown that monodisperse polymer-protein hybrids, composed of lipase B protein and a maleimide functionalized polystyrene polymer, self-assemble in water [75]. The covalent conjugation was possible by complete reduction of the single disulfide bridge Cys293–Cys311 exposed at the surface of the enzyme followed by reaction with the maleimide-functionalized polymer. In order to obtain well-defined structures, the coupling reaction was carried out at the air-water interface using a Langmuir-Blodgett trough. Dispersion of the resulting bioconjugates revealed the formation of giant amphiphiles (rods and bundles of rods) with diameter ranging between 25 and 30 nm.
The construction of a bioconjugate based on BSA and polystryrene was reported in 2008 [76]. The polystyrene provides an amphiphilic character to the bioconjugate, thus leading to the formation of the block copolymer type amphiphile, which was characterized by SEC (size exclusion chromatography), UV spectroscopy, MALDI-TOF and TEM (transmission electron microscopy). TEM images confirmed the formation of spherical aggregates, similar to those observed for conventionally synthesized giant amphiphiles with diameters of 20 to 100 nm. The versatility of this technique on the formation of giant amphiphiles was applied to other proteins, including human serum albumin and reduced human calcitonin.
In a more recent work, the same group presented the combination of ATRP with “click”, copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC), for the in situ formation of novel multifunctional bioconjugates [77]. They claimed that this approach should avoid independent polymer synthesis and help with the control of bioconjugate polydispersity and purification. BSA was chosen as a model protein, taking advantage of its accessible Cys34 amino acid that allowed for specific conjugation. Their novel strategy gave rise to three different amphiphilic bioconjugates (Figure 3) that were synthesized by three different synthetic routes (Route A, B and C). Route A consists of a one pot approach, in which they utilized prop-2-ynyl methacrylate as a monomer in the presence of an azide (benzyl azide or azido-triethylene glycol) to achieve simultaneous CuAAC and living radical polymerization. A sequential ATRP and CuAAC were applied in Route B, as well as Route C. However, in the case of Route C an intermediate deprotection step is included and trimethylsilyl protected propargyl methacrylate was used as a monomer. Polymerization reactions were performed in the presence of a fluorescent dye carboxyfluorescein (CF) to demonstrate the ability of the resulting amphiphiles to self-assemble and form nanocontainers, thus encapsulating CF within its interior. Their investigations supported the efficiency of Route A over Routes B and C in the formation of bioconjugates. Gel electrophoresis and TEM, along with other characterization techniques, validated the formation of well-defined giant amphiphiles from I, II, and III (Figure 3), with sizes ranging from 30 to 150 nm. Confocal Microscopy (CFM) corroborated the statistical encapsulation of CF fluorophore within the amphiphiles.
Figure 3.

Giant amphiphile formation from three different protein-polymer conjugates. (2 column fitting image)
The development of a polymersome nanoreactor assembly system that specifically placed enzymes at three different locations in a well-designed manner was reported in 2009 [78]. The porous polymersomes consisted of block copolymers of isocyanopetides and styrene, and were capable of incorporating glucose oxidase (GOx) in the lumen, Candida antartica lipase B (CalB) in the bilayer membrane and horseradish peroxidase (HRP) on the surface. In order to incorporate HRP on acetylene-based anchors on the polymersome surface, the lysine amino acids of the protein were first modified with azido functional groups by direct azido transfer reaction. More than 90% of the polymersome surface was successfully modified with HRP protein, keeping the polymersome morphology unaffected. 25% of GOx was incorporated in the lumen and the bilayer membrane incorporated approximately 17% of CalB enzyme. These nanoreactors were capable of performing a three-cascade enzyme catalysis reaction, in which CaIB converted glucose acetate to glucose, and in a second step, was then oxidized by GOx to gluconolactone. The produced hydrogen peroxide was used by HRP to oxidize 2,2′ azinobis(3-ethylbenzothiazoline-6-sulfonic acid). TEM images revealed the presence of polymersomes with diameters ranging from 90 to 180 nm.
Proteins can also be conjugated to polymers in order to use them as part of the macromolecular scaffold (Structure II in Figure 2) for drug delivery. For instance, a novel denatured bovine serum albumin (dBSA) platform for the delivery of hydrophobic small molecule camptothecin drug (CPT) has been developed [79]. Denaturation of the protein would enhance the CPT adsorption and loading, on the other hand, denaturation would also decrease its solubility in water. To overcome any issues with solubility, the protein was conjugated with methoxy(propylene glycol) benzaldehyde (m-PEG-CHO) via reaction of CHO groups and protein lysine amino acids followed by reduction of sodium cyanoborohydride. Self-assembly of core-shell micelles of hydrophobic denatured BSA and hydrophilic PEG groups was targeted in aqueous solution. The lipophilic CPT was physically adsorbed into the hydrophobic core. Successful protein conjugation was characterized by nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy (FT-IR). The micelle formation was confirmed by TEM and DLS. The hydrodynamic diameter of mPEG-dBSA assembly was 238 nm and interestingly, CPT loaded mPEG-dBSA showed a smaller size of 150 nm. This smaller size is possibly due to a compaction of the nanoparticle upon drug loading. It was demonstrated that intracellular internalization of dBSA-conjugated nanoparticles was more efficient than the protein itself.
3.2 Formation of Multivalent Protein Conjugate Nanostructures
Covalent conjugation of proteins on the surface of polymeric beads has been reported for the development of an efficient protein delivery carrier [80]. Here, the polymeric beads were coated with glucose molecules that serve to facilitate the cellular uptake of the carrier, since glucose can establish interactions with the cell membrane [81]. Small and monodisperse polymeric beads were obtained in a single step by dispersion polymerization. Glucose-coated beads were prepared using 6-O-glucosylmethacrylate, acrylic acid and styrene as the reagents. Glucose was ligated to methacrylate by CalB, followed by polymerization with styrene and acrylic acid in aqueous methanol to form 150 nm beads. Free amines on EGFP were conjugated with the available acrylic acid functional group by EDC coupling. Protein conjugation to the polymeric beads provides an alternative to genetic hybridization of protein transduction domains to the target protein. Nonetheless, this process can be affected due to the small glucose size relative to the large protein size. Although the surface-bound protein bead can slightly shield the glucose molecule, it was found that the glucose unit could still interact with the cell membrane to facilitate transduction. EGFP was successfully delivered into mES and HeLa cells by the glucose coated polymeric beads.
In 2011, a semi-synthetic multivalent collagen binding protein was obtained by covalent chemical ligation of a recombinant CNA35 protein-thioester with cysteine functionalized divalent (AB2) and tetravalent (AB4) dendrimers [82]. This dendritic system has been previously shown to have great multivalent properties (Structure III in Figure 2), since control over valence, spacer length and attachment of bio-orthogonal reactive functional groups is possible [83]. Native chemical ligation of a 2-mercaptoethanesulfonate (MESNA)-functionalized wild type CNA35 and a weak binding variant CNA35-Y175K to a first generation cysteine functionalized poly(propylene imine) (PPI) dendrimer was achieved (Figure 4) using an IMPACT system described in a previous report [84]. An eightfold molar excess of CNA35-MESNA was incubated overnight with AB2 and AB4 in order to push the reaction to completion. The reaction was performed in the presence of a catalyst, (4-carboxylmethyl) thiophenol (MPAA), which has previously been shown to successfully catalyze the reaction for the preparation of multivalent CNA35 micelles and liposomes [84]. The branched structures of (CNA35)2 were observed on SDS-PAGE as a band at 75 kDa and interestingly, protein ligation using AB4 resulted in two ligation products, namely (CNA35)3 and (CNA35)4, that appeared at higher molecular weights. Unfortunately, AB4 did not exhibit complete conversion however, it did represent an improvement over a previously reported GFP-dendrimer conjugation [85]. These CNA35 dendrimers were tested against collagen mimicking surfaces in order to test the main design elements required for effective multivalent targeting of collagen. This system showed remarkable collagen binding properties with an enhanced affinity of “non-binding” collagen binding protein (CNA35-Y175K).
Figure 4.

Chemical structure representation of AB2-CNA and AB4-CNA35 conjugates. (2 column fitting image)
3.3 Protein Conjugates Formed from Crosslinked Nanostructures
Direct conjugation to nanoassemblies has also been achieved as an alternative approach for providing more stable protein conjugates (Structure IV in Figure 2). In 2003, a single protein encapsulation was attempted in a multistep procedure within a nanoparticle [86]. The protein encapsulation strategy consisted of an enzyme surface amino group modification with acryoyl chloride to yield vinyl groups, followed by vinyl polymer growth from the enzyme surface. An orthogonal polymerization, by silanol condensation reactions, led to the crosslinking of the attached polymers, thus creating a polymeric network surrounding -chymotrypsin (ChT). This modification was shown to provide enhanced enzyme stability at an insignificant increase in mass transfer resistance.
In a similar, but simpler, approach another research group [87] fabricated a two step procedure including i) protein surface acryloylation to generate vinyl groups and ii) in situ polymerization using acrylamide as the monomer and N,N′-methylene bisacrylamide as the crosslinker in an aqueous solution for single protein (horseradish peroxidase, HRP) encapsulation, as presented in Scheme 8. With repetition of the second step, the size and shape of the modified enzyme can be tuned, by adding monomers and crosslinkers to react with the primary nanogel, giving the desired product. Nanogel–protein conjugates, prepared with a) monomer/HRP molar ratio of 400 (NG-A) and b) monomer/HRP molar ratio of 800 (NG-B), were characterized by TEM. NG-A was determined to be ellipsoidal with size 11 nm × 9 nm. HRP is elliptical with dimensions of 6.5 nm × 5.4 nm × 4.3 nm, suggesting that each NG-A contains a single enzyme with a polymer shell of thickness around 2.4 nm. On the other hand, NG-B shows a diameter of 17 nm with a more uniform spherical shape and a shell thickness of 5 nm. Biocatalytic activity of the encapsulated enzyme was examined against free HRP by investigating Km and kcat values. It was observed that free enzyme Km and kcat values (0.297 mM and 2187 s−1) were similar to the encapsulated HRP (0.307 mM and 2037 s−1) in NG-B. These results suggest that the thin polymer shell does not significantly affect the enzyme kinetics of HRP. In addition, the enzyme presented enhanced thermal stability due to multiple covalent attachments to the polymer, hindering the thermal fluctuations, which usually lead to enzyme deactivation.
Scheme 8.

Encapsulation of single HRP protein in nanogel. (single column fitting image)
In 2010, the same group designed another single-protein nanocapsule shell covalently anchored to the protein surface [88]. In this work the protein was also covalently modified with polymerizable vinyl groups. Subsequent polymerization was performed by the addition of either neutral (acrylamide) or positively charged (2-dimethylaminoethyl methacrylate) monomer, followed by the addition of a non-degradable or a degradable crosslinker. Enhanced green fluorescent protein (EGFP), horseradish peroxidase (HRP), bovine serum albumin (BSA), superoxide dismutase (SOD), and caspase-3 (CAS) were used as model proteins for encapsulation in the non-degradable nanocapsule (n-Protein) or in the degradable nanocapsule (de-nProtein). Nanocapsules (NCs) were characterized by TEM showing 15 nm spherical particles, which were also confirmed by dynamic light scattering (DLS) measurements. HRP protein was labeled with Au nanoparticles and encapsulated in the NCs in order to quantify encapsulated protein per nanocapsule. Single protein encapsulation was observed; since the protein radius is 5 nm, the average shell thickness was determined to be around 5 nm as well, creating a thin polymeric skin surrounding the protein. Intracellular protein delivery was performed resulting in long-term stability for the non-degradable NCs compared to the degradable NCs, in which the protein polymeric shell was degraded under acidic conditions, enabling the protein to be active once inside the cell. This method was utilized for multiple concurrent protein delivery, showing great potential for synergistic protein therapy. In vivo experiments in mice were performed in order to investigate nanocapsule stability. Mice organs showed intense fluorescence after n-EGFP injection at 8 hours, prolonging to 50 hours after injection. Activity and cytotoxicity was assessed in vivo as well, demonstrating that the delivered protein is active once released presenting low toxicity.
4. Concluding Remarks
In this review, we have summarized the key advances in polymer-protein and nanostructure-protein bioconjugates. Thanks in part to the application of synthetic techniques such as ATRP and RAFT polymerization, well-controlled linear polymer-protein bioconjugates have been developed. Advances and variations of these methods have also lead to a variety of designs such as semi-, homo-, heterotelechelic and branched bioconjugates. Albeit elegant, these methods do lack the simplicity needed for elaborating these methods to a broad range of proteins and polymeric scaffolds. Similarly, much effort has been put on finding more efficient methods for successful protein conjugation on stimuli (or non stimuli) responsive polymeric structures. This inspiration for this design has been in the area of protein delivery. However, factors that control the delivery of the proteins into cells, release of these proteins in response to a stimulus, bioavailability of these proteins and biodegradability of these polymers are yet to be carefully addressed. Considering the vibrant activity in the area of biopolymers, nanostructured polymer assemblies, and bioconjugation strategies, this area of research is well poised to make a significant impact.
Highlights.
The review highlights recent advances in protein-polymer conjugation.
The review presents different models for covalent conjugation of proteins with linear polymers.
The review examines advances in the development of protein conjugates using nanostructures.
The review also examines the protential future work in the polymer-protein bioconjugation field.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Neutsch L, Eggenreich B, Herwig E, Marchetti-Deschmann M, Allmaier G, Gabor F, Wirth M. Lectin Bioconjugates Trigger Urothelial Cytoinvasion – A Glycotargeted Approach for Improved Intravesical Drug Delivery. Eur J Pharm Biopharm. 2012;82(2):367–375. doi: 10.1016/j.ejpb.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 2.Hadley KB, Sato PH. Catalytic Activity of Administered Gulonolactone Oxidase Polyethylene-glycol Conjugates. Enzyme. 1989;42(4):225–234. doi: 10.1159/000469036. [DOI] [PubMed] [Google Scholar]
- 3.Veronese FM. Peptide and Protein PEGylation: A Review of Problems and Solutions. Biomaterials. 2001;22(5):405–417. doi: 10.1016/s0142-9612(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 4.Schiavon O, Caliceti P, Ferruti P, Veronese FM. Therapeutic Proteins: A Comparison of Chemical and Biological Properties of Uricase Conjugated to Linear or Branched Poly(ethylene glycol) and poly(N -acryloylmorpholine) IL Farmaco. 2000;55(4):264–269. doi: 10.1016/s0014-827x(00)00031-8. [DOI] [PubMed] [Google Scholar]
- 5.Hartmann-Petersen R, Gordon C. Quantifying Protein-Protein Interactions in the Ubiquitin Pathway by Surface Plasmon Resonance. Methods Enzymol. 2005;399:164–177. doi: 10.1016/S0076-6879(05)99011-3. [DOI] [PubMed] [Google Scholar]
- 6.Sapsford KE, Medintz IL, Golden JP, Deschamps JR, Uyeda HT, Mattoussi H. Surface-immobilized Self-assembled Protein-based Quantum Dot Nanoassemblies. Langmuir. 2004;20(18):7720–7728. doi: 10.1021/la049263n. [DOI] [PubMed] [Google Scholar]
- 7.Zimmermann J, Kwak M, Hermann A. Amphiphilic DNAblock Copolymers: Nucleic Acid-Polymerhybrid Materials for Diagnostics and Biomedicine. Methods Mol Biol. 2011;751:239–266. doi: 10.1007/978-1-61779-151-2_15. [DOI] [PubMed] [Google Scholar]
- 8.Baumann P, Tanner P, Onaca O, Palivan CG. Bio-decorated Polymer Membranes: A New Approach in Diagnostics and Therapeutics. Polymers. 2011;3:173–192. [Google Scholar]
- 9.Savariar EN, Ghosh S, Gonzalez DC, Thayumanavan S. Disassembly of Non-covalent Amphiphilic Polymers with Proteins and Utility in Pattern Sensing. J Am Chem Soc. 2008;130(16):5416–5417. doi: 10.1021/ja800164z. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Toro DC, Savariar EN, Thayumanavan S. Fluorescence Patterns from Supramolecular Polymer Assembly and Disassembly for Sensing Metallo and Nonmetalloproteins. J Am Chem Soc. 2009;131(22):7708–7716. doi: 10.1021/ja900579g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyer C, Huang X, Whittaker MR, Bulmus V, Davis TP. An Overview of Protein-Polymer Particles. Soft Matter. 2011;7(5):1599–1614. [Google Scholar]
- 12.Rodthongkum N, Ramireddy R, Thayumanavan S, Vachet RW. Selective Enrichment and Sensitive Dtection of Peptide and Protein Biomarkers in Human Serum Using Polymeric Reverse Micelles and MALDI-MS. Analyst. 2012;137(4):1024–1030. doi: 10.1039/c2an16089g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodthongkum N, Chen Y, Thayumanavan S, Vachet RW. MALDI-MS Signal Enhancement of Peptides after Selective Extraction with Polymeric Reverse Micelles. Anal Chem. 2010;82(9):3686–3691. doi: 10.1021/ac1000256. [DOI] [PubMed] [Google Scholar]
- 14.Azagarsamy MA, Gomez-Escudero A, Yesilyurt V, Vachet RW, Thayumanavan S. Amphiphilic Nanoassemblies for the Detection of Peptides and Proteins Using Fluorescence and Mass Spectrometry. Analyst. 2009;134(4):635–649. doi: 10.1039/b818484d. [DOI] [PubMed] [Google Scholar]
- 15.Wang EA. Bone Morphonetic Proteins (BMPs): Therapeutic Potential in Healing Bony Defect. Trends Biotechnol. 1993;11(9):379–383. doi: 10.1016/0167-7799(93)90096-R. [DOI] [PubMed] [Google Scholar]
- 16.Löfbloom J, Feldwisch J, Tolmachev V, Carlsson J, Stahl S, Frejd FY. Affibody Molecules: Engineered Proteins for Therapeutic, Diagnostic and Biotechnological Applications. FEBES Lett. 2010;584(12):2670–2680. doi: 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Baldwin SP, Saltzman WM. Materials for Protein Delivery in Tissue Engineering. Adv Drug Delivery Rev. 1998;33(1–2):71–86. doi: 10.1016/s0169-409x(98)00021-0. [DOI] [PubMed] [Google Scholar]
- 18.Kalia J, Raines RT. Advances in Bioconjugation. Curr Org Chem. 2010;14(2):138–147. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broyer RM, Grover GN, Maynard HD. Emerging Synthetic Approaches for Protein-Polymer Conjugations. Chem Commun. 2011;47(8):2212–2226. doi: 10.1039/c0cc04062b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cha T, Guo A, Zhu XY. Enzymatic Activity on a Chip: The Critical Role of Protein Orientation. Proteomics. 2005;5(2):416–419. doi: 10.1002/pmic.200400948. [DOI] [PubMed] [Google Scholar]
- 21.Veronese FM, Pasut G. PEGylation, successful Approach to Drug Delivery. Discov Today. 2005;10(21):1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 22.Bontempo D, Heredia KL, Fish BA, Maynard HD. Cysteine-Reactive Polymers Synthesized by Atom Transfer Radical Polymerization for Conjugation to Proteins. J Am Chem Soc. 2004;126(47):15372–15373. doi: 10.1021/ja045063m. [DOI] [PubMed] [Google Scholar]
- 23.Lundblad RL. Techniques in Protein Modification. CRC Press; Boca Raton, FL: 1995. [Google Scholar]
- 24.Chilkoti A, Chen G, Stayton PS, Hoffman AS. Site-specific Conjugation of a Temperature-sensitive polymer to a Geneticallu-engineered protein. Biocong Chem. 1994;5(6):504–507. doi: 10.1021/bc00030a004. [DOI] [PubMed] [Google Scholar]
- 25.Olsen BD, Kornfield JA, Tirrell DA. Yielding Behavior in Injectable Hydrogels from Telechelic Proteins. Macromol. 2010;43(21):9094–9099. doi: 10.1021/ma101434a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maskarinec SA, Tirrell DA. Protein Engineering Approaches to Biomaterials Design. Curr Opin Biotechnol. 2005;16(4):422–426. doi: 10.1016/j.copbio.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Rana S, Yehand Y-C, Rotello VM. Engineering the Nanoparticle-Protein Interface: Application and Possibilities. Curr Opin Chem Biol. 2010;14(6):828–834. doi: 10.1016/j.cbpa.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson S. Small-molecule-Protein Conjugation Procedures. Methods Mol Med. 2004;94:255–265. doi: 10.1385/1-59259-679-7:255. [DOI] [PubMed] [Google Scholar]
- 29.Grover GN, Maynard HD. Protein-polymer Conjugates: Synthetic Approaches by Controlled Radical Polymerizations and Interesting Applications. Curr Opin Chem Biol. 2010;14(6):818–827. doi: 10.1016/j.cbpa.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Droumaguet B, Nicolas J. Recent Advances in the Design of Bioconjugates from Controlled/living Radical Polymerization. Polym Chem. 2010;1(5):563–598. [Google Scholar]
- 31.Kalia J, Raines RT. Advances in Bioconjugation. Curr Org Chem. 2010;14(2):138–147. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Broyer RM, Grover GN, Maynard HD. Emerging Synthetic Approaches for Protein-Polymer Conjugations. Chem Commun. 2011;47(8):2212–2226. doi: 10.1039/c0cc04062b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ratner M. Progress With PEGylation. Nature Biotechnol. 1990;8(4):279–281. [Google Scholar]
- 34.Kodera Y, Matsushima A, Hiroto M, Nishimura H, Ishii A, Ueno T, Inada Y. PEGylation of Proteins and Bioactive Substances for Medical and Technical Applications. 1998;23(7):1233–1271. [Google Scholar]
- 35.Joralemon MJ, McRae S, Emrick T. PEGylated Polymers for Medicine: From Conjugation to Self-assembled Systems. Chem Comm. 2010;46(9):1377–1393. doi: 10.1039/b920570p. [DOI] [PubMed] [Google Scholar]
- 36.Pasut G, Veronese FM. State of the Art in PEGylation: The Great Versatility Achieved After Forty Years of Research. J Controlled Release. 2012;161(2):461–472. doi: 10.1016/j.jconrel.2011.10.037. [DOI] [PubMed] [Google Scholar]
- 37.Harris JM, Martin NE, Modi M. PEGylation- A Novel Process for Modifying Pharmacokinetics. Clin Pharmacokin. 2001;40(7):539–551. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- 38.Tao L, Giuseppe M, Lecolley F, Haddleton DM. Aldehyde Terminally Functional Methacrylic Polymers from Living Radical Polymerization: Application in Protein Conjugation “Pegylation”. J Am Chem Soc. 2004;126(41):13220–13221. doi: 10.1021/ja0456454. [DOI] [PubMed] [Google Scholar]
- 39.Grover GN, Lee J, Matsumoto NM, Maynard HD. Aminooxy and Pyridyl Disulfide Telechelic Poly(poly(ethylene glycol)acrylate) by RAFT Polymerization. Macromolecules. 2012;45(12):4958–4965. doi: 10.1021/ma300575e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lai J, Filla D, Shea R. Functional Polymers from Novel Carboxyl-terminated Trithiocarbones as Highly Efficient RAFT Agents. Macromolecules. 2002;35(18):6754–6756. [Google Scholar]
- 41.Iskin B, Yilmaz G, Yagci Y. Telechelic Polymers by Visible Light Induced Radical Coupling. Macromol Chem Phys. 2013;214(1):94–98. [Google Scholar]
- 42.Bdkhe RB, Stafslien SJ, Cilz N, Daniels J, Thompson SEM, Callow ME, Callow JA, Webster DC. Polyurethanes with amphiphilic Surfaces made Using Telechelic Functional PDMS Having Orthogonal Acid Functional Groups. Prog Org Coat. 2012;75(1–2):38–48. [Google Scholar]
- 43.Heredia KL, Bontempo D, Ly T, Byers JT, Halstenberg S, Maynard HD. In Situ Preparation of Protein-“Smart” Polymer Conjugates with Retention of Bioactivity. J Am Chem Soc. 2005;127(48):16955–16960. doi: 10.1021/ja054482w. [DOI] [PubMed] [Google Scholar]
- 44.Osserman EF, Canfield RE, Begchok S. Lysozyme. New York: Academic press; 1974. [Google Scholar]
- 45.Treetarnmathurot B, Dieudonné L, Ferguson E, Schmaljohann D, Duncan R, Wiwattanapatapee R. Dextrin-trypsin and ST-HPMA-trypsin conjugates: Enzyme Activity, Autolysis and Thermal Stability. Int J Pharm. 2009;373(1–2):68–76. doi: 10.1016/j.ijpharm.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 46.Tao L, Chen G, Zhao L, Xu J, Huang E, Liu A, Marquis CP, Davis TP. Protein Release from Biodegradable PolyHPMA-Lysozyme Conjugates Resulting in Bioactivity Enhancement. Chem Asian J. 2011;6(6):1398–1404. doi: 10.1002/asia.201000729. [DOI] [PubMed] [Google Scholar]
- 47.Mariananyagam NJ, Sunde M, Matthews JM. The Power of Two: Protein Dimerization in Biology. Trends Biochem Sci. 2004;29(11):618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 48.You Y-Z, Manickam DS, Zhou Q-H, Oupicky D. A versatile Approach to Reducible Vinyl Polymers Via Oxidation of Telechelic Polymers Prepared by Reversible Addition Fragmentation Chain Transfer Polymerization. Biomacromolecules. 2007;8(6):2038–2044. doi: 10.1021/bm0702049. [DOI] [PubMed] [Google Scholar]
- 49.Bertozzi CR, Bednarski MD. The Synthesis of Heterobifunctional Linkers for the Conjugation of Ligands to Molecular Probes. J Org Chem. 1991;56(56):4326–4329. [Google Scholar]
- 50.Iyer SS, Anderson AS, Reed S, Swanson B, Schmidt JG. Synthesis of Orthogonal End Functionalized Oligoethylene Glycols of Defined Lengths. Tetrahedron Lett. 2004;45(22):4285–4288. [Google Scholar]
- 51.Tao L, Kaddis CS, Loo RRO, Grover GN. Synthetic Approach to Homodimeric Protein-polymer Conjugates. Chem Commun. 2009;(16):2148–2150. doi: 10.1039/b822799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kramer RH, Karpen JW. Spanning Binding Sites on Allosteric Proteins with Polymer-linked Ligand Dimers. Nature. 1998;395(6703):710–713. doi: 10.1038/27227. [DOI] [PubMed] [Google Scholar]
- 53.Mather BD, Viswananthan K, Miller KM, Long TE. Michael Addition Reactions in macromolecular Design for Emerging Technologies. Prog Polym Sci. 2006;31(5):487–531. [Google Scholar]
- 54.Yang J, Luo K, Pan H, Kope ková P, Kopeek J. Synthesis of Biodegradable Multiblock Copolymers by Click Coupling of RAFT-generated Heterotelechelic polyHPMA Conjugates. React Funct Polym. 2011;71(3):294–302. doi: 10.1016/j.reactfunctpolym.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kopeek J. Polymer-drug Conjugates: Origins, Progress to Date and Future Directions. Adv Drug Deliver Rev. 2012 doi: 10.1016/j.addr.2012.10.014. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masfeld U, Winter A, Hager MD, Hoogenboom R, Günther W, Schubert US. Orthogonal Self-assembly of Stimuli-responsive Supramolecular Polymers Using One-Step Prepared Heterotelechelic Building Blocks. Polym Chem. 2013;4(1):113–123. [Google Scholar]
- 57.Heredia KL, Tao L, Grover GN, Maynard HD. Heterotelechelic Polymers for Capture and Release of Protein-polymer Conjugates. Polym Chem. 2010;1(2):168–170. doi: 10.1039/B9PY00369J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu J, Liu H, Bulmus V, Tao L, Boyer C, Davis T. A Simple Methodology for the Synthesis of Heterotelechelic Protein-Polymer-Polymer-Biomolecule Conjugates. J Polym Sci Pol Chem. 2010;48(6):1399–1405. [Google Scholar]
- 59.Boyer C, Liu J, Bulmus V, Davis TP, Barner-Kowollik B, Stenzel MH. Direct Synthesis of Well-defined Heterotelechelic Polymers for Bioconjugations. Macromolecules. 2008;41(15):5641–5650. [Google Scholar]
- 60.Heredia KL, Grover GN, Tao GN, Tao L, Maynard HD. Synthesis of Heterotelechelic Polymers for Conjugation of Two Different Proteins. Macromolecules. 2009;42(7):2360–2367. doi: 10.1021/ma8022712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tao L, Liu J, Davis TP. Branched Polymer-protein Conjugates Made from Mid-Chain-Functional P(HPMA) 2009;10(10):2847–2851. doi: 10.1021/bm900678r. [DOI] [PubMed] [Google Scholar]
- 62.Veronese FM, Monfardini C, Caliceti P, Schiavon O, Scrawen MD, Beer D. Improvement of Pharmacokinetic, Immunological and Stability Properties of Asparaginase by Conjugation to Linear and Branched Monomethoxy Poly(ethylene glycol) J Controlled Rel. 1996;40(3):199–209. [Google Scholar]
- 63.Gao W, Liu W, Christensen T, Zalutsky MR, Chilkoti A. In Situ Growth of a PEG-like Polymer from the C terminus of an Intein Fusion Protein Improves Pharmacokinetics and Tumor Accumulation. PNAS. 2010;107(38):16432–16437. doi: 10.1073/pnas.1006044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brocchini S, Godwin A, Balan S, Choi J-W, Zloh M, Shaunak S. Disulfide Bridge Based PEGylation of Protein. Adv Drug Deliver Rev. 2008;60(1):3–12. doi: 10.1016/j.addr.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 65.Nojima Y, Suzuki Y, Kazuma I, Shiga T, Iwata A, Fujimoto T, Yoshida K, Shimizu H, Takeuchi T, Sato A. Development of Poly(ethylene glycol) Conjugated Lactoferrin for Oral Administration. Bioconjugate Chem. 2008;19(11):2253–2259. doi: 10.1021/bc800258v. [DOI] [PubMed] [Google Scholar]
- 66.Tao L, Kaddis CS, Loo RRO, Grover GN, Loo JA, Maynard HO. Synthesis of Maleimide-End-Functionalized Star Polymers and Multimeric Protein-Polymer Conjugates. Macromolecules. 2009;42(21):8028–8033. doi: 10.1021/ma901540p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the Enhanced Permeability and Retention Effect for Tumor Targeting. Drug Discov Today. 2006;11(17–18):812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 68.Lee Y, Ishii T, Cabral H, Kim HJ, Seo J-H, Nishiyama N, Oshima H, Osada K, Kataoka K. Change-Conversional Polyionic Complex Micelles-Efficient Nanocarriers for Protein Delivery into Cytoplasm. Angew Chem Int. 2009;48(29):5309–5312. doi: 10.1002/anie.200900064. [DOI] [PubMed] [Google Scholar]
- 69.Verma A, Simard JM, Worrall JWE, Rotello VM. Tunable Reactivation of Nanoparticle-Inhibited -Galactosidase by Glutathione at Intracellular Concentrations. J Am Chem Soc. 2004;126(43):13987–13991. doi: 10.1021/ja046572r. [DOI] [PubMed] [Google Scholar]
- 70.Akagi T, Kaneko T, Kida T, Akashi M. Multifunctional Conjugation of Proteins on/into Bio-Nanoparticles Prepared by Amphiphilic Poly( – Glutamic Acid) J Biomater Sci Polymer Edn. 2006;17(8):875–892. doi: 10.1163/156856206777996871. [DOI] [PubMed] [Google Scholar]
- 71.Gonzalez-Toro D, Ryu J-H, Chacko R, Zhuang J, Thayumanavan S. Concurrent Binding and Delivery of Proteins and Lipophilic Small Molecules Using Polymeric Nanogels. J Am Chem Soc. 2012;134(16):6964–6967. doi: 10.1021/ja3019143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sandanaraj BS, Vutukuri DR, Simard JM, Klaikherd A, Hong R, Rotello VM, Thayumanavan S. Noncovalent Modification of Chymotrypsin Surface Using an Amphiphilic Polymer Scaffold: Implications in Modulating Protein Function. J Am Chem Soc. 2005;127(30):10693–10698. doi: 10.1021/ja051947+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sandanaraj SB, Bayraktar H, Krishnamoorthy K, Knapp M, Thayumanavan S. Recognition and Modulation of Cytochrome c’s Redox Properties using an Amphiphilic Homopolymer Langmuir. 2007;23(7):3891–3897. doi: 10.1021/la063063p. [DOI] [PubMed] [Google Scholar]
- 74.Velonia K. Protein-polymer Amphiphilic Chimeras: Recent Advances and Future Challenges. Polym Chem. 2010;1(7):944–952. [Google Scholar]
- 75.Velonia K, Rowan AE, Nolte RJM. Lipase Polystyrene Giant Amphiphiles. J Am Chem Soc. 2002;124(16):4224–4225. doi: 10.1021/ja017809b. [DOI] [PubMed] [Google Scholar]
- 76.Le Droumaguet B, Velonia K. In Situ ATRP-Mediated Hierarchical Formation of Giant Amphiphile Bioreactors. 2008;47(33):6263–6266. doi: 10.1002/anie.200801007. [DOI] [PubMed] [Google Scholar]
- 77.Daskalaki E, Le Droumaguet B, Gérard D, Velonia K. Multifunctional Giant Amphiphiles via Simultaneous Copper(I)-catalized Azide-alkyne Cycloadition and Living Radical Polymerization. Chem Commun. 2012;48:1586–1588. doi: 10.1039/c1cc15075h. [DOI] [PubMed] [Google Scholar]
- 78.Van Dongen SFM, Nallani M, Cornelissen JJLM, Nolte RJM, van Hest JCM. A Three-Enzyme Cascade Reaction Through Positional Assembly of Enzymes in a Polymersome Nanoreactor. Chem Eur J. 2009;15(5):1107–1114. doi: 10.1002/chem.200802114. [DOI] [PubMed] [Google Scholar]
- 79.Zhang L, Lu Z, Li X, Deng Y, Zhang F, Ma C, He N. Methoxy Poly(ethylene flycol) Conjugated Denatured Bovine Serum Albumin Micelles for Effective Delivery of Camptothecin. Polym Chem. 2012;3(8):1958–1961. [Google Scholar]
- 80.Jung S, Hug S, Cheon Y-P, Park S. Intracellular Protein Delivery by Glucose-Coated Polymeric Beads Chem Commun. 2009;(33):5003–5005. doi: 10.1039/b906268h. [DOI] [PubMed] [Google Scholar]
- 81.Freisleben HJ, Fürstenberger HJ, Deisinger S, Freisleben KB, Wiernsperger N, Zimmer G. Interaction of Glucose and Metformin with Isolated Red Cell Membrane. Arzneimittelforschung. 1996;46(8):773–778. [PubMed] [Google Scholar]
- 82.Breuken M, Lempens EHM, Temming RP, Helms BA, Meijer EW, Merkx M. Collagen Targeting Using Multivalent Protein-functionalized Dendrimers. Bioorgan Med Chem. 2011;19(3):1062–1071. doi: 10.1016/j.bmc.2010.07.058. [DOI] [PubMed] [Google Scholar]
- 83.Lempens EHM, Helms BA, Bayles AR, Merkx M, Meijer EW. A versatile, Modular Plarform for Multivalent Peptide Ligands Based on a Dendritic Wedge. Eur J Org Chem. 2010;2010(1):111–119. [Google Scholar]
- 84.Reulen SWA, Dankers PYW, Bomans PHH, Meijer EW, Merkx M. Collagen Targeting Using Protein-Functionalized Micelles: The Strength of Multiple Weak Interactions. J Am Chem Soc. 2009;131(21):7304–7312. doi: 10.1021/ja807723p. [DOI] [PubMed] [Google Scholar]
- 85.Van Baal L, Malda H, Synowsky SA, Van Dongen JLJ, Hackeng TM, Merkx M, Meijer EW. Multivalent Peotide and Protein Dendrimers Using Native Chemical Ligation. Angew Chem Int Ed. 2005;44(32):5052–5057. doi: 10.1002/anie.200500635. [DOI] [PubMed] [Google Scholar]
- 86.Kim J, Grate JW. Single-Enzyme Nanoparticles Armored By a Nanometer- Scale Organic/Inorganic Network. Nano Lett. 2003;3(9):1219–1222. [Google Scholar]
- 87.Yan M, Ge J, Zheng L, Ouyang P. Encapsulation of Single Enzyme in Nanogel with Enhanced Biocatalytic Activity and Stability. J Am Chem Soc. 2006;128(34):11008–11009. doi: 10.1021/ja064126t. [DOI] [PubMed] [Google Scholar]
- 88.Yan M, Du J, Gu Z, Liang M, Hu Y, Zhang W, Priceman S, Wu L, Zhou ZH, Liu Z, Segura T, Tang Y, Lu Y. A Novel Intracellular Protein Delivery Platform Based on Single-Protein Nanocapsules. Nat Nanotechnol. 2010;5(1):48–53. doi: 10.1038/nnano.2009.341. [DOI] [PubMed] [Google Scholar]