

Abstract
A proper DNA damage response (DDR), which monitors and maintains the genomic integrity, has been considered to be a critical barrier against genetic alterations to prevent tumor initiation and progression. The representative tumor suppressor p53 plays an important role in the regulation of DNA damage response. When cells receive DNA damage, p53 is quickly activated and induces cell cycle arrest and/or apoptotic cell death through transactivating its target genes implicated in the promotion of cell cycle arrest and/or apoptotic cell death such as p21WAF1, BAX, and PUMA. Accumulating evidence strongly suggests that DNA damage-mediated activation as well as induction of p53 is regulated by posttranslational modifications and also by protein-protein interaction. Loss of p53 activity confers growth advantage and ensures survival in cancer cells by inhibiting apoptotic response required for tumor suppression. RUNX family, which is composed of RUNX1, RUNX2, and RUNX3, is a sequence-specific transcription factor and is closely involved in a variety of cellular processes including development, differentiation, and/or tumorigenesis. In this review, we describe a background of p53 and a functional collaboration between p53 and RUNX family in response to DNA damage.
1. Introduction
The initial chromatin-associated molecular event upon DNA damage is the activated ataxia telangiectasia mutated- (ATM-) mediated phosphorylation of histone variant H2AX (γH2AX), which marks the sites of DNA damage (nuclear foci) [1]. Then a large nuclear adaptor protein termed mediator of DNA damage response 1 (MDC1)/nuclear factor with BRCT domain 1(NFBD1) associates with the sites of DNA damage and facilitates the recruitment of DNA repair machinery including MRN (MRE11, Rad50, and NBS1) complex onto nuclear foci to repair damaged DNA [2–5]. When cells receive the repairable DNA damage, cell cycle arrest takes place to save time to correctly repair damaged DNA, and then cells with repaired DNA reenter into the normal cell cycle (cell survival). In contrast, when cells receive the severer DNA damage, which cannot be repaired, cells with seriously damaged DNA undergo apoptotic cell death and are then eliminated from tissues (cell death). Thus, the appropriate DNA damage response plays an important role to maintain genomic integrity to avoid genomic aberrations such as deletions and mutations, which result in genomic instability and finally induce tumor formation [6–8].
p53 has been initially identified as a 53 KDa of nuclear protein which tightly associated with oncogenic simian virus 40 (SV40) large T antigen [9–13]. Structural analysis revealed that p53 is composed of three functional domains including NH2-terminal acidic transactivation domain (TA, amino acid residues 1–45), central sequence-specific DNA-binding domain (DB, amino acid residues 102–292), and COOH-terminal oligomerization domain (OD, amino acid residues 319–359) [6–8], suggesting that p53 could act as a nuclear transcription factor. Indeed, p53 had an ability to bind to salmon sperm DNA in vitro [14]. Tetramerization, which is mediated by its oligomerization domain, is essential for the ability of p53 to positively regulate gene expression [15]. In addition to these three characteristic domains, p53 contains three nuclear localization signals (NLS, amino acid residues 305–322, 369–375, and 379–384) [16], a Leu-rich nuclear export signal (NES, amino acid residues 339–352) [17], and a Pro-rich domain (amino acid residues 63–97) [18]. Subsequent studies demonstrated that p53 has an oncogenic potential and has a capacity to promote tumor growth [19]. Intriguingly, p53 was easily detectable in a variety of tumor-derived cell lines, implying that p53 is abundantly expressed in these cancerous cells [20]. Based on these observations, initially isolated p53 came to be classified as an oncogene product [21]. However, this classical point of view has been challenged by the findings demonstrating that the initially identified p53 is a mutant form of p53 [22]. A number of studies clearly showed that, in contrast to mutant form of p53, wild-type p53 is able to suppress aberrant cell growth of transformed cells as well as tumors [23, 24]. Of note, p53-deficient mice frequently developed spontaneous tumors [25], indicating strongly that wild-type p53 acts as a tumor suppressor.
Extensive mutation searches revealed that nearly half of human tumors carry p53 mutations except human neuroblastoma, rhabdomyosarcoma, and melanoma [26]. el-Deiry et al. described that cyclin-dependent protein kinase (CDK) inhibitor termed wild-type p53-activated fragment 1 (p21WAF1) is one of p53-inducible gene products, and proposed that p53 recognizes and binds to a consensus sequence motif made of tandem 10 bp elements (RRRCWWGYYY) separated by 1–13 bp found within the promoter regions of p53-target genes [27, 28]. In accordance with their findings, accumulating evidence suggests that p53 is a sequence-specific nuclear transcription factor [29, 30]. To date, numerous p53-inducible gene products have been identified including proapoptotic BAX (Bcl2-associated X protein), NOXA (Latin for damage), PUMA (p53-upregulated modulator of apoptosis) and p53AIP1 (p53-regulated apoptosis-inducing protein 1) [31–34] and gene products inducing cell cycle arrest such as p21WAF1 and 14-3-3σ [35]. Importantly, 95% of p53 mutations have been detected within the genomic region encoding its central sequence-specific DNA-binding domain [26]. Among them, 20% of mutations accumulate within six hot-spots (amino acid residues 175, 245, 248, 249, 273, and 282), which are most targeted in p53 gene. These mutations disrupt the native conformation of the sequence-specific DNA-binding domain of wild-type p53 and result in the loss of its sequence-specific DNA-binding activity [30]. Several lines of evidence indicated that the sequence-specific DNA-binding ability of p53 is tightly linked to its proapoptotic activity [30], suggesting that blocking p53-dependent sequence-specific transcription is a critical event in tumorigenesis. Mutant forms of p53 lose the activity to prevent uncontrolled cell growth and protect cells from genomic alterations [36]. Thus, mutant forms of p53 lack its critical function to maintain genomic integrity in response to DNA damage.
Human runt-related transcription factor (RUNX) family is composed of three members including RUNX1, RUNX2, and RUNX3. RUNX family is highly conserved in their runt homology domain, which is involved in the sequence-specific DNA binding and heterodimerization with the common co-factor CBFβ [37]. In addition to runt domain, RUNX family also possesses the other functional subdomains such as a large transactivation domain in COOH-terminal part and an inhibitory domain at COOH-terminal end of the transactivation domain [37]. A growing body of evidence demonstrated that each of RUNX family members has a distinct biological function. For example, RUNX1 is required for the establishment of the hematopoietic stem cells and is a frequent target of chromosomal gene translocations in hematopoietic malignancies [38–40]. On the other hand, RUNX2 plays an essential role in the promotion of both osteoblast and terminal chondrocyte differentiation and is also responsible for bone formation and mineralization in vivo [41, 42]. Since RUNX3-deficient mice displayed hyperplasia in gastric mucosa caused by reduced apoptosis and the stimulated growth of the gastric epithelial cells, RUNX3 has been considered to be a tumor suppressor for human gastric cancer [43]. Although the functional contribution of RUNX family members to the regulation of DNA damage response has been elusive, we have found for the first time that RUNX1 as well as RUNX3 acts as a coactivator for p53 in response to DNA damage, whereas RUNX2 represses p53-dependent apoptotic cell death following DNA damage. Based on our recent findings, RUNX family might participate in the regulation of p53-mediated DNA damage response.
2. Induction of p53 in Response to DNA Damage
Since p53 plays a critical role in growth suppression and disruption of balanced p53 activity as well as amount would lead to tumor development, under normal physiological conditions, p53 is kept at extremely low level and thus barely detectable. Upon multiple cellular stresses including DNA damage, oncogene activation, hypoxia, nucleotide imbalance and oxidative damage, p53 is rapidly induced to largely accumulate in cell nucleus through sequential post-translational modifications such as phosphorylation (Ser-15, Ser-20, and Ser-46) and acetylation (Lys-370, Lys-372, Lys-373, Lys-381, and Lys-392) [6–8]. These chemical modifications convert p53 from latent form to active one via the dynamic conformation shift. DNA damage-mediated induction of p53 is mainly regulated at protein level but not at transcription level. RING-finger type E3 ubiquitin protein ligase Murine double minute 2 (MDM2), which has an oncogenic potential, binds to NH2-terminal portion of p53, catalyses ubiquitination of six Lys residues (Lys-370, Lys-372, Lys-373, Lys-381, Lys-382, and Lys-386) at its COOH-terminal region, and then promotes its proteolytic degradation through proteasome [44–46]. Since MDM2 is one of p53-induced gene products, MDM2 participates in a negative autoregulatory feedback loop, which controls p53 expression level [47]. It is likely that MDM2 monitors the amount as well as the activity of p53 in response to DNA damage, thereby prohibiting inappropriate apoptotic cell death by overactive p53. As NH2-terminal phosphorylation of p53 facilitates the dissociation of MDM2 from p53 and also COOH-terminal acetylation inhibits MDM-mediated p53 ubiquitination following DNA damage, p53 becomes stable in response to DNA damage. Recently, Meek and Hupp described that, in response to DNA damage, ATM and CHK (checkpoint kinase) phosphorylate multiple Ser residues within or close to RING-finger domain of MDM2 thereby destabilizing MDM2 [48]. Thus, the intracellular amount of MDM2 available appears to be critical in determining the expression level of p53. Additionally, MDM2 masks the important amino acid residues of NH2-terminal transactivation domain of p53, and therefore the sequence-specific transactivation ability of p53 is significantly attenuated by MDM2 [49].
3. Cell Survival or Cell Death following DNA Damage
Biological consequences in response to DNA damage might be dependent on the degree and/or the nature of DNA damage. When DNA damage is severe and repair is impossible, activated p53 exerts its strong proapoptotic function to remove cells with seriously damaged DNA through apoptotic cell death. Under these conditions, p53 transactivates pro-apoptotic key target genes including BAX, PUMA, NOXA, and p53AIP1 [6–8]. Among them, PUMA is a direct activator of BAX [50]. The collaboration of these pro-apoptotic gene products contributes to the disruption of mitochondrial membrane potential, thereby releasing cytochrome c from mitochondrial intermembrane space. Then the release of cytochrome c causes apoptotic protease activating factor 1 (Apaf1) oligomerization, resulting in apoptosome formation. This complex, in turn, recruits and activates procaspase-9, which then activates executioner caspases-3 and caspase-7, which is a critical step in p53-dependent pro-apoptotic pathway [51, 52].
In contrast, when cells receive the repairable DNA damage, p53 induces G1/S and/or G2/M cell cycle arrest through the transactivation of p21 WAF1, p53R2 (p53-inducible ribonucleotide reductase small subunit), 14-3-3σ and GADD45 (Growth arrest and DNA damage 45) implicated in the promotion of cell cycle arrest and DNA repair [53, 54]. After DNA repair is completed, cells reenter into normal cell cycle to faithfully transmit genetic information to their daughter cells. During this repair process, mispaired DNA bases are replaced with correct bases by mismatch repair (MMR), and small chemical alterations of DNA bases are repaired by base excision repair (BER) through excision of the damaged base [55, 56]. We have previously found that MDC1/NFBD1 attenuates ATM-dependent phosphorylation of p53 at Ser-15 during the early phase of DNA damage response, suggesting that MDC1/NFBD has an ability to inhibit pro-apoptotic activity of p53 and save time to repair damaged DNA [57]. At the late phase of DNA damage response, the expression level of MDC1/NFBD1 was reduced, p53 dissociated from MDC1/NFBD1, and p53 exerted its pro-apoptotic activity. MDC1/NFBD1-deficient mice exhibited chromosome instability and DNA repair defects. In addition, it has been shown that p53 transrepresses some genes such as anti-apoptotic Bcl-2 [58, 59], indicating that the concerted action of a whole set of p53-induced genes as well as p53-repressed genes triggers a specific biological consequence following DNA damage. Therefore, it is likely that p53 stands at the crossroad between cell survival and cell death in response to DNA damage (Figure 1). However, the molecular basis for the choice between cell cycle arrest and apoptotic cell death induction by p53 is not well understood.
Figure 1.
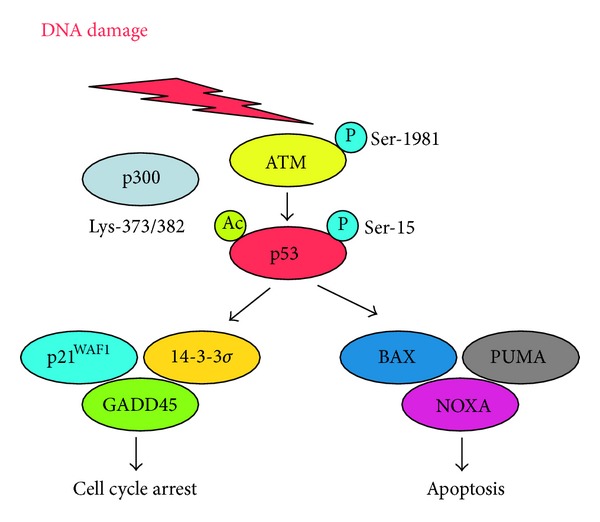
p53-dependent DNA damage response. Upon DNA damage, p53 is phosphorylated at Ser-15 and acetylated at Lys-373/382 by ATM and p300, respectively. Activated form of p53 induces cell cycle arrest and/or apoptotic cell death.
4. Protein-Protein Interaction
In addition to the posttranslational modifications, transcriptional as well as pro-apoptotic activity of p53 is also regulated positively or negatively by protein-protein interaction. Certain cellular proteins affect the modification status of p53 following DNA damage. The early study demonstrated that nuclear nonreceptor tyrosine kinase c-Abl negatively regulates cell growth [60]. Subsequent analysis showed that c-Abl, associates with p53 and enhances its sequence-specific transcriptional activation and mutant c-Abl which no longer binds to p53, fails to increase the transcriptional activity of p53 [61]. Additionally, Yuan et al. revealed that c-Abl is required for p53-dependent apoptotic cell death in response to DNA damage [62]. Alternatively, Samuels-Lev et al. found that apoptosis stimulating protein of p53 (ASPP), which contains ankyrin repeats, SH3 domain and Pro-rich domains, interacts with p53 and enhances its sequence-specific transactivation as well as its proapoptotic activity [63]. According to their observations, ASPP stimulated p53-dependent apoptotic cell death in response to DNA damage. Recently, Gillotin and Lu described that ASPP forms a complex with p300 histone acetyltransferase and selectively regulates the transcriptional activity of p53 [64]. Yang et al. reported that 14-3-3σ is a direct transcriptional target of p53 and interacts with p53 to increase its protein stability through blocking MDM2-mediated ubiquitination of p53 [35]. Consistent with these observations, 14-3-3σ has been shown to enhance the transcriptional activity of p53. Since 14-3-3σ is one of p53-responsive gene products, 14-3-3σ creates a positive auto-regulatory loop in which p53 transactivates 14-3-3σ and in turn 14-3-3σ enhances p53 activity. Recently, Kim et al. demonstrated that tumor suppressor wilms tumor gene on X chromosome (WTX) associates with DNA-binding domain of p53 and elevates its CBP (CREB-binding protein)/p300-mediated acetylation level at Lys-373/382 [65]. Based on their results, WTX was able to increase the transcriptional and pro-apoptotic activities of p53 through the regulation of the interaction between p53 and CBP [65, 66].
Protein-protein interaction does not always enhance p53 activity. As mentioned above, MDM2 binds to NH2-terminal transactivation domain of p53 and strongly blocks its transcriptional as well as pro-apoptotic ability [49]. We have previously described that oncogenic polo-like kinase 1 (Plk1) whose expression level is significantly higher in various human tumor tissues as compared with their corresponding normal ones, associates with p53 and inhibits its activity through phosphorylation [67]. Alternatively, it has been shown that silent mating type information regulation 2 homolog 1 (SIRT1) interacts with p53 and inhibits p53 through deacetylation of p53 [68]. Consistent with these observations, the catalytically impaired SIRT1 increased p53-target gene expression after DNA damage [69]. Recently, Jang et al. described that SIRT1 promotes the human liver cancer survival [70] and Cha et al. reported that the expression level of SIRT1 is a poor prognostic indicator for human gastric carcinomas [71].
In general, subcellular localization of p53 might be one of critical determinants for p53 activity. For example, it has been well known that human neuroblastomas express nonfunctional p53 without mutations, which might be due to its abnormal cytoplasmic retention [72]. Intriguingly, Nikolaev et al. discovered a large cytoplasmic protein termed p53-associated, Parlin-like cytoplasmic protein (Parc), which associated with cytoplasmic p53 in neuroblastoma cells [73, 74]. According to their findings, Parc bound to the majority of cytoplasmic p53 and acted as a cytoplasmic anchor protein for p53. In a good agreement with this notion, silencing of Parc induced nuclear access of p53 and promoted apoptotic cell death in neuroblastoma cells. However, knockdown of Parc in hepatocellular cells had an undetectable effect on cytoplasmic p53 [75], suggesting that Parc could act as a cytoplasmic anchor protein for p53 in a neuroblastoma-specific manner. In addition to Parc, SIRT1 has been shown to block nuclear translocation of cytoplasmic p53 in response to the endogenous reactive oxygen species (ROS) [76].
5. Mutant Form of p53
As mentioned above, p53 is frequently mutated in human various tumor tissues. Most of p53 mutations are detectable within the genomic region encoding its sequence-specific DNA-binding domain (exons 5–8), and thus mutant forms of p53 lack the sequence-specific transactivation ability. In a sharp contrast to wild-type p53, mutant forms of p53 display a greatly extended half-life, which might be attributed to the escape from MDM2-mediated ubiquitin/proteasome-dependent proteolytic degradation system [6–8]. Recently, Wiech et al. described that heat shock protein 70 (HSP70) partially inhibits proteasomal degradation of mutant p53 [77]. Furthermore, mutant forms of p53 exhibit a dominant-negative behavior toward wild-type p53 [6–8]. When wild-type p53 and mutant form of p53 are coexpressed in certain cancerous cells, mutant form of p53 strongly prohibits the tumor suppressive activity of wild-type p53 (Figure 2). Indeed, cancerous cells carrying p53 mutations sometimes display the chemoresistant phenotypes, which might be at least in part due to the presence of mutant p53. Consistent with these observations, many patients with p53 mutations have an increased resistance to conventional chemotherapy and poorer prognosis than those who have wild-type p53 or no p53 protein [78–81].
Figure 2.
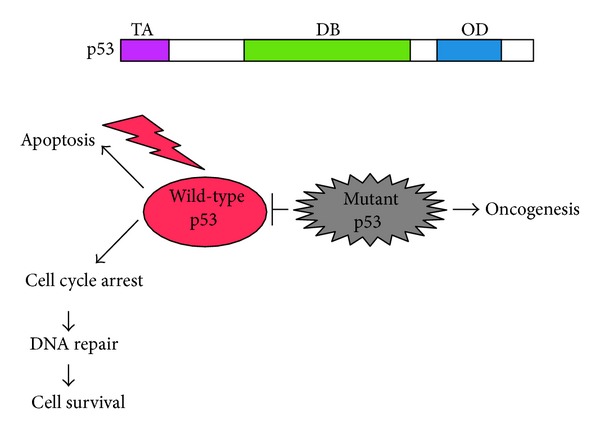
Structure of p53 and dominant-negative behavior of mutant p53 toward wild-type p53. p53 is composed of three functional domains including NH2-terminal transactivation domain (TA) followed by central sequence-specific DNA-binding domain (DB) and COOH-terminal oligomerization domain (OD) (upper panel). Lower panel shows the dominant-negative behavior of mutant p53 against wild-type p53.
In addition to the dominant-negative activity of mutant p53 toward wild-type p53, a growing body of evidence suggests that mutant p53 acquires the alternative gain-of-function activities. Although mutant p53 has been considered to be transcriptionally inactive due to the fact that most p53 mutants fail to transactivate p53-inducible genes, it has been shown that mutant p53 has an ability to regulate the expression of the specific set of genes implicated in tumor initiation and maintenance such as MDR-1, c-Myc, c-fos, and HSP70 [82–85]. Among them, MDR-1 confers multidrug-resistant phenotype. These findings imply that mutant p53 retains the transactivation potential, whereas its target genes are completely different from those of wild-type p53. Indeed, most promoters activated by mutant p53 lack the sequences similar to the consensus p53-responsive element, suggesting that mutant p53 might regulate transcription through the response elements that are distinct from wild-type p53-target sequence. Of note, these promoters transactivated by various p53 mutants show no sequence homology, indicating that sequence recognition by p53 mutants might be dependent on their specific conformations [86].
Nuclear factor Y (NF-Y) is a heterotrimeric transcription factor, which recognizes CCAAT consensus motif, and has been shown to regulate CCAAT-containing promoters including E2F-1, cyclin A, cdc25C, and MDR-1 in response to DNA damage [87]. Di Agostino et al. have described that NF-Y associates with mutant p53, and this complex increases the expression of cell cycle-related cyclin A, cyclin B1, cdk1, and cdc25C following DNA damage [88]. According to their results, mutant p53/NF-Y complex was recruited onto NF-Y-target promoters together with histone acetyltransferase p300. In contrast, it has been reported that the expression of these cell cycle-related genes is repressed by wild-type p53/NF-Y complex in response to DNA damage [88]. Under their experimental conditions, wild-type p53 formed a complex with NF-Y on CAAT-containing promoters, and this complex recruited histone deacetylase (HDAC) and released histone acetyltransferase. Therefore, it is likely that mutant p53 binds to the similar transcription factors of wild-type p53 and causes the aberrant transcriptional regulation of their target gene expression.
6. Possible Therapeutic Strategies
Dysfunction of a proper DNA damage response mediated by wild-type p53 results in the accumulation of various gene mutations and induces genomic instability, thereby promoting tumor formation. As mentioned above, mutant forms of p53 play a critical role in the disruption of p53-dependent appropriate DNA damage response. In contrast to the short half-life of wild-type p53, mutant forms of p53 are extremely stable and have a prolonged half-life. Furthermore, mutant forms of p53 display a dominant-negative behavior toward wild-type p53. Thus, the intracellular balance between mutant p53 and wild-type p53 might be a major determinant of cell fate in response to DNA damage. Since wild-type p53 is quickly degraded by MDM2, extensive efforts have been performed to identify the small compound(s), which could inhibit the interaction between p53 and MDM2. A small molecule termed nutlin, which specifically occupied p53-binding pocket on the surface of MDM2, blocked the interaction between p53 and MDM2 and liberated p53 from MDM2-mediated degradation system, leading to the stabilization, accumulation, and activation of p53 in cancerous cells with wild-type p53 [89, 90]. Nutlin inhibited tumor growth in a nongenotoxic manner in xenografted tumor mice [91]. This nongenotoxic effect of nutlin might be attractive from a therapeutic standpoint. Since the conventional antitumor drugs introduce DNA damage in tumor tissues as well as their surrounding normal ones, it is possible that these treatments might have a potential for secondary malignancies. Surprisingly, Aziz et al. found that, upon repeated exposure of nutlin to certain cancerous cells expressing wild-type p53, a small population of nutlin-resistant cells expands and finally acquires somatic p53 mutations [92].
The previous studies demonstrated that abnormal conformation of certain mutant forms of p53 is reversible under the specific conditions [93], indicating that certain mutant p53 could be reactivated by conformation change. Bykov et al. have extensively screened a library of small compounds in order to find out compound(s) that could restore wild-type function to mutant p53 [94]. According to their results, they finally identified one small compound termed p53 reactivation and induction of massive apoptosis (PRIMA-1), which was able to restore sequence-specific transactivation function and the active conformation to mutant p53 (Figure 3). Furthermore, treatment of PRIMA-1 exhibited a significant anti-tumor effect in mice. Their results raise a possibility that the combination of PRIMA-1 and the conventional chemotherapeutic drugs might be a novel strategy to treat mutant p53-carrying malignant cancers. Bykov et al. found a remarkable synergistic effect in vitro between PRIMA-1Met and various antitumor drugs including cisplatin (CDDP), adriamycin (ADR), and camptothecin (CPT) [95]. PRIMA-1Met, which was more active than the original compound, is a methylated form of PRIMA-1. Among them, combination of PRIMAMet and cisplatin produced a significantly smaller volume of tumors than those of PRIMAMet or cisplatin treatment alone.
Figure 3.
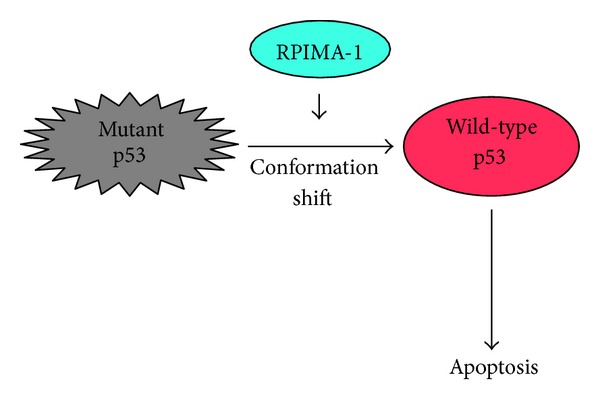
PRIMA-1 reactivates mutant p53. Small chemical compound termed PRIMA-1 has an ability to reactivate mutant p53 through the conformation change.
Recently, Wassman et al. utilized a computational method to identify a small molecule(s) which could open p53 core domain flanked by loop L1 and sheet S3 (L1/S3 reactivation pocket) [96]. Finally, they found stictic acid as a p53 reactivation compound. Based on their results, stictic acid enhanced luciferase activity driven by p21 WAF1 promoter in cells expressing R175H mutant of p53.
7. RUNX1 Acts as a Positive Regulator for p53-Dependent Apoptotic Cell Death in Response to DNA Damage
Human runt-related transcription factor (RUNX) family is composed of three members including RUNX1, RUNX2, and RUNX3 [37]. The highly conserved runt domain has been shown to be responsible for both sequence-specific DNA binding and heterodimerization with CBF-β [37]. RUNX1 is the most frequent target for chromosomal translocation in human acute myeloid leukemia (AML), generating the oncogenic fusion proteins such as RUNX1/ETO [38–40]. To date, more than ten RUNX1 fusion proteins have been identified [97]. RUNX1 has been identified as a gene located at a breakpoint of the chromosomal translocation t(8;21) [38–40]. In addition to the chromosomal translocation, RUNX1 point mutations are found in sporadic as well as familial myeloid leukemia and also in AML. Subsequent studies revealed that most of RUNX1 mutations are detectable within the genomic region encoding its runt domain (nearly 80%) [98, 99]. These runt domain mutants lack sequence-specific DNA-binding activity; however, they retain an ability to form a heterodimer with CBF-β. RUNX1 mutant binds to CBF-β more efficiently than wild-type RUNX1. Since CBF-β protects RUNX1 from ubiquitin/proteasome-dependent degradation system, RUNX1 mutant/CBF-β heterodimer is much more stable than wild-type RUNX1/CBF-β hetero-dimer. Thus, RUNX1 mutant inhibits sequence-specific transcriptional activity of wild-type RUNX1, thereby acting as a dominant-negative inhibitor toward wild-type RUNX1 [100, 101]. Although several lines of evidence strongly suggest that RUNX1 might act as a tumor suppressor, the precise molecular mechanisms of how RUNX1 could exert its tumor-suppressive activity have been elusive.
Numerous evidences strongly suggest that RUNX1 is required for normal blood development. For example, RUNX1-deficient mice displayed a remarkable defect in hematopoiesis [38, 39]. In accordance with these observations, RUNX1 stimulated the transcription of a number of myeloid- and lymphoid-related genes [102, 103]. In addition to the hematopoiesis-specific genes RUNX1 has been shown to be also involved in the regulation of cell cycle-related genes, such as p21 WAF1 [104]. Intriguingly, it has been reported that RUNX1 induces senescence-like growth arrest in primary murine fibroblasts, and this response is lost in cells lacking functional p53 [105, 106]. Recently, Satoh et al. demonstrated that loss of function mutant of RUNX1 attenuates DNA damage-mediated repair response in hematopoietic stem/progenitor cells [107], indicating that RUNX1 might be required for the proper DNA damage response. These results prompted us to investigate whether RUNX1 could participate in p53-dependent apoptotic cell death in response to DNA damage.
According to our recent results [108], RUNX1 together with p53 accumulated in human osteosarcoma-derived U2OS cell nucleus following ADR exposure, formed a stable complex, and was recruited onto p53-activated promoters including p21 WAF1 and BAX, suggesting that RUNX1 might affect the transcriptional activity of p53. Knockdown of RUNX1 resulted in a significant reduction of ADR-mediated apoptotic cell death in association with a remarkable downregulation of p53-responsive gene expression. It was worth noting that RUNX1 associates with p300 and p300-mediated acetylation of p53 at Lys-373/382 following ADR treatment is reduced in RUNX1 knockdown cells. Hence, it is conceivable that RUNX1 acts as a molecular bridge or a scaffolding protein for p53-p300 binding, thereby enabling p300-mediated acetylation of p53 at Lys-373/382 in response to DNA damage.
8. RUNX3 Acts as a Positive Regulator for p53-Dependent Apoptotic Cell Death in Response to DNA Damage
RUNX3 has been shown to be involved in the formation of variety of a human cancers [37]. The initial studies revealed that RUNX3-deficient mice develop the hyperplasia in their gastric mucosa, which might be due to the increased proliferation as well as diminished apoptotic cell death [43]. Intriguingly, RUNX3 is located on human chromosome 1p36, a region which has long been suggested to be a tumor suppressor locus in a variety of human cancers. Although RUNX3 is infrequently mutated in various human cancers, R122C mutation found within its conserved runt domain, which was identified in a gastric cancer patient, abolished the tumor-suppressive function of RUNX3 in nude mice [43]. The expression level of RUNX3 in human cancers is kept at extremely low level, and the higher RUNX3 expression level in human cancers is closely associated with a favorable prognosis with reduced recurrence and better survival rates in patients. On the other hand, lower expression level of RUNX3 is associated with tumor progression and poor prognosis in various cancers [109–112]. Subsequent studies demonstrated that the combination of hemizygous deletion of RUNX3 and the hypermethylation of RUNX3 promoter region contributes to the silencing of RUNX3 expression in various human cancers [37].
Since RUNX3 is a sequence-specific transcription factor, its nuclear localization is essential for its transcriptional function. In this regard, the mislocalization of RUNX3 might be one of the molecular mechanisms behind the inactivation of RUNX3. Indeed, it has been shown that RUNX3 is frequently detectable in cytoplasm of various human cancers including gastric cancer, colorectal cancer, and breast cancer [113]. Thus, one allele of RUNX3 is silenced through promoter hypermethylation, while the remaining wild-type allele is inactivated through the abnormal cytoplasmic localization of its gene product.
RUNX3 activity is highly associated with transforming growth factor β (TGF-β) signaling, which promotes both cell cycle arrest and apoptotic cell death [114, 115]. Previous studies showed that gastric epithelial cells derived from RUNX3-deficient mice are highly resistant to the growth-suppressive and apoptotic cell death-inducing effects of TGF-β [43]. Consistent with these observations, Ito et al. found that RUNX3, a downstream of the tumor-suppressive TGF-β pathway, antagonizes the oncogenic Wnt pathway in intestinal carcinogenesis, thereby inhibiting the transcriptional activity of β-catenin/ T-cell factor 4 (TCF4) [116]. In addition, Yano et al. reported that RUNX3 is required for transcriptional upregulation of proapoptotic BH-3 only protein Bcl-2-interacting mediator of cell death (Bim) during TGF-β-induced apoptotic cell death [117]. However, a possible involvement of RUNX3 in the regulation of DNA damage response remains to be unknown.
Recently, we have found for the first time that RUNX3 knockdown significantly attenuates ADR-mediated apoptotic cell death in p53-proficient U2OS and human lung carcinoma A549 cells but not in p53-deficient human lung carcinoma H1299 and human osteosarcoma SAOS-2 cells [118]. Of note, knockdown of RUNX3 resulted in a massive reduction of ADR-dependent phosphorylation level of p53 at Ser-15 in association with a remarkable downregulation of various p53-target gene expressions, and forced expression of RUNX3 elevated the ADR-mediated p53 phosphorylation level at Ser-15 in its upstream protein kinase ATM-dependent manner. Under the unstressed conditions, RUNX3 was detected in both cell nucleus and cytoplasm, whereas RUNX3 was induced to translocate into cell nucleus following ADR exposure. Since RUNX3 formed a stable complex with p53 and activated ATM in the presence of ADR, it is likely that RUNX3 assists ADR-mediated phosphorylation of p53 at Ser-15 in response to ADR, indicating that, like RUNX1, RUNX3 acts as a positive regulator for p53 following DNA damage.
9. RUNX2 Acts as a Negative Regulator for p53-Dependent Apoptotic Cell Death in Response to DNA Damage
RUNX2 has been considered to be one of the master regulators for osteoblast differentiation and essential for bone formation as well as mineralization in vivo [41, 119]. In accordance with this notion, RUNX2 has an ability to transactivate a number of osteogenic markers such as type 1 collagen, osteopontin, and osteocalcin [120]. In a sharp contrast to RUNX1 and RUNX3, oncogenic property of RUNX2 has been postulated in several human cancers. It has been shown that RUNX2 gene amplification is frequently observed in human osteosarcoma [121]. Additionally, accumulating evidence demonstrated that the dysregulation of RUNX2 expression is frequently detectable in a variety of human cancers and higher expression level of RUNX2 is closely correlated with poor clinical outcome of the patients [122–124]. However, the precise molecular mechanism(s) how RUNX2 could contribute to the initiation and progression of cancers remains elusive. Of note, Blyth et al. described that RUNX2 provides a strong anti-apoptotic signal even in the presence of functional p53, indicating that RUNX2 neutralizes p53 [125]. Westendorf et al. revealed that RUNX2 represses p53-target gene p21 WAF1 expression [126]. Thus, it is likely that there is a functional interaction between RUNX2 and p53, and we have then investigated whether RUNX2 could participate in the regulation of p53-dependent DNA damage response.
Under our experimental conditions, we have found the complex formation between p53 and RUNX2 in ADR-treated U2OS cells but not in untreated cells. RUNX2/p53 complex was recruited onto the various p53-responsive promoters including p21 WAF1 and BAX in response to ADR. Forced expression of RUNX2 downregulated the transcription of p53-inducible genes, and RUNX2 knockdown further elevated the expression levels of p53-target genes in response to ADR as compared with those in ADR-treated cells expressing the endogenous RUNX2. Moreover, knockdown of RUNX2 significantly enhanced ADR-mediated apoptotic cell death in U2OS cells, whereas RUNX2 knockdown had a negligible effect on p53-deficient H1299 cells (Figure 4). Unlike RUNX1 and RUNX3, RUNX2 showed an undetectable effect on ADR-mediated phosphorylation as well as acetylation status of p53. Intriguingly, we have found that RUNX2/p53 complex contains HDAC6 (histone deacetylase 6), and HDAC6-specific chemical inhibitor termed tubacin treatment enhances ADR-mediated induction of p53-responsive gene expression, indicating that deacetylase activity of HDAC6 is required for RUNX2-mediated downregulation of p53-target genes. Taken together, our results implicate that, unlike RUNX1 and RUNX3, RUNX2 acts as a negative regulator for p53 in response to DNA damage and might be an attractive novel molecular target for improved therapeutic outcome [127].
Figure 4.

Knockdown of RUNX2 enhances ADR-mediated apoptotic cell death in U2OS cells. U2OS cells were transiently transfected with control siRNA or with siRNA targeting RUNX2. Twenty-four hours after transfection, cells were exposed to ADR or left untreated. Twenty-four hours after ADR exposure, cells were observed by phase-contrast microscopy.
10. Future Perspective
Maintenance of genomic integrity by the proper DNA damage response is critical to prevent tumorigenesis. Tumor suppressor p53 is the most studied key player which monitors and checks the genomic integrity during DNA damage response and eliminates cells with seriously damaged DNA through apoptotic cell death. RUNX family members are the novel players, which participate in the regulation of p53-dependent DNA damage response. RUNX1 and RUNX3 enhance proapoptotic activity of p53, whereas RUNX2 prohibits p53-dependent apoptotic cell death following DNA damage (Figure 5). Among them, RUNX2 might be an attractive molecular target to improve the conventional chemotherapy. Since siRNA-mediated knockdown of RUNX2 significantly enhanced the antitumor effect of ADR through the activation of wild-type p53, it is likely that the constitutive silencing of RUNX2 expression and/or the inhibition of the transcriptional activity of RUNX2 might be a promising strategy to enhance the antitumor effect of the conventional chemotherapy. To our surprise, knockdown of RUNX2 in certain cancerous cells carrying p53 mutation further promoted apoptotic cell death in the presence of well-known antitumor drugs (unpublished observations). Therefore, we postulate that the silencing of RUNX2 might be effective on the conventional chemotherapy of malignant tumors regardless of their p53 status.
Figure 5.
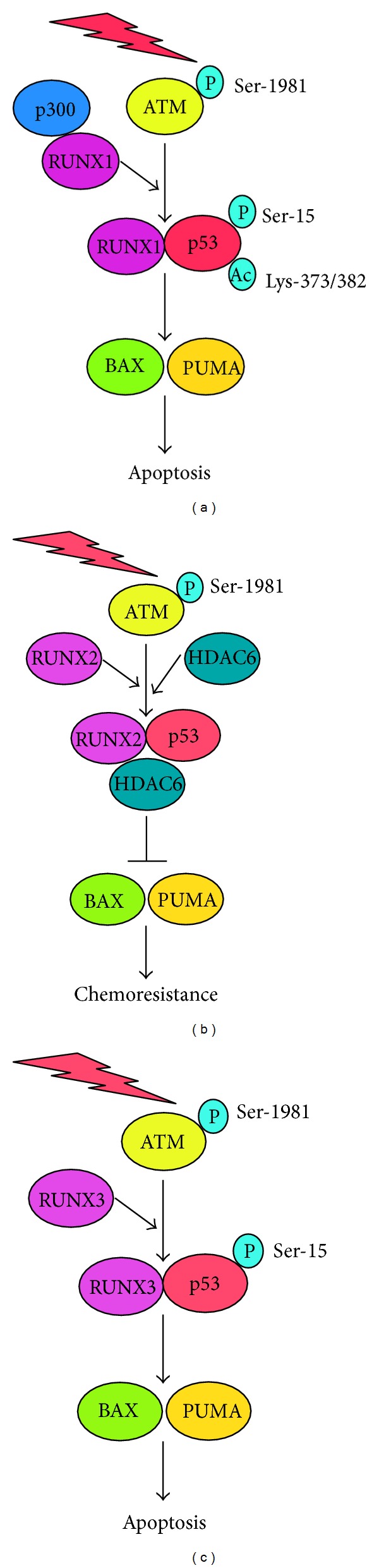
Distinct regulatory role of RUNX family on p53 in response to DNA damage. Under our experimental conditions, RUNX1 as well as RUNX3 acts as a positive regulator for p53 in response to DNA damage, whereas DNA damage-induced proapoptotic activity of p53 is strongly abrogated by RUNX2.
Acknowledgment
Work in the authors' laboratories was supported in part by a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- 1.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Current Biology. 2000;10(15):886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg M, Stucki M, Falck J, et al. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421(6926):952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- 3.Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421(6926):957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- 4.Stewart GS, Wang B, Bignell CR, Taylor AMR, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421(6926):961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 5.Xu X, Stern DF. NFBD1/MDC1 regulates ionizing radiation-induced focus formation by DNA checkpoint signaling and repair factors. FASEB Journal. 2003;17(13):1842–1848. doi: 10.1096/fj.03-0310com. [DOI] [PubMed] [Google Scholar]
- 6.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nature Reviews Cancer. 2002;2(8):594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 7.Sionov RV, Haupt Y. The cellular response to p53: the decision between life and death. Oncogene. 1999;18(45):6145–6157. doi: 10.1038/sj.onc.1203130. [DOI] [PubMed] [Google Scholar]
- 8.Prives C, Hall PA. The p53 pathway. The Journal of Pathology. 1999;187:112–126. doi: 10.1002/(SICI)1096-9896(199901)187:1<112::AID-PATH250>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 9.DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(5):2420–2424. doi: 10.1073/pnas.76.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. Journal of Virology. 1979;31(2):472–483. doi: 10.1128/jvi.31.2.472-483.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40 transformed cells. Nature. 1979;278(5701):261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 12.Linzer DIH, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40 transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17(1):43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 13.Melero JA, Stitt DT, Mangel WF, Carroll RB. Identification of new polypeptide species (48-55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology. 1979;93(2):466–480. doi: 10.1016/0042-6822(79)90250-2. [DOI] [PubMed] [Google Scholar]
- 14.Steinmeyer K, Deppert W. DNA binding properties of murine p53. Oncogene. 1988;3(5):501–507. [PubMed] [Google Scholar]
- 15.Shaulian E, Zauberman A, Milner J, Davies EA, Oren M. Tight DNA binding and oligomerization are dispensable for the ability of p53 to transactivate target genes and suppress transformation. EMBO Journal. 1993;12(7):2789–2797. doi: 10.1002/j.1460-2075.1993.tb05940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaulsky G, Goldfinger N, Ben-Ze’ev A, Rotter V. Nuclear accumulation of p53 protein is mediated by several nuclear localization signals and plays a role in tumorigenesis. Molecular and Cellular Biology. 1990;10(12):6565–6577. doi: 10.1128/mcb.10.12.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO Journal. 1999;18(6):1660–1672. doi: 10.1093/emboj/18.6.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruaro EM, Collavin L, Del Sal G, et al. A proline-rich motif in p53 is required for transactivation-independent growth arrest as induced by Gas1. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(9):4675–4680. doi: 10.1073/pnas.94.9.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eliyahu D, Michalovitz D, Oren M. Overproduction of p53 antigen makes established cells highly tumorigenic. Nature. 1985;316(6024):158–160. doi: 10.1038/316158a0. [DOI] [PubMed] [Google Scholar]
- 20.Dippold WG, Jay G.f DeLeo AB, Khoury G, Old LJ. p53 transformation-related protein: detection by monoclonal antibody in mouse and human cells. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(3):1695–1699. doi: 10.1073/pnas.78.3.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lane DP. p53 in Paris, an oncogene comes of age. Oncogene. 1987;1(3):241–242. [PubMed] [Google Scholar]
- 22.Takahashi T, Nau MM, Chiba I, et al. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989;246(4929):491–494. doi: 10.1126/science.2554494. [DOI] [PubMed] [Google Scholar]
- 23.Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(22):8763–8767. doi: 10.1073/pnas.86.22.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker SJ, Markowitz S, Fearon ER, Willson JKV, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249(4971):912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- 25.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 26.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253(5015):49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 27.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 28.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nature Genetics. 1992;1(1):45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 29.Tokino T, Thiagalingam S, el-Deiry WS, Waldman T, Kinzler KW, Vogelstein B. p53 tagged sites from human genomic DNA. Human Molecular Genetics. 1994;3(9):1537–1542. doi: 10.1093/hmg/3.9.1537. [DOI] [PubMed] [Google Scholar]
- 30.Pietenpol JA, Tokino T, Thiagalingam S, el-Deiry WS, Kinzler KW, Vogelstein B. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(6):1998–2002. doi: 10.1073/pnas.91.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80(2):293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 32.Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288(5468):1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 33.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Molecular Cell. 2001;7(3):673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 34.Oda K, Arakawa H, Tanaka T, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by ser-46-phosphorylated p53. Cell. 2000;102(6):849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 35.Yang H-Y, Wen Y-Y, Chen C-H, Lozano G, Lee M-H. 14-3-3σ positively regulates p53 and suppresses tumor growth. Molecular and Cellular Biology. 2003;23(20):7096–7107. doi: 10.1128/MCB.23.20.7096-7107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lavigueur A, Maltby V, Mock D, Rossant J, Pawson T, Bernstein A. High incidence of lung, bone, and lymphoid tumors in transgenic mice overexpressing mutant alleles of the p53 oncogene. Molecular and Cellular Biology. 1989;9(9):3982–3991. doi: 10.1128/mcb.9.9.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito Y. RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Advances in Cancer Research. 2008;99:33–76. doi: 10.1016/S0065-230X(07)99002-8. [DOI] [PubMed] [Google Scholar]
- 38.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 39.Wang Q, Stacy T, Binder M, Marín-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(8):3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(23):10431–10434. doi: 10.1073/pnas.88.23.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 42.Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 43.Li Q-L, Ito K, Sakakura C, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109(1):113–124. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- 44.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 45.Kubbutat MHG, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 46.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Letters. 1997;420(1):25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 47.Juven T, Barak Y, Zauberman A, George DL, Oren M. Wild type p53 can mediate sequence-specific transactivation of an internal promoter within the mdm2 gene. Oncogene. 1993;8(12):3411–3416. [PubMed] [Google Scholar]
- 48.Meek DW, Hupp TR. The regulation of MDM2 by multisite phosphorylation-Opportunities for molecular-based intervention to target tumours? Seminars in Cancer Biology. 2010;20(1):19–28. doi: 10.1016/j.semcancer.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 49.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes and Development. 1993;7(7):1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 50.Luo X, He Q, Huang Y, Sheikh MS. Transcriptional upregulation of PUMA modulates endoplasmic reticulum calcium pool depletion-induced apoptosis via Bax activation. Cell Death and Differentiation. 2005;12(10):1310–1318. doi: 10.1038/sj.cdd.4401659. [DOI] [PubMed] [Google Scholar]
- 51.Soengas MS, Alarcón RM, Yoshida H, et al. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284(5411):156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- 52.Fadeel B, Ottosson A, Pervaiz S. Big wheel keeps on turning: apoptosome regulation and its role in chemoresistance. Cell Death and Differentiation. 2008;15(3):443–452. doi: 10.1038/sj.cdd.4402265. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka H, Arakawa H, Yamaguchi T, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404(6773):42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 54.Kazantsev A, Sancar A. Does the p53 up-regulated Gadd45 protein have a role in excision repair? Science. 1995;270(5238):1003–1006. [PubMed] [Google Scholar]
- 55.Tom S, Ranalli TA, Podust VN, Bambara RA. Regulatory roles of p21 and apurinic/apyrimidinic endonuclease 1 in base excision repair. Journal of Biological Chemistry. 2001;276(52):48781–48789. doi: 10.1074/jbc.M109626200. [DOI] [PubMed] [Google Scholar]
- 56.Spencer DMS, Bilardi RA, Koch TH, et al. DNA repair in response to anthracycline-DNA adducts: a role for both homologous recombination and nucleotide excision repair. Mutation Research. 2008;638(1-2):110–121. doi: 10.1016/j.mrfmmm.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 57.Nakanishi M, Ozaki T, Yamamoto H, et al. NFBD1/MDC1 associates with p53 and regulates its function at the crossroad between cell survival and death in response to DNA damage. Journal of Biological Chemistry. 2007;282(31):22993–23004. doi: 10.1074/jbc.M611412200. [DOI] [PubMed] [Google Scholar]
- 58.Ho JSL, Ma W, Mao DYL, Benchimol S. p53-dependent transcriptional repression of c-myc is required for G 1 cell cycle arrest. Molecular and Cellular Biology. 2005;25(17):7423–7431. doi: 10.1128/MCB.25.17.7423-7431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bourgarel-Rey V, Savry A, Hua G, et al. Transcriptional down-regulation of Bcl-2 by vinorelbine: identification of a novel binding site of p53 on Bcl-2 promoter. Biochemical Pharmacology. 2009;78(9):1148–1156. doi: 10.1016/j.bcp.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 60.Sawyers CL, McLaughlin J, Goga A, Havlik M, Witte O. The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell. 1994;77(1):121–131. doi: 10.1016/0092-8674(94)90240-2. [DOI] [PubMed] [Google Scholar]
- 61.Goga A, Liu X, Hambuch TM, et al. p53 dependent growth suppression by the c-Abl nuclear tyrosine kinase. Oncogene. 1995;11(4):791–799. [PubMed] [Google Scholar]
- 62.Yuan Z-M, Huang Y, Ishiko T, Kharbanda S, Weichselbaum R, Kufe D. Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(4):1437–1440. doi: 10.1073/pnas.94.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samuels-Lev Y, O’Connor DJ, Bergamaschi D, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Molecular Cell. 2001;8(4):781–794. doi: 10.1016/s1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- 64.Gillotin S, Lu X. The ASPP proteins complex and cooperate with p300 to modulate the transcriptional activity of p53. FEBS Letters. 2011;585(12):1778–1782. doi: 10.1016/j.febslet.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 65.Kim WJ, Rivera MN, Coffman EJ, Haber DA. The WTX tumor suppressor enhances p53 acetylation by CBP/p300. Molecular Cell. 2012;45(5):587–597. doi: 10.1016/j.molcel.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dai C, Gu W. WTX: an unexpected regulator for p53. Molecular Cell. 2012;45(5):581–582. doi: 10.1016/j.molcel.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ando K, Ozaki T, Yamamoto H, et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. Journal of Biological Chemistry. 2004;279(24):25549–25561. doi: 10.1074/jbc.M314182200. [DOI] [PubMed] [Google Scholar]
- 68.Luo J, Nikolaev AY, Imai S-I, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107(2):137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 69.Solomon JM, Pasupuleti R, Xu L, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Molecular and Cellular Biology. 2006;26(1):28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jang KY, Noh SJ, Lehwald N, et al. SIRT1 and c-Myc promote liver tumor cell survival and predict poor survival of human hepatocellular carcinomas. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045119.e45119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cha EJ, Noh SJ, Kwon KS, et al. Expression of DBC1 and SIRT1 is associated with poor prognosis of gastric carcinoma. Clinical Cancer Research. 2009;15(13):4453–4459. doi: 10.1158/1078-0432.CCR-08-3329. [DOI] [PubMed] [Google Scholar]
- 72.Moll UM, LaQuaglia M, Benard J, Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(10):4407–4411. doi: 10.1073/pnas.92.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nikolaev AY, Li M, Puskas N, Qin J, Gu W. Parc: a cytoplasmic anchor for p53. Cell. 2003;112(1):29–40. doi: 10.1016/s0092-8674(02)01255-2. [DOI] [PubMed] [Google Scholar]
- 74.Kastan MB, Zambetti GP. Parc-ing p53 in the cytoplasm. Cell. 2003;112(1):1–2. doi: 10.1016/s0092-8674(02)01281-3. [DOI] [PubMed] [Google Scholar]
- 75.Yamamoto H, Ozaki T, Nakanishi M, et al. Oxidative stress induces p53-dependent apoptosis in hepatoblastoma cell through its nuclear translocation. Genes to Cells. 2007;12(4):461–471. doi: 10.1111/j.1365-2443.2007.01065.x. [DOI] [PubMed] [Google Scholar]
- 76.Han M-K, Song E-K, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2(3):241–251. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wiech M, Olszewski MB, Tracz-Gaszewska Z, Wawrzynow B, Zylicz M, Zylicz A. Molecular mechanism of mutant p53 stabilization: the role of HSP70 and MDM2. PLoS One. 2012;7 doi: 10.1371/journal.pone.0051426.e51426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene. 1999;18(2):477–485. doi: 10.1038/sj.onc.1202314. [DOI] [PubMed] [Google Scholar]
- 79.Bristow RG, Peacock J, Jang A, Kim J, Hill RP, Benchimol S. Resistance to DNA-damaging agents is discordant from experimental metastatic capacity in MEF ras-transformants-expressing gain of function MTp53. Oncogene. 2003;22(19):2960–2966. doi: 10.1038/sj.onc.1206405. [DOI] [PubMed] [Google Scholar]
- 80.Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG., Jr. Chemosensitivity linked to p73 function. Cancer Cell. 2003;3(4):403–410. doi: 10.1016/s1535-6108(03)00078-3. [DOI] [PubMed] [Google Scholar]
- 81.Scian MJ, Stagliano KER, Anderson MAE, et al. Tumor-derived p53 mutants induce NF-κB2 gene expression. Molecular and Cellular Biology. 2005;25(22):10097–10110. doi: 10.1128/MCB.25.22.10097-10110.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chin K-V, Ueda K, Pastan I, Gottesman MM. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science. 1992;255(5043):459–462. doi: 10.1126/science.1346476. [DOI] [PubMed] [Google Scholar]
- 83.Frazier MW, He X, Wang J, Gu Z, Cleveland JL, Zambetti GP. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Molecular and Cellular Biology. 1998;18(7):3735–3743. doi: 10.1128/mcb.18.7.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Preuss U, Kreutzfeld R, Scheidtmann KH. Tumor-derived p53 mutant C174Y is a gain-of-function mutant which activates the fos promoter and enhances colony formation. International Journal of Cancer. 2000;88:162–171. doi: 10.1002/1097-0215(20001015)88:2<162::aid-ijc3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 85.Tsutsumi-Ishii Y, Tadokoro K, Hanaoka F, Tsuchida N. Response of heat shock element within the human HSP70 promoter to mutated p53 genes. Cell Growth and Differentiation. 1995;6(1):1–8. [PubMed] [Google Scholar]
- 86.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes & Development. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mantovani R. A survey of 178 NF-Y binding CCAAT boxes. Nucleic Acids Research. 1998;26(5):1135–1143. doi: 10.1093/nar/26.5.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Di Agostino S, Strano S, Emiliozzi V, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10(3):191–202. doi: 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 89.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 90.Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3(4):419–421. [PubMed] [Google Scholar]
- 91.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends in Molecular Medicine. 2007;13(1):23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 92.Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene. 2011;30(46):4678–4686. doi: 10.1038/onc.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Selivanova G, Wiman KG. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene. 2007;26(15):2243–2254. doi: 10.1038/sj.onc.1210295. [DOI] [PubMed] [Google Scholar]
- 94.Bykov VJN, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nature Medicine. 2002;8(3):282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 95.Bykov VJN, Zache N, Stridh H, et al. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene. 2005;24(21):3484–3491. doi: 10.1038/sj.onc.1208419. [DOI] [PubMed] [Google Scholar]
- 96.Wassman CD, Baronio R, Demir O, et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nature Communications. 2013;4(article 1407) doi: 10.1038/ncomms2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.de Braekeleer E, Douet-Guilbert N, Morel F, le Bris M-J, Férec C, de Braekeleer M. RUNX1 translocations and fusion genes in malignant hemopathies. Future Oncology. 2011;7(1):77–91. doi: 10.2217/fon.10.158. [DOI] [PubMed] [Google Scholar]
- 98.Osato M, Yanagida M, Shigesada K, Ito Y. Point mutations of the RUNX1/AML1 gene in sporadic and familial myeloid leukemias. International Journal of Hematology. 2001;74(3):245–251. doi: 10.1007/BF02982056. [DOI] [PubMed] [Google Scholar]
- 99.Osato M. Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene. 2004;23(24):4284–4296. doi: 10.1038/sj.onc.1207779. [DOI] [PubMed] [Google Scholar]
- 100.Imai Y, Kurokawa M, Izutsu K, et al. Mutations of the AML1 gene in myelodysplastic syndrome and their functional implications in leukemogenesis. Blood. 2000;96(9):3154–3160. [PubMed] [Google Scholar]
- 101.Langabeer SE, Gale RE, Rollinson SJ, Morgan GJ, Linch DC. Mutations of the AMLI gene in acute myeloid leukemia of FAB types M0 and M7. Genes Chromosomes and Cancer. 2002;34(1):24–32. doi: 10.1002/gcc.10031. [DOI] [PubMed] [Google Scholar]
- 102.Takahashi A, Satake M, Yamaguchi-Iwai Y, et al. Positive and negative regulation of granulocyte-macrophage colony-stimulating factor promoter activity by AML1-related transcription factor, PEBP2. Blood. 1995;86(2):607–616. [PubMed] [Google Scholar]
- 103.Taniuchi I, Osato M, Egawa T, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111(5):621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- 104.Lutterbach B, Westendorf JJ, Linggi B, Isaac S, Seto E, Hiebert SW. A mechanism of repression by acute myeloid leukemia-1, the target of multiple chromosomal translocations in acute leukemia. Journal of Biological Chemistry. 2000;275(1):651–656. doi: 10.1074/jbc.275.1.651. [DOI] [PubMed] [Google Scholar]
- 105.Linggi B, Müller-Tidow C, van de Locht L, et al. The t(8;21) fusion protein, AML1-ETO, specifically represses the transcription of the p14ARF tumor suppressor in acute myeloid leukemia. Nature Medicine. 2002;8(7):743–750. doi: 10.1038/nm726. [DOI] [PubMed] [Google Scholar]
- 106.Wotton SF, Blyth K, Kilbey A, et al. RUNX1 transformation of primary embryonic fibroblasts is revealed in the absence of p53. Oncogene. 2004;23(32):5476–5486. doi: 10.1038/sj.onc.1207729. [DOI] [PubMed] [Google Scholar]
- 107.Satoh Y, Matsumura I, Tanaka H, et al. C-terminal mutation of RUNX1 attenuates the DNA-damage repair response in hematopoietic stem cells. Leukemia. 2012;26(2):303–311. doi: 10.1038/leu.2011.202. [DOI] [PubMed] [Google Scholar]
- 108.Wu D, Ozaki T, Yoshihara Y, Kubo N, Nakagawara A. Runt-related transcription factor 1 (RUNX1) stimulates tumor suppressor p53 protein in response to DNA damage through complex formation and acetylation. The Journal of Biological Chemistry. 2013;288:1353–1364. doi: 10.1074/jbc.M112.402594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sakakura C, Miyagawa K, Fukuda K-I, et al. Frequent silencing of RUNX3 in esophageal squamous cell carcinomas is associated with radioresistance and poor prognosis. Oncogene. 2007;26(40):5927–5938. doi: 10.1038/sj.onc.1210403. [DOI] [PubMed] [Google Scholar]
- 110.He J-F, Ge M-H, Zhu X, et al. Expression of RUNX3 in salivary adenoid cystic carcinoma: implications for tumor progression and prognosis. Cancer Science. 2008;99(7):1334–1340. doi: 10.1111/j.1349-7006.2008.00826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nomoto S, Kinoshita T, Mori T, et al. Adverse prognosis of epigenetic inactivation in RUNX3 gene at 1p36 in human pancreatic cancer. British Journal of Cancer. 2008;98(10):1690–1695. doi: 10.1038/sj.bjc.6604333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sasahira T, Kurihara M, Yamamoto K, Bhawal UK, Kirita T, Kuniyasu H. Downregulation of runt-related transcription factor 3 associated with poor prognosis of adenoid cystic and mucoepidermoid carcinomas of the salivary gland. Cancer Science. 2011;102(2):492–497. doi: 10.1111/j.1349-7006.2010.01787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lau QC, Raja E, Salto-Tellez M, et al. RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer. Cancer Research. 2006;66(13):6512–6520. doi: 10.1158/0008-5472.CAN-06-0369. [DOI] [PubMed] [Google Scholar]
- 114.Ito Y, Miyazono K. RUNX transcription factors as key targets of TGF-β superfamily signaling. Current Opinion in Genetics and Development. 2003;13(1):43–47. doi: 10.1016/s0959-437x(03)00007-8. [DOI] [PubMed] [Google Scholar]
- 115.Fukamachi H, Ito K. Growth regulation of gastric epithelial cells by Runx3. Oncogene. 2004;23(24):4330–4335. doi: 10.1038/sj.onc.1207121. [DOI] [PubMed] [Google Scholar]
- 116.Ito K, Lim AC-B, Salto-Tellez M, et al. RUNX3 attenuates β-catenin/T cell factors in intestinal tumorigenesis. Cancer Cell. 2008;14(3):226–237. doi: 10.1016/j.ccr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 117.Yano T, Ito K, Fukamachi H, et al. The RUNX3 tumor suppressor upregulates bim in gastric epithelial cells undergoing transforming growth factor β-induced apoptosis. Molecular and Cellular Biology. 2006;26(12):4474–4488. doi: 10.1128/MCB.01926-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yamada C, Ozaki T, Ando K, et al. RUNX3 modulates DNA damage-mediated phosphorylation of tumor suppressor p53 at Ser-15 and acts as a Co-activator for p53. Journal of Biological Chemistry. 2010;285(22):16693–16703. doi: 10.1074/jbc.M109.055525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 120.Welch RD, Jones AL, Bucholz RW, et al. Effect of recombinant human bone morphogenetic protein-2 on fracture healing in a goat tibial fracture model. Journal of Bone and Mineral Research. 1998;13(9):1483–1490. doi: 10.1359/jbmr.1998.13.9.1483. [DOI] [PubMed] [Google Scholar]
- 121.Lau CC, Harris CP, Lu X-Y, et al. Frequent amplification and rearrangement of chromosomal bands 6p12-p21 and 17p11.2 in osteosarcoma. Genes Chromosomes and Cancer. 2004;39(1):11–21. doi: 10.1002/gcc.10291. [DOI] [PubMed] [Google Scholar]
- 122.Brubaker KD, Vessella RL, Brown LG, Corey E. Prostate cancer expression of runt-domain transcription factor Runx2, a key regulator of osteoblast differentiation and function. Prostate. 2003;56(1):13–22. doi: 10.1002/pros.10233. [DOI] [PubMed] [Google Scholar]
- 123.Kayed H, Jiang X, Keleg S, et al. Regulation and functional role of the Runt-related transcription factor-2 in pancreatic cancer. British Journal of Cancer. 2007;97(8):1106–1115. doi: 10.1038/sj.bjc.6603984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Endo T, Ohta K, Kobayashi T. Expression and function of Cbfa-1/Runx2 in thyroid papillary carcinoma cells. Journal of Clinical Endocrinology and Metabolism. 2008;93(6):2409–2412. doi: 10.1210/jc.2007-2805. [DOI] [PubMed] [Google Scholar]
- 125.Blyth K, Vaillant F, Hanlon L, et al. Runx2 and MYC collaborate in lymphoma development by suppressing apoptotic and growth arrest pathways in vivo. Cancer Research. 2006;66(4):2195–2201. doi: 10.1158/0008-5472.CAN-05-3558. [DOI] [PubMed] [Google Scholar]
- 126.Westendorf JJ, Zaidi SK, Cascino JE, et al. Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21CIP1/WAF1 promoter. Molecular and Cellular Biology. 2002;22(22):7982–7992. doi: 10.1128/MCB.22.22.7982-7992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ozaki T, Wu D, Sugimoto H, Nagase H, Nakagawara A. Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death and Disease. 2013;4(article e610) doi: 10.1038/cddis.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]