

Abstract
During the past five years, the three-dimensional structures of 14 different G-protein coupled receptors (GPCRs) have been resolved by X-ray crystallography. The most recently published structures, those of the opioid receptors (ORs), are remarkably important in pain modulation, drug addiction, and mood disorders. These structures, confirmed previously proposed key interactions conferring potency and antagonistic properties, including the well-known interaction with Asp138, conserved in all aminergic GPCRs. In addition, crystallization of the opioid receptors highlighted the potential function of the ECL2 and ICL2 loops. We have previously reported a set of potent and selective kappa opioid receptor peptide agonists, of which ff(D-nle)r-NH2 is among the most potent and selective ones. These peptides were identified from the deconvolution of a 6,250,000 tetrapeptide combinatorial library. A derivative of this set is currently the subject of a phase 2 clinical trial in the United States. In this work, we describe comparative molecular modeling studies of kappa-OR peptide agonists with the co-crystallized antagonist, JDTic, and also report structure-activity relationships of 23 tetrapeptides. The overall binding and contact interactions are sound and interactions known to favor selectivity and potency were observed. Additional modeling studies will reveal conformational changes that the kappa-OR undergoes upon binding to these peptide agonists.
Keywords: kappa-opioid receptor, tetrapeptide, molecular modeling, combinatorial chemistry
1. Introduction
Opioid receptors (OR) belong to Family A of the G-Protein Coupled Receptor (GPCR) superfamily. GPCRs activate signal transduction from the outside to the inside of cells. These receptors are therapeutic targets for approximately 40% of current medicinal drugs. Notably, the contributions of Dr. Robert Lefkowitz and Dr. Bryan Kobilka to the GPCR field merited the 2012 Nobel Prize in Chemistry[1]. Opioid receptors in particular, participate in many physiological processes, including pain, drug addiction and mood disorders, among other diseases. Due to the therapeutic and structural relevance of the OR, intense research has been directed to understand their structure and function. The four OR subtypes, named mu, kappa, delta and the nociceptin receptor, have been crystalized this year at 2.8, 2.9, 3.4, and 3.0 Å resolution, respectively [2-5]. These three-dimensional structures allow one to investigate, at the atomic level, the structural features that promote binding affinity, selectivity and ultimately, give insights into the mechanism(s) of activation of these receptors. In addition, these three-dimensional structures can be used as templates for the development of molecular models of other GPCRs. It is expected that molecular models will help fill the gaps in the 3D structures of other GPCRs, as well as be used to investigate activation mechanisms, postulate allosteric sites, develop structure-affinity and structure-function relationships, etc.
A number of molecules have been synthesized [6] and evaluated for opioid receptor binding affinity through binding assays, as well as functional assays. The chemical structures of the ligands, as well as their associated biochemical information, can be easily obtained from public domain sources: for example from the International Union of Basic and Clinical Pharmacology (http://www.iuphar-db.org/), CheMBL, PubChem, among others.
Over the years a number of opioid ligands have been identified with the deconvolution of mixture-based combinatorial libraries at the Torrey Pines Institute for Molecular Studies (TPIMS) [7-9]. Efforts have been made to integrate this mixture-based approach with computational screening [10]. Chemoinformatic studies by means of molecular scaffolds, molecular properties, and structural fingerprints show the diversity of these libraries and their uniqueness, based on: a) the partial overlap with the structural space of drugs, b) the presence of scaffolds not contained in other compound collections [11], and c) the increased molecular complexity as compared to compound libraries commonly used in high-throughput screening (HTS) programs [12].
For opioid receptor binding affinity, the scaffolds explored in these libraries include bicyclic guanidines, piperazines, triamines, and peptides of different lengths [13-15]. A particularly useful study consisted of the deconvolution of a 6,250,000 tetrapeptide combinatorial library. This library allowed the identification of potent and selective ligands for the kappa OR, with the general sequence ffir-NH2 [16]. A derivative of this set is currently the subject of a phase 2 clinical trial.
Molecular models of small molecules [17] and peptides [18] bound to opioid receptors, are described elsewhere. Conformational preferences of cyclic peptides have also been explored [19]. With the aim of investigating the mode of action of opioid-binding peptides, cyclic analogues (e. g. pep10) have been synthesized and evaluated by Pogozheva et al [20]. For the mu-OR, potent and selective cyclic peptides were obtained [21]. However, for the kappa-OR, potency was accomplished, but selectivity remained elusive for these cyclic peptides. The binding mode derived by Pogozheva et al [20] of a potent disulfur cyclic peptide fulfills pharmacophoric features known to be important for OR binding affinity. Our understanding of GPCR function is increasing due to the crystal structures now available. It can be hypothesized that structural features that are common among the available crystal structures could be translated across other GPCRs. An interesting example is the comparison of the crystal structures of the agonist-bound β-adrenoceptors in an inactive state [22]. The structures of the thermostabilized receptors bound to a partial agonist or a full agonist were nearly identical. Moreover, the structures of the receptors were very similar to the structures when bound to antagonists. A similar goal was pursued and achieved for the β-adrenergic receptor (β2AR) where the agonist-bound receptor remained in the inactive form [23]. This approach can be translated to other GPCRs. For example, the report of the crystallographic structure of the kappa-OR is complemented with the molecular modeling of agonists and antagonists into the binding pocket, including salvinorin A (SalA) analogues. SalA is a natural product described in 1982 [24] and pharmacologically characterized in 2004 [25]. The unique feature of SalA and analogues, compared to all previously known opioid ligands, is that it lacks a charged or polar nitrogen atom, known to be essential for OR binding recognition.
In this work, using the inactive state of the crystallographic structure of the kappa-OR, we explored and compared the conformational preferences of the side chains in the binding site when bound to the crystallographic ligand (an antagonist), to a potent non-selective kappa-OR cyclic peptide agonist, and to our previously reported potent and kappa-OR selective tetrapeptide agonist. Structure-activity relationship studies of 24 analogues of our kappa-OR agonists are in agreement with our model.
2. Results and Discussion
A summary of the relevant interactions involved in binding affinity, selectivity and activation of the kappa-OR, is shown in Table 1; these interactions have been identified mainly from mutagenesis studies [3].
Table 1.
Important interactions in the kappa-OR orthosteric binding site
| Interacting residues | Type of interaction | Notes |
|---|---|---|
| Potency | ||
| Asp138 | Salt bridge | Conserved residue among
aminergic GPCRs |
| Selectivity to kappa-OR | ||
| Val108 | Hydrophobic | Based on JDTic
Interactions consistent with mutagenesis and SAR studies |
| Val118 | Hydrophobic | |
| Ile294 | Hydrophobic | |
| Tyr312 | Hydrophobic / polar | |
| Selectivity to kappa-OR | ||
| Glu297 | Salt bridge at the entrance of the binding pocket |
Based on morphinans. Message
- address concept. Interactions consistent with mutagenesis studies |
| Ile294 | Hydrophobic | Based on morphinans.
Interactions consistent with mutagenesis studies |
| Glu209 | Polar | Based on morphinans.
Interactions consistent with mutagenesis studies. |
| Ser211 | Polar | Located at the ECL2,
interactions found in nor-BNI |
| Agonism/antagonism | ||
| Trp287 | Hydrophobic | Key residues in the
message-address concept. |
| His291 | Hydrophobic | |
| ECL2 | Deemed important for
ligand recognition and selectivity. |
|
| D/ERY motif | Salt bridge Arg3.5 to: Thr2736.34 (kappa); Asp/Glu6.30 (mu, delta) |
Ionic lock of TM3 and TM6
thought to stabilize the inactive conformation. Located at the cytoplasmic end of TM3. |
| NPXXY motif |
Molecular switch
associated with GPCR activation. Located at the cytoplasmic side of TM7. |
Three kappa-OR ligands were investigated in this study; JDTic [3], pep10 [20], and ff(D-nle)r-NH2 [16]. In order to gain some insight on the flexibility of each ligand-receptor complex, we performed a conformational search of the side chains of all the residues within 4Å to each ligand. During the searches, the ligand and the neighboring residues were free to move. A summary of residues neighboring each ligand and ligand-receptor interactions are shown in Tables 2 and 3, respectively.
Table 2.
Residues within 4Å to ligand; key residues are in bold, residues important for kappa-OR selectivity are underlined
| hydrophobic | acidic | basic | Polar | |
| JDTic X-Ray structure |
Val108,
Val118,
Trp124, Val134, Ile135, Met142, Ile209, Val230, Trp287, Ile294, Ile316 |
Asp138 | Lys227, His291 |
Thr111, Gln115, Tyr139, Cys210, Tyr312, Gly319, Tyr320 |
| Pep10 |
Val118,
Trp124, Val 134, Ile135, Met142, Ile208, Val230, Ile290, Ile294, Leu309 |
Asp138,
Glu209, Glu297 |
Lys227, His291 |
Tyr139, Cys210, Tyr312 |
| ff(D-Nle)r-NH2 | Trp124, Ile135, Met142, Leu212, Met226, Val230, Trp287, Ile290, Ile294, Ile316 |
Asp138,
Glu209, Asp223, Glu297 |
Arg202, Lys227, His291 |
Tyr139, Cys210,
Ser211, Tyr219, Tyr312 |
Table 3.
Ligand-receptor interactions observed for pep10 and ff(D-nel)r-NH2
| Ligand * | Receptor * | Interactions | distance | E (kcal/mol) | Importance |
| pep10 | |||||
| N 1 | ID2 Asp138 (A) | H-donor | 2.54 | −1.0 | |
| N 1 | OD2 Asp138 (A) | ionic | 3.22 | −8.4 | |
| 6-ring | CA Glu209 (A) | pi-H | selectivity | ||
| 6-ring | CG Glu209 (A) | pi-H | |||
| SG 81 | NZ Lys227 (A) | H-acceptor | 3.22 | −1.7 | |
| OH 12 | ND1 His291 (A) | H-acceptor | 2.89 | −0.9 | |
| O 79 | OE2 Glu297 (A) | H-donor | 2.67 | −1.4 | selectivity |
| 6-ring | CG1Ile294 (A) | pi-H | |||
| O 27 | OH Tyr312 (A) | H-acceptor | 2.67 | −3.8 | selectivity |
| ff(D-nle)r-NH2 | |||||
| N1 1 | O Asp138 (A) | H-donor | 2.75 | −0.9 | |
| N1 1 | OD1 Asp138 (A) | ionic | 2.80 | −0.9 | |
| N1 1 | OD2 Asp138 (A) | ionic | 2.80 | −8.3 | |
| N23 23 | OH Tyr139 (A) | H-donor | 2.98 | −0.5 | |
| N69 69 | OE1 Glu209 (A) | H-donor | 2.62 | −1.1 | selectivity |
| N72 72 | OE2 Glu209 (A) | H-donor | 2.63 | −1.4 | |
| C70 70 | OE1 Glu209 (A) | ionic | 2.80 | −2.2 | |
| C70 70 | OE2 Glu209 (A) | ionic | 2.80 | −1.8 | |
| O65 65 | NZ Lys227 (A) | H-acceptor | 2.80 | −1.0 | |
| N86 86 | OE2 Glu297 (A) | H-donor | 3.21 | −1.1 | selectivity |
| O26 26 | OH Tyr312 (A) | H-acceptor | 2.76 | −1.1 | selectivity |
PDB atom nomenclature
2.1 Conformational search of JDTic-kappa-OR binding site
The SAR, binding interactions and pharmacophoric features of the kappa-OR co-crystalized ligand, JDTic, are described elsewhere [3]. Here we explored the conformational freedom of JDTic into the inactive state of the kappa-OR resulting in 11 conformations. Notably, except for Tyr312, Gln115 and Thr139, the conformations of the residues around JDTic remained nearly identical to those observed in the crystal structure. A graphical representation of the different conformations and the relative orientations of JDTic are shown in Figure S1A. The previously reported V-shape of JDTic observed when bound to the kappa-OR [3] was maintained in the conformational search. These conformations can be clustered into two slightly different orientations. During the conformational search, we deleted structured water molecules to explore the effect of water-mediated stabilization. Interestingly, the two binding poses obtained for JDTic differed from the reported orientation observed in the kappa-OR crystal structure, (see Figure S1B in Supplementary material) with a larger displacement on the isoquinoline side of JDTic. For pose1, shown in Figure S1C, the hydroxyl group of the isoquinoline group is able to engage in a hydrogen bonding interaction with Tyr139. In doing so, the interaction with Asp138 is now through the amide nitrogen atom, instead of the amines in the piperidine and the isoquinoline moieties, as reported [3]. The displacement obtained in pose 2, shown in Figure S1D, is more dramatic. In this pose, the isoquinoline ring makes contacts with Asp223, and the carbonyl group maintains a hydrogen bond interaction with Tyr312. Although these two putative orientations make favorable polar interactions, in both cases the interaction of the amine groups with Asp138 and the overall orientation of JDTic in the binding pocket are compromised. This underscores the importance of structured water molecules.
2.2 Conformational search of pep10-kappa-OR binding site
The conformationally constrained cyclic peptide, pep10, derived by Pogozheva et al. [20] was translated to the coordinates of the kappa-OR crystal structure. The resulting complex was then subjected to conformational search within the binding site, producing 29 conformers. The conformational space spanned by pep10 and representative conformers are shown in Figure 1A and 1B. While the hydroxyphenyl group of pep10 located at the bottom of the pocket remained unchanged, in terms of both conformation and orientation, during the conformational search, the other two phenyl rings spanned a large number of rotamers. This is an indication of the tightness at the bottom of the pocket as well as the favorable orientation of the hydroxyphenyl group of pep10. An example of the relative orientation of JDTic with pep10 is shown in Figure 1C.
Figure 1.
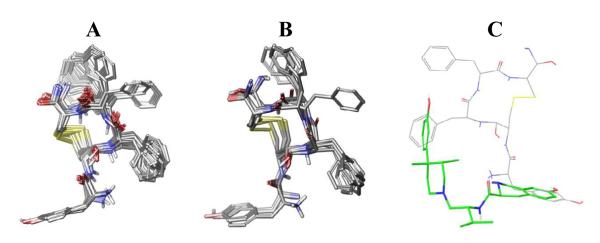
A) Conformations of pep10 from the conformational search, B) representative conformations after conformer clustering procedure. C) Relative orientation of JDTic (green) and pep10 (grey) into kappa-OR binding pocket. The structure of JDTic corresponds to the one in the crystal structure (PDBID: 4DJH).
As expected, the positively charged amine group makes a salt bridge with Asp138, and the hydroxyphenyl group located at the bottom of the pocket forms a hydrogen bond with His291. These two interactions serve to anchor pep10 into this region. In this model, pep10 is able to make hydrogen bond interactions with Tyr312 and Glu297. These residues have been regarded as important for kappa selectivity; substitution of Tyr312 for Trp7.35, found in the mu-OR results in the loss of this polar interaction. However, pep10 binds tightly to the mu-OR. Therefore, absence of an H-bond interaction in mu was not sufficient, in and of itself, to impair the binding affinity of pep10 to mu-OR. In turn; Glu297 has been found to be relevant to kappa selectivity, particularly for morphinans. Although this interaction would enhance binding affinity to kappa-OR, it is absent in the kappa-OR crystal structure bound to JDTic, suggesting that it is not a requirement for achieving kappa selectivity.
2.3 Conformational search of ff(D-nle)r-NH2-kappa-OR binding site
Initially, automated docking was attempted for ff(D-nle)r-NH2 (using GLIDE). The resulting binding modes did not show the expected interactions, e. g. the salt bridge with Asp138. One of the reasons that makes automated docking of this tetrapeptide a challenging task is its high flexibility; this tetrapeptide contains 22 rotatable bonds. In contrast, typical small molecules in docking studies have up to 10 rotatable bonds [12]. In order to develop an initial orientation of ff(D-nle)r-NH2 in the binding pocket, we used pep10 as a reference and performed a conformational search similar to that performed for JDTic and pep10. For comparison, the relative orientation of ff(D-nle)r-NH2 and JDTic is shown in Figure 2A, and to pep10, shown in Figure 2B. The relatively good alignment of ff(D-nle)r-NH2 and pep10 and the presence of important interactions within the kappa-OR binding site gave a level of confidence to the binding model of ff(D-nle)r-NH2. To note, while ff(D-nle)r-NH2 is a selective kappa binder, pep10 is not. Therefore, it is expected that interactions promoting selectivity to kappa-OR should be present in ff(D-nle)r-NH2 but absent in pep10. The conformational search of ff(D-nle)r-NH2 within the kappa-OR binding site produced 34 very similar conformers. The orientation obtained allows the characteristic salt bridge with Asp138 and the localization of a phenyl ring at the bottom of the pocket. A number of other favorable interactions were also present, and are summarized in Table 3. Similarly to pep10, ff(D-nle)r-NH2 makes H-bond interactions to Lys227, Glu297 and Tyr312. However, while pep10 interacts weakly with Glu209 via π-H contacts, ff(D-nle)r-NH2 makes strong H-bond and ionic interactions with this residue, located in the ECL2. Polar interactions with Glu209 appear to be promoting the kappa-OR subtype selectivity of ff(D-nle)r-NH2. It is worth stressing that peptide-binding receptors crystallized to date (opioid receptors and CXCR4) reveal that the ECL2 forms a β-hairpin structure, believed important for recognition and selectivity [26].
Figure 2.

Relative orientation of ff(D-nle)r-NH2 (grey thick sticks) to A) JDTic (green) and B) pep10 (tin sticks) into the kappa-OR binding pocket. The structure of JDTic corresponds to the one in the crystal structure (PDBID: 4DJH). Orientation C) and contact interactions D) of ff(D-nle)r-NH2 into the kappa-OR binding site.
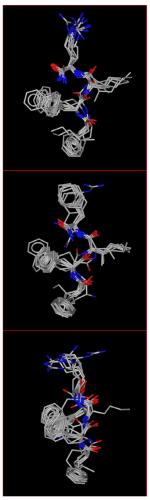
Lastly, our conformational search of the side chains in the binding pocket shows that ff(D-nle)r-NH2 favors the side chain rotation of Trp287, located at the bottom of the pocket. Experimental studies [27] indicate that Trp6.48 is a key residue in GPCR activation. Figure 3 shows the orientation of Trp287 in the crystallographic structure (green) and in the ff(D-nle)r-NH2 model. In the presence of JDTic, the isopropyl group locks the orientation of Trp287 making hydrophobic interactions. Meanwhile, ff(D-nle)r-NH2 favors the side chain rotation of Trp287, most likely enhancing its agonist behavior. This subtle structural difference is the subject of discussion in regard to kappa-OR agonists versus antagonists [3]. Activation of GPCRs conveys changes in the orientation of the transmembrane helices (Supplementary Table S1). For example, the all-atom rmsd for rhodopsin when comparing the dark or inactive state (PDBID: 1U19) to the light or activated state (PDBID:3PQR) is 3.07 Å. Changes in the binding pocket are less pronounced (rmsd 1.75 Å). Moreover, the all-atom rmsd after alignment of TMs 1, 2, and 4 (thought to move less upon activation) increases to 6.5 Å. The same trend is observed when comparing the kappa-OR crystal structure with a publicly available homology model of kappa-OR in a modeled activated state (Supplementary Table S1). This illustrates how small changes in the binding pocket lead to large changes in the conformation of the receptor.
Figure 3. Effect of ff(D-nle)r-NH2 in Trp287.

Panel A) shows the overall structure of the kappa-OR, showing the binding model of ff(D-nle)r-NH2, relevant residues are labeled. B) and C) Comparison of the orientation of Trp in the presence of the antagonist crystallographic ligand, JDTic, shown in green, and the agonist tetrapeptide ff(D-nle)r-NH2, shown in gray.
2.4 Structure activity relationships for ff(D-nle)r-NH2 analogues
The model obtained for ff(D-nle)r-NH2 was then used as a template to analyze tetrapeptides obtained from the same combinatorial library [16]. We have previously reported the side chains and the corresponding Ki values for each tetrapeptide, they are summarized in Table 4 [16]. Clearly, incorporation of a tryptophan side chain in the R3 position conducted to peptides with Ki values greater than 1000nM. Another clear pharmacophoric feature is the presence of an arginine group in the R4 position, this group conducted to the best kappa-OR binders of the series.
Table 4.
Ki values for tetrapeptides studied here and 3D similarity to reference peptide ff(D-nel)r-NH2
| Ki (nM) | R1 | R2 | R3 | R4 | 3D similarity to mol1 | |
|---|---|---|---|---|---|---|
| 1 | 1.2 | f | f | nle | r | 1 |
| 2 | 1.5 | nle | nal | i | r | 0.623 |
| 3 | 2.3 | nle | nal | nle | r | 0.561 |
| 4 | 2.4 | f | f | i | r | 0.807 |
| 5 | 3.6 | f | nal | i | r | 0.646 |
| 6 | 4.2 | nle | f | nle | r | 0.588 |
| 7 | 7.1 | f | nal | nle | r | 0.715 |
| 8 | 9.3 | nle | f | i | r | 0.647 |
| 9 | 26 | f | f | nle | cha | 0.774 |
| 10 | 27 | nle | nal | nle | cha | 0.542 |
| 11 | 33 | f | nal | nle | cha | 0.601 |
| 12 | 53 | f | nal | i | cha | 0.596 |
| 13 | 56 | nle | nal | i | cha | 0.562 |
| 14 | 64 | f | f | i | cha | 0.745 |
| 15 | 71 | nle | f | nle | cha | 0.587 |
| 16 | 341 | nle | f | i | cha | 0.564 |
| 17 | 1140 | f | f | W | r | 0.598 |
| 18 | 1150 | f | nal | W | r | 0.514 |
| 19 | 1760 | nle | nal | W | r | 0.514 |
| 20 | 4550 | nle | f | W | r | 0.452 |
| 21 | 6000 | f | nal | W | cha | 0.53 |
| 22 | 10000 | nle | f | W | cha | 0.566 |
| 23 | 10000 | f | f | W | cha | 0.555 |
| 24 | 10000 | nle | nal | W | cha | 0.495 |
Taking ff(D-nle)r-NH2 as a reference, 3D overlays of each peptide were performed, similarity values are summarized in Table 4. Overlays are graphically represented in Table 5, along with average Ki and 3D similarity values divided into three groups. These overlays are based on common pharmacophoric features to the reference peptide and do not consider any spatial restrains from the receptor. Not surprisingly the most active tetrapeptides (molecules 2-8) have overall higher similarity to ff(D-nle)r-NH2 compared to the two other groups; molecules 9-16 and 17-24.
Table 5.
3D overlay to reference peptide ff(D-nel)r-NH2 classified into three groups by activity
| Molecules aligned to molecule 1 | Average Ki (nM) |
Average 3D similarity
to molecule 1 |
|
| 2-8 Common pharmacophore to active peptides: R4=r and R3≠W |
|
3.95 | 0.71 |
| 9-16 Common pharmacophore to middle active peptides: R4=cha and R3≠W |
83.87 | 0.60 | |
| 17-24 Common pharmacophore to inactive peptides R3=W regardless R1,R2 and R4 |
5575 | 0.53 | |
The importance of the R3 and R4 positions can be graphically and numerically compared with the tree shown in Figure 4. Based on Ki values, the first split corresponds to the R3 position, where the average Ki values for peptides with R3=W is 5575 and for R3≠W is 43.9. The next split corresponds to the R4 position, where in all cases (nodes 4-7), the arginine side chain is preferred. The average Ki values are lower when R4=r than R4=D-cha. The last split (terminal nodes 8-11) corresponded to R3 and R1 for nodes 6 and 7, respectively. For node 6, molecules 9-16, nle consistently conducted to better kappa-OR binders. However, for the most active tetrapeptides (node 7) the average Ki for R1=r or R1=nle did not differ significantly: 3.6 and 4.3nM, respectively.
Figure 4. Classification tree for 24 tetrapeptides based on Ki values.

Each split classifies molecules based on Ki values. Nodes are identified by ID numbers. Average and variance of Ki values at each node are denoted by Mu and Var values. Number of members on each node is denoted by N.
The drastic drop in activity when R3=W shows that this position cannot allocate this substituent while maintaining the biologically significant orientation of the remaining groups into the kappa-OR binding pocket. Importantly, peptides with the arginine group at the R4 position, and with R3≠W, not only were the most active but also the most selective ones [16]. According to our model, described above, the presence of an arginine group in the R4 position allows strong H-bond and ionic interactions with Glu209, which are important for kappa-OR selectivity, in agreement with our model.
This study exemplifies the feasibility of modeling a kappa-OR peptide agonist into the crystallographic structure obtained in the inactive state. The overall binding and contact interactions are sound, and the hypothesis is supported by crystallographic structures of agonist-bound receptors obtained in the inactive state [22, 23].
3. Conclusions
Molecular modeling studies were conducted for the kappa-OR co-crystallized antagonist and for selective and non-selective peptide agonists using the recently published crystallographic structure of the kappa-OR (in the active state). The orientation of the ligands in the kappa-OR binding site is in agreement with interactions known to promote binding affinity and selectivity. In particular, strong interactions with Glu209, located in the ECL2, were observed for the selective peptide agonist. This interaction appears to be enhancing kappa selectivity. In addition, the selective peptide agonist studied here, ff(D-nle)r-NH2, promoted rotation of the Trp287 side chain, known to be involved in GPCR activation. Structure-activity relationships derived for 23 tetrapeptides obtained from the same combinatorial library underscored the importance for an arginine side chain towards Glu209 and the drop in activity when incorporating a tryptophan group at the R3 position. The knowledge-driven modeling approach employed here produced sound models and can be expanded to conformational studies of other relevant opioid receptor ligands.
4. Methods
Data collection
Atomic coordinates of rhodopsin in the inactive (PDBID: 1U19) and active (PDBID: 3PQR) states, as well as of the kappa-OR in the inactive form (PDBID: 4DJH) were downloaded from the Protein Data Bank (PDB). A homology model of the kappa-OR and the atomic coordinates of the peptide modeled by Pogozheva et al (pep10) were downloaded from http://mosberglab.phar.umich.edu/resources/. Translation to the same coordination frame of the active and inactive form of each receptor and rmsd calculations were performed with Chimera [28]. The missing intracellular loop in the X-ray structure of the kappa-OR was introduced and modeled with Maestro [29].
Conformational searches
The conformational searches were performed in Macromodel (Schrödinger Inc.) using the OPLS-2005 force field and GB/SA water model (in the presence of the receptor). The ligands were free to rotate, translate and change conformation during the conformational search. Residues within a radius of 4Å to the ligand were also free to change conformation. For all the residues in this layer, constrained-atom mutual interactions were calculated. An additional shell of 5Å was set from the first layer; residues on this layer were not allowed to move. The remainder of the protein was ignored. The energy minimization was performed using the Polak-Ribier Conjugate Gradient (PRCG) method [30]. Default settings were employed for convergence (gradient, threshold of 0.05) for up to 500 iterations. The conformational search was sampled using the “mixed torsional/large-scale-low-mode” method. Torsion sampling options were set to intermediate, retaining mirror-image conformations. The maximum number of steps was 200. The energy window for saving structures was 21.0 kJ/mol. Redundant conformers were eliminated using a maximum atom deviation cutoff of 0.5Å. The resulting complexes were clustered based on atomic rmsd of the ligands and neighboring residues within 4Å of the ligand. The similarity rmsd matrix was clustered using a complete linkage method, and the number of clusters was chosen using the Kelley penalty function [31, 32]. The centroid of each cluster was selected to represent each cluster. The final complexes were analyzed with Maestro 9.3 [29].
3D alignments and similarity values
3D overlays were performed within MarvinSketch 5.11.4 by ChemAxon. The “align by pharmacophore type” option was used and the flexible alignment was lunched for 40 initial conformations. This 3D alignment protocol searches for matching of H-bond donors, acceptors, aromatic and hydrophobic groups, as well as positive and negative charges, the similarity values correspond to the Tanimoto coefficient.
Supplementary Material
Highlights.
* Agonist and antagonist tetrapeptides were analyzed in the inactive conformation of the kappa-OR crystal structure
* Model developed highlights the importance of polar and H-bond interactions of selective kappa-OR tetrapeptides with Glu209, located in the ECL2.
* Rotation of Trp287, important for GPCR’s activation, was observed in the model of our tetrapeptide agonist.
Acknowledgements
This work was supported by the State of Florida, Executive Officer of the Governor’s Department of Economic Opportunity and funded, in part, by NIDA DA031370. We would like to thank ChemAxon (http://www.chemaxon.com) and StatSoft for kindly providing an academic license of their software. KMM thanks DGAPA-UNAM (PAPIIT IA200513-2).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1. [21 Nov 2012];The Nobel Prize in Chemistry 2012, N. o. http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2012/
- 2.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–U170. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu HX, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–U369. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–U171. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson AA, Liu W, Chun E, Katritch V, Wu HX, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–U150. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarado C, Guzman A, Diaz E, Patino R. Synthesis of Tramadol and Analogues. J. Mex. Chem. Soc. 2005;49:324–327. [Google Scholar]
- 7.Houghten RA, Pinilla C, Giulianotti MA, Appel JR, Dooley CT, Nefzi A, Ostresh JM, Yu YP, Maggiora GM, Medina-Franco JL, Brunner D, Schneider J. Strategies for the use of mixture-based synthetic combinatorial libraries: Scaffold ranking, direct testing, in vivo, and enhanced deconvolution by computational methods. J. Comb. Chem. 2008;10:3–19. doi: 10.1021/cc7001205. [DOI] [PubMed] [Google Scholar]
- 8.Houghten RA, Pinilla C, Appel JR, Blondelle s. E., Dooley CT, Eichler J, Nefzi A, Ostresh JM. Mixture-based synthetic combinatorial libraries. J. Med. Chem. 1999;42:3743–3778. doi: 10.1021/jm990174v. [DOI] [PubMed] [Google Scholar]
- 9.Pinilla C, Appel JR, Borras E, Houghten RA. Advances in the use of synthetic combinatorial chemistry: mixture-based libraries. Nat. Med. 2003;9:118–122. doi: 10.1038/nm0103-118. [DOI] [PubMed] [Google Scholar]
- 10.Yongye AB, Pinilla C, Medina-Franco JL, Giulianotti MA, Dooley CT, Appel JR, Nefzi A, Scior T, Houghten RA, Martinez-Mayorga K. Integrating computational and mixture-based screening of combinatorial libraries. J. Mol. Model. 2011;17:1473–1482. doi: 10.1007/s00894-010-0850-1. [DOI] [PubMed] [Google Scholar]
- 11.Singh N, Guha R, Giulianotti MA, Pinilla C, Houghten RA, Medina-Franco JL. Chemoinformatic Analysis of Combinatorial Libraries, Drugs, Natural Products, and Molecular Libraries Small Molecule Repository. J. Chem. Inf. Model. 2009;49:1010–1024. doi: 10.1021/ci800426u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez-Vallejo F, Giulianotti MA, Houghten RA, Medina-Franco JL. Expanding the medicinally relevant chemical space with compound libraries. Drug Discovery Today. 2012;17:718–726. doi: 10.1016/j.drudis.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Mayorga K, Medina-Franco JL, Giulianotti MA, Pinilla C, Dooley CT, Appel JR, Houghten RA. Conformation-opioid activity relationships of bicyclic guanidines from 3D similarity analysis. Bioorg. & Med. Chem. 2008;16:5932–5938. doi: 10.1016/j.bmc.2008.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yongye AB, Appel JR, Giulianotti MA, Dooley CT, Medina-Franco JL, Nefzi A, Houghten RA, Martinez-Mayorga K. Identification, structure-activity relationships and molecular modeling of potent triamine and piperazine opioid ligands. Bioorg. & Med. Chem. 2009;17:5583–5597. doi: 10.1016/j.bmc.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yongye AB, Martinez-Mayorga K. Molecular Aspects of Opioid Receptors and Opioid Receptor Painkillers. In: Racz Gabor B., Noe CE., editors. Pain Management - Current Issues and Opinions. InTech; 2012. [Google Scholar]
- 16.Dooley CT, Ny P, Bidlack JM, Houghten RA. Selective ligands for the mu, delta, and Kappa opioid receptors identified from a single mixture based tetrapeptide positional scanning combinatorial library. J. Biol. Chem. 1998;273:18848–18856. doi: 10.1074/jbc.273.30.18848. [DOI] [PubMed] [Google Scholar]
- 17.Gentilucci L, Tolomelli A, De Marco R, Artali R. Molecular Docking of Opiates and Opioid Peptides, a Tool for the Design of Selective Agonists and Antagonists, and for the Investigation of Atypical Ligand-Receptor Interactions. Curr. Med. Chem. 2012;19:1587–1601. doi: 10.2174/092986712799945030. [DOI] [PubMed] [Google Scholar]
- 18.Eguchi M. Recent advances in selective opioid receptor agonists and antagonists. Med. Res. Rev. 2004;24:182–212. doi: 10.1002/med.10059. [DOI] [PubMed] [Google Scholar]
- 19.Yongye AB, Li YM, Giulianotti MA, Yu YP, Houghten RA, Martinez-Mayorga K. Modeling of peptides containing D-amino acids: implications on cyclization. J. Comp.-Aid. Mol. Des. 2009;23:677–689. doi: 10.1007/s10822-009-9295-y. [DOI] [PubMed] [Google Scholar]
- 20.Przydzial MJ, Pogozheva ID, Ho JC, Bosse KE, Sawyer E, Traynor JR, Mosberg HI. Design of high affinity cyclic pentapeptide ligands for κ-opioid receptors. J. Pep. Res. 2005;66:255–262. doi: 10.1111/j.1399-3011.2005.00295.x. [DOI] [PubMed] [Google Scholar]
- 21.Mosberg HI, Fowler CB. Development and validation of opioid ligand-receptor interaction models: the structural basis of mu vs. delta selectivity. J. Pep. Res. 2002;60:329–335. doi: 10.1034/j.1399-3011.2002.21061.x. [DOI] [PubMed] [Google Scholar]
- 22.Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AGW, Schertler GFX, Tate CG. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SGF, Choi HJ, DeVree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ortega A, J.F. B, Manchad PS. Salvinorin, a New trans-Noclerodane Diterpene from Salvia divinorum (Labiatae) J. Chem. Soc. Perkin Trans. 1982:2505–2508. [Google Scholar]
- 25.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: A potent naturally occurring nonnitrogenous k opioid selective agonist. Proc. Natl. Acad. Sci. USA. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang JB, Johnson PS, Wu JM, Wang WF, Uhl GR. Human kappa-opiate receptor 2nd extracellular loop elevates dynorphins affinity for human mu/kappa chimeras. J. Biol. Chem. 1994;269:25966–25969. [PubMed] [Google Scholar]
- 27.Standfuss J, Edwards PC, D’Antona A, Fransen M, Xie GF, Oprian DD, Schertler GFX. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471:656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 29.Maestro 9.1. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 30.Polak E, Ribiere G. Note on Convergence of Conjugate Direction Methods Revue Francaise D Informatique de Recherche Operationnelle. 1969;3:35. [Google Scholar]
- 31.Kelley LA, Gardner SP, J. SM. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally related subfamilies. Prot. Engineering. 1996;9:1063–1065. doi: 10.1093/protein/9.11.1063. [DOI] [PubMed] [Google Scholar]
- 32.Yongye AB, Bender A, Martinez-Mayorga K. Dynamic clustering threshold reduces conformer ensemble size while maintaining a biologically relevant ensemble. J. Comput.-Aided Mol. Des. 2010;24:675–686. doi: 10.1007/s10822-010-9365-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.