

Abstract
Wolf-Hirschhorn syndrome (WHS) is a genetic disease with characteristic facial features and developmental disorders. Of interest, loss of the MMSET gene (also known as WHSC1) is considered to be responsible for the core phenotypes of this disease. Patients with WHS also display antibody deficiency, although the underlying cause of this deficiency is unclear. Recent studies suggest that the histone methyltransferase activity of MMSET plays an important role in the DNA damage response by facilitating the recruitment of 53BP1 to sites of DNA damage. We hypothesize that MMSET also regulates class switch recombination (CSR) through its effect on 53BP1. Here we show that MMSET indeed plays an important role in CSR through its histone methyltransferase activity. Knocking down MMSET expression impaired 53BP1 recruitment as well as the germline transcription of the Igh switch regions, resulting in defective CSR but no effect on cell growth and viability. These results suggest that defective CSR caused by MMSET deficiency could be a cause of antibody deficiency in WHS patients.
Introduction
Human antibody molecules are comprised of heavy chains and light chains with both constant (C) and variable (V) regions that are encoded by different genes. During early B cell development in primary lymphoid organs, the genes encoding the heavy-chain and light chain V regions are assembled from component V, diversity (D) and joining (J) gene segments by the process of V(D)J recombination(1). The productive assembly of heavy chains and light chains generates B cells that express immunoglobulin M (IgM) on its surface. These B cells subsequently migrate to the secondary lymphoid organs such as spleen and lymph nodes, where upon encountering antigen, they can switch expression of immunoglobulin (Ig) class (isotype) from IgM to IgG, IgE, or IgA to mediate different isotype-specific effector functions essential for normal immunity. This process requires an additional round of genetic alteration, termed class-switch recombination (CSR)(2-5).
CSR is initiated by activation induced cytidine deaminase (AID), which deaminates deoxycytidines on both DNA strands within Igh switch (S) regions (5, 6). The deaminated S regions are then processed by proteins of the base excision repair pathway (including uracil DNA glycosylase, APE1 etc.,) and mismatch repair pathway (including Msh2/Msh6, Mlh1/Pms2, Exo1 etc.,) to ultimately generate DNA double-stranded breaks (DSBs) (7). Finally, the AID-induced DSBs are repaired predominantly by the nonhomologous end-joining (NHEJ) pathway. In NHEJ-deficient cells, DSBs are repaired by an alternative end-joining pathway (A-EJ) (8-10). Of interest, we have recently reported that B cells can be induced to acquire heightened DNA repair activity upon receipt of signals from CD4 T cells. We termed this process somatic hyperrepair, which we believe is necessary to protect the well-being of B cells during the processes of somatic hypermutation and CSR (11).
Proteins required for CSR include Ku, DNA-PK, ATM, Mre11-Rad50-Nbs1, γH2AX, RNF8, 53BP1, MDC1, and XRCC4-ligase IV (12-19). All of these proteins are important for faithful joining of S regions, and in their absence aberrant recombination and chromosomal translocations involving S regions occur. Among these proteins, 53BP1 deficiency affects CSR the most, as CSR is reduced more than 90% in cultured 53BP1−/− splenic B cells relative to wild-type B cells (20-22). This is not due to decreased cell proliferation or reduced germline (GL) transcripts at the switch regions. 53BP1-deficient cells do not have a dramatic increase in general chromosome instability, unlike ATM−/− and H2AX−/− cells, but a much higher proportion of the chromosomal aberrancies in 53BP1−/− cells involve the Igh locus, suggesting that 53BP1 has a distinct role at this locus (22).
53BP1 is also a well-known mediator of the cellular response to DNA damage (39). 53BP1 localizes to DNA breaks via two mechanisms, one by interaction with methylated histone H4 Lys20 (H4K20), and another via ubiquitylation of H2A-type histones mediated by a novel E3 ubiquitin ligase RNF8. We and others recently found that the histone methyltransferase MMSET could affect H4K20 methylation and thereby 53BP1 recruitment at the sites of DNA damage (23, 24). Interestingly, loss of the MMSET gene at chromosome 4p is linked to Wolf-Hirschhorn syndrome (WHS) (25, 26). MMSET is considered one of the causative genes because this gene is deleted in every known case of WHS (25). WHS is a genetic disease with characteristic craniofacial features and developmental disorders including microcephaly, growth and mental retardation, muscle hypotonia, seizures, and congenital heart defects (25). MMSET+/- mice show varying degrees of congenital heart disease, growth retardation, and craniofacial defects similar to those seen in WHS patients (27). Patients with WHS also display antibody deficiency, especially IgA and IgG isotypes, although the underlying cause of this deficiency is unclear (28). Based on our recent finding of a role for MMSET in the DNA damage response, the goal of this study was to test the hypothesis that MMSET is important for CSR.
Methods
In vitro cell culture
CH12F3 cells and the subclone C2 were maintained in vitro and CSR assays performed as previously described(29). In brief, cells were maintained in RPMI 1640 supplemented with 10% FBS, 10 mM 2-mercaptoethanol, and 5% NCTC-109 (Invitrogen). The subclone C2 cells were used in most of the experiments. For the CSR assay, cells were stimulated with 1 ng/ml of recombinant human TGF-β1 (R&D Systems), 10 ng/ml of recombinant murine IL-4 (R&D Systems), and 250 ng/ml recombinant murine CD40 ligand (PerproTech) or 2 μg/ml of functional-grade purified anti-murine CD40 antibody (BD) for 72 hrs, and then analyzed by flow cytometry.
siRNA, shRNA lentiviral infection and antibody information
To knockdown 53BP1 in CH12F3 B cells, we used siRNA against 53BP1 that was previously described(30). Two independent murine 53BP1- targeting sequences were used: #1: TGGTCATCCAATGGCTAC and #2: GCCAGGTTCTGGAAGAAGA. Lentiviral shRNA constructs for MMSET, and nonsilencing negative control shRNA were purchased from Origene (Catalog No. TF517851). The target sequences for murine MMSET are: #2:5’-TGGATATTTGAGAAGAGCCTTGTTGCTTT-3’; and #3: 5’-ATGTCAATAGAGGAGCGGAAAGCCAAATT-3’. For viral infection, CH12F3-2 cells were transduced with lentivirus by spin inoculation at 800 × g for 30 min at room temperature in the presence of 8 μg/mL of polybrene. Cells were then incubated for 3 days, after which positively transduced clones were obtained by puromycin selection. For growth curve analysis, CH12F3-2 cells were diluted to a concentration of 1 × 105 cells/mL and aliquoted in triplicate in T25 cell culture flasks. At various time points, the numbers and viabilities were analyzed on a Vi-Cell Analyzer (Beckman Coulter).
Antibodies used in this study include the following: anti-γ-H2AX (Upstate Biotechnology; Cat# 07-164), anti-53BP1(Novus; Cat# NB100-304), anti-H4K20me2(Active Motif; Cat#39173), anti-H3K4me3(Upstate, Cat# 17-614), anti-H3K36me2(Upstate, Cat# 07-369), anti-MMSET (Abcam, Cat# ab75359), and anti- AID (Cell Signaling; Cat#4959).
Flow cytometric analyses
For analyzing CSR from IgM (IgM+/IgA-) to IgA (IgM-/IgA+), CH12F3-2 cells were intracellularly stained with PE-conjugated anti-murine IgA clone 11–44-2 (eBiosciences, Cat# 12-5994-82), using Cytofix/Cytoperm and Perm/Wash buffers (BD Biosciences), and assessed for membrane IgM expression using FITC-conjugated anti-murine IgM (eBiosciences; Cat# 11-5890-82). Cells were then analyzed on a FACS Calibur (BD Biosciences) and the data were analyzed using FlowJo software (TreeStar).
ChIP
ChIP assays were performed as previously described (23, 31). Briefly, 2 × 106 cells were fixed by adding formaldehyde (1% final concentration) for 10 min at room temperature and reactions were quenched by adding glycine (final concentration 0.125 M). Cell lysates were sonicated to reduce the DNA length and the soluble chromatin fraction was obtained after centrifugation. This fraction was diluted in ChIP dilution buffer (2 ml) and precleared using Protein A agarose slurry (Upstate). Precleared lysates were incubated with 4 μg of anti-γ-H2AX, anti-53BP1, anti-H4K20me2, anti-H3K4me3, anti-H3K36me2/3, or IgG overnight at 4°C and complexes were recovered using protein G-Sepharose (Amersham). Precipitates were washed several times with high salt washing buffer and two times with lithium chloride washing buffer. The bound immunocomplex was then reverse cross-linked in elution buffer by heating at 65°C for 10-12 h. Samples were treated with RNase A and proteinase K, respectively, and DNA was ethanol precipitated after a phenol chloroform extraction. Precipitated DNA was dissolved in 50 μl of TE and subjected to PCR with or without serial dilution using the following chromatin immunoprecipitation primers:
Sμ1F: 5’- GCTTCTAAAATGCGCTAAACTGAGGTGATT-3’
Sμ1R: 5’-GTTTAGCTCTATTCAACCTAG-3’
Sμ2F: 5’-AAAGAGACATTTGTGTGTCTTTGAGTACCG-3’
Sμ2R: 5’ATTGGTTAACAGGCAACATTTTTCTTTTAC-3’
Sμ3F: 5’GCTAAACTGAGGTGATTACTCTGAGGTAAG-3’
Sμ3R: 5’GTTTAGCTTAGCGGCCCAGCTCATTCCAGT-3’
CμF: 5’-CTGTCGCAGAGATGAACCCCA-3’
CμR: 5’-ATCCTTTGTTCTCGATGGTCACCGG-3’
SαF: 5’-GTGATTCAGGGAGCAAGAGC-3’
SαR: 5’-TCTAGCCTGGGAGTCTCCTG-3’
Sequencing of switch junctions
Cμ-Cα switch junction sequences from parental as well as MMSET knockdown CH13F3 cells were amplified by PCR. The PCR products were then subcloned and sequenced. Specifically, genomic DNAs, isolated from about 105 CIT induced CH12F3 cells by proteinase K digestion and ethanol precipitation, were amplified by PCR with primers M1 (5’-TAGTAAGCGAGGCTCTAAAAAGCAT-3′) and A1 (5’-CAGCAGTGAGTTTAACAATCC-3′) and nested PCR with primers M2 (5’-GCTTGAGCCAAAATGAAGTAGACT-3′) and A2 (5’-CCTCAGTGCAACTCTATCTAGGTC T-3′). The final PCR products were then subcloned into pCR®II TOPO TA vector (Invitrogen) and subsequently sequenced. A total of 20 sequences from MMSET depleted cells and 22 sequences from parental cells were analyzed. Switch junction sequences were aligned and analyzed for microhomology using the MACAW program (NIH). The sizes of sequence microhomology of the two groups were compared for statistical significance using an unpaired two-tailed T test.
Quantitative PCR
Quantitative PCR (qPCR) was performed on a 7500 RT–PCR System (Applied Biosystems) using the SYBR Green detection system with the following program: 95°C for 5min, 1 cycle; 95°C for 45s and 62°C for 45s, 40 cycles. As an internal control for the normalization of the specific fragments amplified, a locus outside the region of the DSB was amplified, in this case FKBP5, using the input control sample as template. The internal control (FKBP5) primers were as follows: forward, 5′-CAGTCAAGCAATGGAAGAAG-3′; reverse, 5′-CCCGTGCCACCCCTCAGTGA-3′.
After qPCR amplification, the FKBP5 input controls for non-CIT treatment (no class switch) and CIT treatment (class switch) were used to normalize the unswitched and switched samples, respectively. After normalization, the relative levels of the indicated proteins on a specific Igh region were calculated by comparison of unswitched and switched samples to their respective IgG controls. Each experiment was independently performed at least three times and the s.e.m. values were calculated from at least three independent experiments.
RT-PCR Analysis
Total RNA was extracted from CH12F3 cells using a PARIS kit (Applied Biosystems). qRT-PCR was performed using Brilliant II SYBR Green qRT-PCR Master Mix kit (Agilent Technologies). The primers for RT-PCR analysis of germline transcripts (GLTs) are as follows:
μGLT forward: 5′- CTCTGGCCCTGCTTATTGTTG-3′
μGLT reverse: 5′- AATGGTGCTGGGCAGGAAGT-3′
αGLT forward: 5′- CCAGGCATGGTTGAGATAGAGATAG-3′
αGLT reverse: 5′- GAGCTGGTGGGAGTGTCAGTG-3′
RESULTS
MMSET binds to regions of the Igh locus and H4K20me2 increases during CSR
Histone posttranslational modifications such as H2A phosphorylation, and H4k20 and H3K4 methylation, have been shown to be important for CSR (32-36). To determine whether the histone methyltransferase activity of MMSET is involved in CSR, we used the CH12F3 cell line capable of switching from IgM to IgA expression upon stimulation with CD40 ligand, interleukin 4 and transforming growth factor-β (CIT)(29). As shown in Figure 1A, 3 days after stimulation with CIT, about 21% of the CH12F3 cells underwent CSR from IgM to IgA as indicated by the loss of IgM and simultaneous gain of IgA expression.
Figure 1. Induction of H4K20 methylation and recruitment of MMSET to regions of theIghlocus during CSR. A.
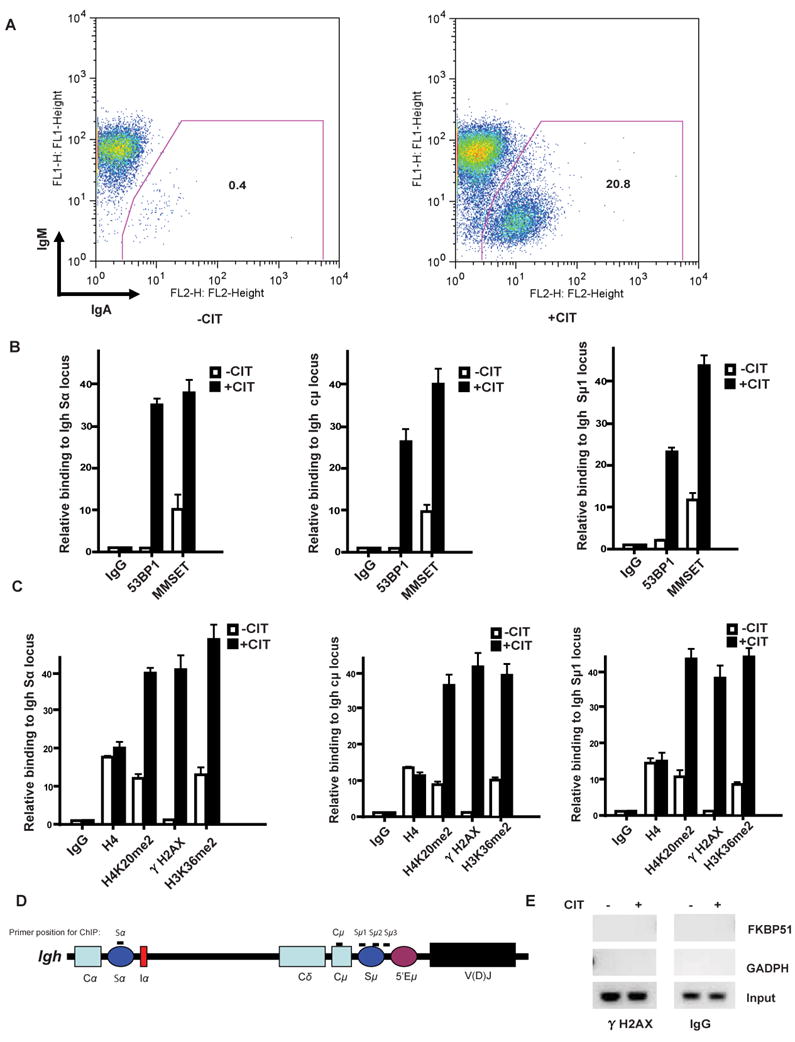
Flow cytometry analysis of CH12F3 cells stimulated with CD40L, interleukin 4, and transforming growth factor-β (CIT) for 3 days and stained with anti-IgM and anti-IgA antibodies. Simultaneous gain of IgA and loss of IgM expression signifies an effective CSR. Numbers indicate the percentage of total live IgA+ CH12F3 B cells. B, MMSET ChIP assay for regions of the Igh locus (Sα, left panel, Cμ, middle panel, and Sμ1, right panel) of the CH12F3 cell line with or without CIT stimulation. qPCR analyses of the indicated ChIP samples were performed, where the Y axis represents the relative enrichment of the indicated proteins compared to the IgG control (after normalized with a PCR internal control to a locus other than the Igh region). Three independent experimental replicates were performed for each experiments (± s.e.m., n=3). C, H4K20me2 and H3K36me2 ChIP assay for the indicated regions (Sα, left panel, Cμ, middle panel and Sμ1, right panel) of the Igh locus in the CH12F3 cell line stimulated with or without CIT for 24hrs. D. Schematic diagram of the position of the PCR products for the ChIP assays employed in this study. E, ChIP assay of γ-H2AX at non-Ig gene loci (GADPH and FKBP51) was performed after 24 hours of CIT stimulation of CH12F3-2 cells.
We next tested whether MMSET localizes to the S regions following CSR induction. After CSR induction with CIT, chromatin was immunoprecipitated from the cells using antibodies directed against MMSET and qPCR was used to determine the relative abundance of MMSET at Igh switch regions, while conventional PCR gave a visual representation of the relative accumulation of these proteins at the DSB sites. As shown in Figure 1B and Supplementary Figure 1A, similar to 53BP1, MMSET accumulated at the Igh Sμ1, Cμ and Sα regions during CSR. Since MMSET is a histone methyltransferase, we examined histone methylation at H3K36 and H4K20 during CSR. Using a ChIP assay, we observed that dimethylated H3K36 (H3K36me2) and H4K20 (H4K20me2) increased after CSR induction at Igh switch regions, as did the γH2AX signal (Figure 1C and Supplementary Figure 1B). This CIT-induced accumulation was specific to the regions of the Igh locus because non-Ig genes, such as glyceraldehyde-3-phosphate dehydrogenase (GADPH) and FKBP51, were not coimmunoprecipitated by antibodies against γH2AX (Fig. 1E). These results imply that MMSET may play a role in CSR.
MMSET facilitates CSR
Although B cells from MMSET deficient adult mice would be ideal for testing our hypothesis, MMSET null mice are embryonic or neonatal lethal (27). Therefore, we alternatively used the CH12F3 murine lymphoma cell line to study the role of MMSET in CSR (29). Cells were infected with lentivirus encoding different shRNAs specific for MMSET, and MMSET knock down cells were isolated using puromycin selection. Cells depleted of 53BP1 were used as a positive control. Compared with cells infected with a scrambled, nonsilencing shRNA construct (control), MMSET knock down cells showed a significant reduction in class switching efficiency (40% and 62% decrease using MMSET sh-2 and sh-3, respectively) (Fig. 2A and Supplementary Figure S2A). To further determine whether the MMSET methyltransferase activity is required for these processes, we mutated the critical residue (F1117) required for MMSET methyltransferase activity (23). We reintroduced shRNA-resistant human wild-type (WT) MMSET or MMSET-F1117A to cells stably transfected with MMSET shRNA. As shown in Figure S2A, while MMSET-WT restored class switch efficiency, MMSET-F1117A did not.
Figure 2. MMSET facilitates CSR. A.

CH12F3 cells were infected with lentivirus expressing the indicated shRNA, and CSR was assessed using flow cytometry analysis of IgM and IgA expression following CIT. Numbers indicate the percentage of total live IgA+ B cells. The right panels show the knockdown efficiency of 53BP1and MMSET by western blot. Data are representative of two independent experiments. B, Evaluation of the effect of MMSET expression on cell growth. Cell numbers of CH12F3 cells expressing either control shRNA or different MMSET-specific shRNAs were counted on 3 consecutive days. C, Evaluation of the effect of MMSET expression on cellular viability of pre-switched CH12F3 cells. Similar to the experiments in B, cell viability of CH12F3 cells expressing either control shRNA or different MMSET-specific shRNAs were analyzed on 3 consecutive days. D, Evaluation of the effect of MMSET expression on cell viability of post-switched CH12F3 cells. Class switched IgA+/IgM-CH12F3 cells expressing either control or MMSET-specific shRNA were sorted, and cell viability was analyzed. E, Western analysis of AID expression in CH12F3 cells pre- and post-MMSET knock down by specific shRNAs. F, ChIP analysis of γH2AX at the Sμ1 region and Sα region of Igh locus in CH12F3 cells transfected with the indicated shRNAs, with or without CIT stimulation. Three independent experimental replicates were performed for each experiment (± s.e.m., n=3).
It is possible that downregulation of MMSET could affect cell growth or viability, which could account for the decrease in class switching efficiency. We next tested whether MMSET affects cell proliferation. As shown in Figure 2B-C, cells depleted of MMSET using shRNAs displayed growth curves and overall cell viability comparable to that of control cells before CSR induction. Cells depleted of MMSET also showed similar cell viability to that of parental cells after CSR (Fig. 2D), suggesting that impaired proliferation is not a primary reason for the dramatic decrease of CSR in MMSET-deficient cells.
AID is essential for CSR, and AID expression is rapidly induced upon CIT induction(1). Since MMSET has been shown to play a role in gene expression (27, 37, 38), it is also possible that MMSET knockdown affected AID expression and hence CSR efficiency. However, knockdown of MMSET had no detectable effect on AID expression at the protein level (Fig. 2E). The production of DNA DSBs at the μ region of Igh locus was also not significantly affected, but DNA DSBs at the Sα region were dramatically decreased, as indicated by the γH2AX level (Fig. 2F, Supplementary Figure 2B). The latter observation might reflect the possibility that the transcriptional level of these two regions is differently regulated by MMSET as we describe below.
MMSET regulates 53BP1 recruitment to regions of the Igh locus
The inability to complete CSR is frequently associated with accumulation of Ig heavy chain-associated breaks, and 53BP1 is essential for repair of Igh breaks (22). Since MMSET regulates 53BP1 recruitment to DSB sites in other somatic cells(23, 24), we hypothesized that MMSET regulates CSR in B cells through its effects on 53BP1. To test whether MMSET regulates 53BP1 recruitment and function during CSR, we examined the accumulation of 53BP1 at the Igh switch regions by ChIP coupled with qPCR assay. Indeed, we found that downregulation of MMSET significantly decreased CIT-induced recruitment of 53BP1 to the Sμ region, but not the level of γH2AX, which lies upstream of MMSET in the DNA repair pathway (Fig. 3A-B). Since H4K20me2 is affected by MMSET and is required for 53BP1 recruitment during the DNA damage response, we next examined the level of H4K20me2 at the Igh switch regions. As shown in Figure 3B and Supplementary Figures 3A and 3B, knockdown of MMSET dramatically decreased H4K20me2 levels at the Igh switch regions.
Figure 3. MMSET regulates H4k20 methylation in regions of theIgHlocus and 53BP1 recruitment. A,B.

ChIP analysis of the indicated proteins at the Igh- Sμ1switch region in CH12F3 cells transfected with the indicated shRNA with or without CIT stimulation. Right panels: qPCR analysis of ChIP samples from left panels, where the Y axis represents the relative enrichment of the indicated proteins compared to the IgG control. Three independent experimental replicates were performed for each experiment (± s.e.m., n=3).
MMSET affects DNA end-joining of the Igh switch regions
Since 53BP1 is critical for end joining at the Igh locus during CSR, MMSET could also affect the end joining of broken switch regions through its effect on 53BP1. To directly assay for alterations in DNA repair, we analyzed switch recombination junctions from successfully class-switched IgA-positive B cells. We found that Sμ-Sα junctions were significantly different between control and MMSET-deficient cells. In control cells, more than 65% of the switch junctions analyzed were direct joins, with no more than 10% of junctions displaying 1 nucleotide (nt) microhomology and only 20% of junctions showing 2 nt microhomology (Fig. 4A). In contrast, in the MMSET-depleted cells, we observed that approximately 20% of the junctions had more than 10 nt of microhomology and another 20% had above 5 nt microhomology. Lastly, less than 10% were direct joins (Fig. 4A). These results suggest that NHEJ is defective in MMSET-depleted cells and DNA end joining during CSR resembles that observed in 53BP1-deficient cells (39).
Figure 4. MMSET regulates DNA end-joining and transcription of theIghswitch region. A.
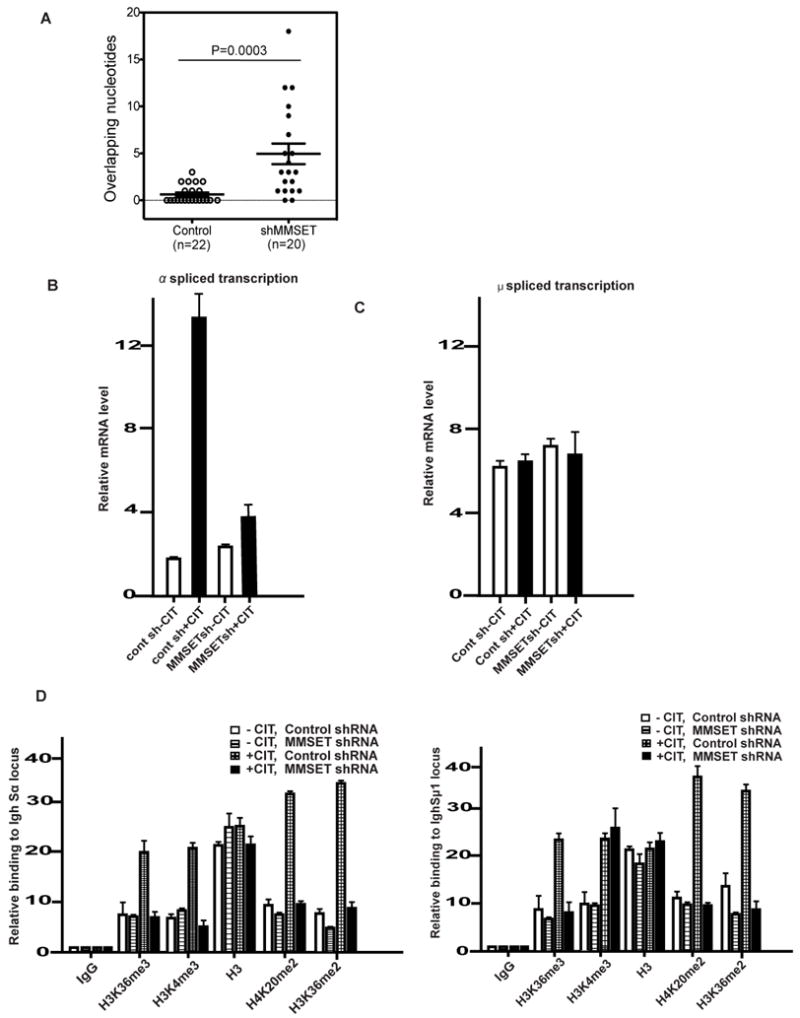
Effect of MMSET on microhomology of Cμ-Cα junctions. Percentage breakdown of clones based on the number of overlapping nucleotides (n=20 for MMSET depleted, n=22 for parental cells). B, RT-qPCR analysis of spliced Igh-Sα switch transcripts in CH12F3 B cells stimulated under the indicated conditions. Data are representative of three independent experiments. C, RT-qPCR analysis of spliced Igh-Sμ1 switch transcripts in CH12F3 B cells stimulated under the indicated conditions. Data are representative of three independent experiments. D, CH12F3 cells were transfected with the indicated shRNA and treated with or without CIT. ChIP analysis was then performed for the indicated histone modifications by the Sα primer (left panel) or Sμ1 primer (right panel) pair. The Y axis represents the relative enrichment of the indicated proteins compared to the IgG control. Data are representative of three independent experiments. (± s.e.m., n=3).
MMSET regulates transcription of the Igh switch region
MMSET may also modulate gene transcription by regulating H3K36 methylation and H4K20 methylation (23, 27, 37, 38). Previous studies have established that transcription of the Igh switch regions could regulate class switch efficiency (2, 5, 34, 35, 40). To investigate whether MMSET regulates transcription of the Igh switch regions, we measured the levels of germline transcripts that were spliced to join the initiating (I) exon located 5′ of the switch region to the constant (C) exons located 3′ of the switch regions from stimulated B cells. We found that MMSET-depleted cells had reduced levels of the Cα germline transcript, but had near-normal Cμ germline transcripts (Fig. 4B-C). This is consistent with the lower levels of H4K4me3, another histone marker linked to transcription initiation regulation at the Igh-Sα region but not at the Igh-Sμ region when MMSET was depleted. (Figure 4D). We additionally found that both H3K36me2 and H4K20me2 decreased at both Igh-Sα and Igh-Sμ regions in the absence of MMSET (Figure 4D and Figure S3B). Therefore, in addition to the reduced end-joining capability, the decreased transcription of Cα germline transcript at the Igh regions in MMSET-depleted cells may also contribute to the reduced levels of CSR.
Discussion
Recent findings, especially studies linking 53BP1 to CSR, suggest that the DNA damage response plays an important role in CSR. Therefore, knowledge gained in DNA damage response pathways has provided many insights into the molecular mechanism of CSR. Similar to the DNA damage response pathway, the induction of DSBs during CSR initiated an ATM-dependent signaling pathway. ATM phosphorylates H2AX, and phosphorylated H2AX (γH2AX) amplifies the DNA damage signal, in part, by recruiting MDC1 and the E3 ubiquitin ligase RNF8 to promote H2A/H2AX ubiquitination critical for subsequent accumulation of 53BP1 to DSBs(13, 15, 20, 21). In support of this model, ATM-, H2AX-, MDC1- and RNF8-deficient mice all exhibit defects in CSR due to DSB repair defects (13, 15, 21). Our recent finding that MMSET regulates 53BP1 accumulation through histone methylation at DSBs prompted us to investigate whether MMSET plays a role in CSR. Our study indicates a significant contribution of MMSET to the CSR reaction. We found that following CSR induction, histone H3K36 and H4K20 methylation are increased in the Igh switch regions. Depletion of MMSET decreases these histone modifications and further attenuates 53BP1 accumulation in these Igh switch regions, resulting in significant defects in CSR.
We found that MMSET not only regulates 53BP1 recruitment and subsequent DNA end joining, but also regulates the germline transcription of Sα. Germline transcription at the Igh locus, known to be critical for CSR, is also defective in MMSET-deficient cells (Fig. 4B). Since 53BP1 is not required for germline transcription at the Igh locus (20, 21), these results suggest that, in addition, MMSET regulates a 53BP1-independent function. MMSET deficiency only affects the downstream α germline transcript, but has no effect on levels of the μ germline transcript. This is reminiscent of the change of H3K4me3 in PTIP-/- cells (34). Thus PTIP driven changes in H3K4me3 levels can regulate the α germline transcript, but has no effect on levels of the μ germline transcript. These observations make it likely that MMSET regulates germline transcription through modulation of H3K36 and H4K20 methylation. Several studies suggest the primary methyltransferase activity of MMSET is toward H3K36, but not H4K20 (27, 41, 42). We and others also observed changes of H4K20 methylation during DNA damage responses (23, 24). It is possible that MMSET regulates H4K20 methylation indirectly. Another possibility is that MMSET specificity could change at the sites of DNA damage, given that the specificity of MMSET is substrate dependent (41). Interestingly, H3K4 methylation also decreased in the Igh-Sα region in MMSET-deficient cells. We therefore suggest there is crosstalk between these histone posttranslational modifications in the Igh-Sα region but not in Igh-Sμ region and this is consistent with previous reports (34,38). Future studies are required to elucidate how MMSET regulates germline transcription at the switch region
Of great interest, MMSET was named as such due to its overexpression as a result of a chromosomal translocation (t(4;14)) in a subset of patients with multiple myeloma (MM), a malignancy of post-CSR plasma cells. Therefore, it is tempting to speculate that aberrant CSR may be directly involved in the initiation of oncogenic transformation of MM. Consistent with this speculation, many MM signature translocations involve IgH loci further suggesting their CSR connection/origin.
Finally, our studies also provide new insights into the etiology of Wolf-Hirschhorn syndrome. Although it has long been recognized that Wolf-Hirschhorn syndrome patients display antibody deficiency, especially IgA and IgG isotypes, the underlying cause(s) of this deficiency, however, has remained unclear (25, 28). Our study showing that MMSET, a gene often deleted in Wolf-Hirschhorn syndrome, regulates CSR provides one possible mechanism for antibody deficiency observed in these patients.
Supplementary Material
Acknowledgments
We thank Dr. Tasuku Honjo for kindly providing the CH12F3 cell line.
Footnotes
This work was supported by Richard Schulze Family Foundation, and NIH (CA130996 and CA108961 to ZL and R01CA136591 to DFJ). This work was also supported by the Fellowship Award from the Fraternal Order of Eagles and specific start-up funding from the Beijing Proteome Research Center (to HP).
Authorship Contributions and Disclosure of Conflicts of Interest
Contribution: HD, TL, and XW performed experiments, analyzed data, and wrote the paper; and YK, DFJ and ZL designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol. 2006;24:541–570. doi: 10.1146/annurev.immunol.23.021704.115830. [DOI] [PubMed] [Google Scholar]
- 2.Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, Manis J, Alt FW. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 3.Papavasiliou FN, Schatz DG. Somatic hypermutation of immunoglobulin genes: merging mechanisms for genetic diversity. Cell. 2002;109(Suppl):S35–44. doi: 10.1016/s0092-8674(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 4.Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, Scharff MD. The biochemistry of somatic hypermutation. Annu Rev Immunol. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- 5.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 7.Saribasak H, Rajagopal D, Maul RW, Gearhart PJ. Hijacked DNA repair proteins and unchained DNA polymerases. Philos Trans R Soc Lond B Biol Sci. 2009;364:605–611. doi: 10.1098/rstb.2008.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan-Hammarstrom Q, Jones AM, Lahdesmaki A, Zhou W, Gatti RA, Hammarstrom L, Gennery AR, Ehrenstein MR. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. J Exp Med. 2005;201:189–194. doi: 10.1084/jem.20040772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franco S, Murphy MM, Li G, Borjeson T, Boboila C, Alt FW. DNA-PKcs and Artemis function in the end-joining phase of immunoglobulin heavy chain class switch recombination. J Exp Med. 2008;205:557–564. doi: 10.1084/jem.20080044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, Rajewsky K, Alt FW. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 11.Wu X, Tschumper RC, Gutierrez A, Jr, Mihalcik SA, Nowakowski GS, Jelinek DF. Selective induction of DNA repair pathways in human B cells activated by CD4+ T cells. PLoS One. 2010;5:e15549. doi: 10.1371/journal.pone.0015549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L, Halaby MJ, Hakem A, Cardoso R, El Ghamrasni S, Harding S, Chan N, Bristow R, Sanchez O, Durocher D, Hakem R. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J Exp Med. 2010;207:983–997. doi: 10.1084/jem.20092437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lahdesmaki A, Taylor AM, Chrzanowska KH, Pan-Hammarstrom Q. Delineation of the role of the Mre11 complex in class switch recombination. J Biol Chem. 2004;279:16479–16487. doi: 10.1074/jbc.M312796200. [DOI] [PubMed] [Google Scholar]
- 15.Ramachandran S, Chahwan R, Nepal RM, Frieder D, Panier S, Roa S, Zaheen A, Durocher D, Scharff MD, Martin A. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc Natl Acad Sci U S A. 2010;107:809–814. doi: 10.1073/pnas.0913790107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med. 2004;200:1103–1110. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reina-San-Martin B, Nussenzweig MC, Nussenzweig A, Difilippantonio S. Genomic instability, endoreduplication, and diminished Ig class-switch recombination in B cells lacking Nbs1. Proc Natl Acad Sci U S A. 2005;102:1590–1595. doi: 10.1073/pnas.0406289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J Exp Med. 2003;197:1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santos MA, Huen MS, Jankovic M, Chen HT, Lopez-Contreras AJ, Klein IA, Wong N, Barbancho JL, Fernandez-Capetillo O, Nussenzweig MC, Chen J, Nussenzweig A. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J Exp Med. 2010;207:973–981. doi: 10.1084/jem.20092308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 21.Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, Nussenzweig MC, Chen J. 53BP1 is required for class switch recombination. J Cell Biol. 2004;165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, Bergsagel PL, Wang L, You Z, Lou Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124–128. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajdu I, Ciccia A, Lewis SM, Elledge SJ. Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc Natl Acad Sci U S A. 2011;108:13130–13134. doi: 10.1073/pnas.1110081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005;21:188–195. doi: 10.1016/j.tig.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Stec I, Wright TJ, van Ommen GJ, de Boer PA, van Haeringen A, Moorman AF, Altherr MR, den Dunnen JT. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum Mol Genet. 1998;7:1071–1082. doi: 10.1093/hmg/7.7.1071. [DOI] [PubMed] [Google Scholar]
- 27.Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, Kaneda Y. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–291. doi: 10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- 28.Hanley-Lopez J, Estabrooks LL, Stiehm R. Antibody deficiency in Wolf-Hirschhorn syndrome. J Pediatr. 1998;133:141–143. doi: 10.1016/s0022-3476(98)70194-5. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura M, Kondo S, Sugai M, Nazarea M, Imamura S, Honjo T. High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int Immunol. 1996;8:193–201. doi: 10.1093/intimm/8.2.193. [DOI] [PubMed] [Google Scholar]
- 30.Tripathi V, Nagarjuna T, Sengupta S. BLM helicase-dependent and -independent roles of 53BP1 during replication stress-mediated homologous recombination. J Cell Biol. 2007;178:9–14. doi: 10.1083/jcb.200610051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Begum NA, Kinoshita K, Kakazu N, Muramatsu M, Nagaoka H, Shinkura R, Biniszkiewicz D, Boyer LA, Jaenisch R, Honjo T. Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch. Science. 2004;305:1160–1163. doi: 10.1126/science.1098444. [DOI] [PubMed] [Google Scholar]
- 32.Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc Natl Acad Sci U S A. 2010;107:22190–22195. doi: 10.1073/pnas.1016923108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, Espejo A, Bedford MT, Nussenzweig A, Busslinger M, Jenuwein T. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008;22:2048–2061. doi: 10.1101/gad.476008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, Filsuf D, Chen HT, Gazumyan A, Yamane A, Cho YW, Sun HW, Ge K, Peng W, Nussenzweig MC, Casellas R, Dressler GR, Zhao K, Nussenzweig A. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science. 2010;329:917–923. doi: 10.1126/science.1187942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwab KR, Patel SR, Dressler GR. Role of PTIP in class switch recombination and long-range chromatin interactions at the immunoglobulin heavy chain locus. Mol Cell Biol. 2011;31:1503–1511. doi: 10.1128/MCB.00990-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell. 2010;40:364–376. doi: 10.1016/j.molcel.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, Martinez-Martinez Y, Chesi M, Bergsagel PL, Zhou MM, Waxman S, Leibovitch BA, Walsh MJ, Licht JD. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111:3145–3154. doi: 10.1182/blood-2007-06-092122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez-Garcia E, Popovic R, Min DJ, Sweet SM, Thomas PM, Zamdborg L, Heffner A, Will C, Lamy L, Staudt LM, Levens DL, Kelleher NL, Licht JD. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211–220. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, Sleckman BP, Nussenzweig A. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4:452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ, 3, Qiao Q, Neubert TA, Xu RM, Gozani O, Reinberg D. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem. 2009;284:34283–34295. doi: 10.1074/jbc.M109.034462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, Li W, Gozani O. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44:609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.