

Abstract
The measles virus (MV) phosphoprotein (P) and V proteins block the interferon (IFN) response by impeding phosphorylation of the signal transducer and activator of transcription 1 (STAT1) by the Janus kinase 1 (JAK1). We characterized how STAT1 mutants interact with P and JAK1 phosphorylation. Certain mutants of the linker, the Src-homology 2 domain (SH2), or the transactivation domain had reduced or abolished phosphorylation through JAK1 after IFN treatment. Other mutants, mainly localized in the linker, failed to interact with P as documented by the lack of interference with nuclear translocation. Thus the functional footprint of P on STAT1 localizes mainly to the linker domain; there is also some overlap with the STAT1 phosphorylation functional footprint on the SH2 domain. Based on these observations, we discuss how the MV-P might operate to inhibit the JAK/STAT pathway.
Keywords: measles virus, phosphoprotein, STAT1, innate immunity, interferon signaling
Introduction
Viruses have evolved mechanisms to evade host innate defenses, including interferon (IFN). The proteins coded by the Paramyxovirus P/C/V gene specifically counteract the IFN response (Fontana et al., 2008). While the C protein blocks IFN induction (Nakatsu et al., 2006; Nakatsu et al., 2008; Toth et al., 2009), the P and V proteins block the phosphorylation of the signal transducer and activator of transcription 1 (STAT1) and the subsequent nuclear translocation of the STAT1/STAT2 complex after type I IFN activation (Caignard et al., 2009; Caignard et al., 2007; Devaux et al., 2007; Palosaari et al., 2003). The P and V proteins share their amino-terminal domain but differ in their carboxy-terminal domains (Cattaneo et al., 1989). A 22-residue segment located in the shared amino-terminal domain of P and V mediates the interaction with the STAT1 protein (Caignard et al., 2009; Devaux et al., 2011; Ramachandran et al., 2008). Homologous segments with similar function are located in the amino terminal domain of the P and V proteins of other Morbilliviruses, like canine distemper virus and rinderpest virus, and of the related Henipaviruses, Nipah virus (NiV) (Ciancanelli et al., 2009; Devaux et al., 2007). However, the domains of STAT1 that interact with MV-P have not been identified.
The α/β IFN signaling pathway is well characterized: upon receptor binding, secreted type I IFN activates the Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), which in turn phosphorylate STAT1 and STAT2. Phosphorylated STAT1 and STAT2 interact with each other through their Src-homology 2 (SH2) domains. The STAT1/STAT2 heterodimer then interacts with the DNA-binding protein IRF-9 (Aaronson and Horvath, 2002). The IRF9/STAT1/STAT2 complex, called interferon-stimulated gene factor 3, translocates into the nucleus and binds to the IFN-stimulated response element present in target promoters. This leads to the transcriptional activation and up-regulation of several hundred genes responsible for the establishment of an antiviral state in infected and surrounding non-infected cells (Darnell, 1997; Der et al., 1998).
The STAT1, a 91kDa protein, consists of 750 residues (Fig. 1A). It contains the N-terminal domain (NTD, residues1-135), coiled-coil domain (CCD, residues 136–317), DNA-binding domain (DBD, residues 318–488), linker domain (Linker, 389–576), SH2 domain (577–683) and transcriptional activation domain (TAD, residues 684–750). While segments and individual residues of MV proteins necessary for IFN control have been mapped (Caignard et al., 2009; Caignard et al., 2007; Ohno et al., 2004; Palosaari et al., 2003; Takeuchi et al., 2003; Yokota et al., 2003), the domains of STAT1 important for the interaction with the MV proteins remain uncharacterized. We sought to characterize how STAT1 interacts with MV-P.
Fig. 1.

STAT1 linear representation, sequence of the carboxy-terminal third of STAT1, and structure of the unphosphorylated STAT1 dimer. (A) Linear representation of STAT1 organization. NTD, N-Terminal domain; CCD, Coiled coil domain; DBD, DNA binding domain (DBD); Linker, linker domain; SH2, SH2 domain; and TAD, transactivation domain. Domains are color coded, and the colors coding is carried over in panel B and C. The black line represents the regions of STAT1 for which 3D structures are available. The PDB identifiers of the structures are indicated on the right. (B) Sequence alignment of residues 475–750 of STAT1 and residues 473–830 of STAT2, performed with the ClustalW program. Residues 509–712 are indicated with a green box. “*”: identical residues. “:” and “.”: conservative and semi-conservative substitutions, respectively. Red boxes: 18 block mutants in non-homologous regions. (C) Crystal structure of the dimeric unphosphorylated form of STAT1 (Mao et al., 2005) (PDB: 1YVL). The linker domain (389–576), SH2 domain (577–683) and TAD (684–750) are represented with light blue, light orange and light pink colors, respectively. The mutagenized residues are shown in red. The rest of the protein is shown in dark or light gray, depending on the subunit. The orange residues represent a co-crystalized phosphopeptide (arrowheads). Short protein segments within the regions of interest (544–550, and 621–625 in the first monomer and 620–622 in the second monomer) are disordered, and thus not visible in the structure.
Both MV- and NiV-P interfere with the phosphorylation of cytoplasmic STAT1 (Caignard et al., 2007; Ciancanelli et al., 2009; Devaux et al., 2007; Shaw et al., 2004) and the residues involved in the interaction are collinear ((Ciancanelli et al., 2009; Devaux et al., 2011) and unpublished data). It is also known that STAT1 residues 509–712 are necessary for binding to NiV-P (Rodriguez et al., 2004). Thus, we focused our genetic analysis of the STAT1 interaction with P on this protein segment, which covers the linker, SH2 and TAD domain. While the linker domain function is not clear, the SH2 domain is required for the recruitment of the STATs to the phosphotyrosine on the receptor, and for the reciprocal SH2-phosphotyrosine interaction of the monomeric STATs to form a dimer (Horvath, 2000). Upon binding of the SH2 domain to the phosphotyrosine of the IFN receptor, STAT1 becomes a substrate for JAK phosphorylation at tyrosine-701 which is required for its dimerization, nuclear translocation and DNA binding (O’Shea et al., 2002; Shuai et al., 1993a). We show here that MV-P interacts with the linker domain of STAT1 and that this interaction interferes with its function.
Results
Targeted mutagenesis of STAT1
Based on the characterization of how STAT1 interacts with NiV-P (Rodriguez et al., 2004), we focused mutagenesis on residues 509–712. Within this segment, we selected residues that are not conserved with STAT2 (Fig. 1B, compare STAT1 and STAT2 sequences). In particular, we identified six regions, containing stretches of 6 or more non-conserved residues, and named them I-VI. Non-conserved segments of 4 or less residues were not considered in this phase of mutagenesis. Within these regions, 18 mutants of 2 to 3 residues were generated (Fig. 1B, red boxes). Seven of these blocks of residues are located in the linker domain, seven in the SH2 domain and four in the TAD. For mutagenesis, two small residues were chosen: alanine to substitute charged and polar residues, and serine to replace apolar residues. Theses changes limit structural interferences that can lead to reduced protein folding and transport. The locations of these blocks of mutants are indicated on the crystal structure of the cytoplasmic unphosphorylated STAT1 dimer (Fig. 1C, red residues, (Mao et al., 2005)).
Residues in the SH2 and TAD domains of STAT1 influence its phosphorylation by JAK1
After the interaction of its SH2 domain with the phosphotyrosine of the IFN receptor, STAT1 becomes a substrate for JAK1 (Shuai et al., 1993a; Shuai et al., 1993b). To assess whether the mutations we introduced in STAT1 affect its expression or phosphorylation, the mutants were transfected in STAT1-deficient U3A cells (Leung et al., 1995; Muller et al., 1993), human fibroblast widely used for the study of STAT1 function in IFN signaling pathway (Horvath et al., 1995; Parisien et al., 2002; Zhong et al., 2005). Expression and phosphorylation after type 1 IFN treatment were characterized by immunoblots (Fig. 2A). Seventeen of the eighteen mutants were expressed; one at minimal level (625EPD), four at lower levels (542FC, 637KEL, 643TF, 645PD), and twelve at similar or higher levels as standard STAT1 (Fig. 2A, STAT1 immunoblot and Fig. 2C, quantification). Mutant 625EPD and the marginally expressed mutant 643TF were not further considered.
Fig. 2.

Phosphorylation of STAT1 mutants and their MV-P interactions. (A) Expression and phosphorylation of STAT1 mutants in the presence or absence of IFN. Immunoblot analyses of U3A cells transfected with the plasmids indicated above each lane. The location (residue numbers) of the mutants in the STAT1 protein is indicated above the gel. Thirty-six hours post transfection, cells were incubated (top three gels) or not (bottom three gels) with IFN for 1h, then cells extracts were processed for SDS-PAGE analysis. Membranes were blotted with antibodies against STAT1 or phospho-STAT1 (P-STAT1), as indicated on the right. Actin is a loading control. The * indicate non-specific bands. Phosphorylation levels in the presence of IFN are indicated with boxes below the bottom gel: white boxes, same as control; cyan boxes, intermediate; dark blue boxes, none; crossed boxes, no expression. (B) Cellular localization of STAT1 mutants in absence (top row) or presence (bottom row) of P. For primary data see Figure 3. Nu: nuclear localization in absence of P; Nu+P: nuclear localization in presence of P; (+) nuclear translocation; (−) no nuclear translocation; NA, not phosphorylated. Yellow indicates mutants affecting the interaction with P, while green indicates mutants affecting both the interaction with P and STAT1-phosphorylation by JAKs. Mutants are color-coded using the same convention in panel D. (C) Quantification of STAT1 mutant expression. Protein bands were quantified using the Typhoon fluorimager and the Imagequant 5.0 software. Expression levels are reported as percentiles of control STAT1. (D) Residues affecting STAT1 phosphorylation or its nuclear translocation in presence of P shown on the crystal structure of phosphorylated STAT1. Mutants with strong or moderately impaired JAK1-dependent phosphorylation are shown in blue and cyan, respectively; mutants with impaired P-interaction are yellow. Mutants with both impaired JAK1-dependent phosphorylation and P-interaction are green. The linker, SH2 and TAD are in light blue, light orange and pink, respectively. Residues indicated with Δ were disordered and thus not visualized on the structure.
In the absence of IFN treatment, none of the mutants were phosphorylated (Fig. 2A, second panel from bottom, P-STAT1 immunoblot). After IFN treatment, ten of the sixteen remaining mutants had reduced levels of phosphorylation, with no phosphorylation of mutant 542FC, 637KEL, 700GY and 702IKT (Fig. 2A, P-STAT1 immunoblot, second panel from top, dark blue boxes at the bottom). These residues are shown in dark blue on the structure of the phosphorylated STAT1 (Fig. 2D). We used this structural template for illustration because it is more complete than the structure of cytoplasmic STAT1 shown in Fig. 1B. The 700GY mutant includes the tyrosine that is phosphorylated by JAK1. Mutants 531ASP, 551NF, 628FHA, 640SAV, 645PD, and 697KGT, which had reduced phosphorylation (Fig. 2A, second panel from top), are distributed over the three domains (Fig. 2A, cyan boxes at the bottom; and Fig. 2D cyan and green residues). These observations validate the experimental system by identifying residues in the SH2 and TAD domains of STAT1 that are required for its efficient phosphorylation by JAK1.
The functional footprint of P on STAT1 localizes mainly to the linker domain
To identify mutations that may affect the interaction between STAT1 and P, we analyzed nuclear translocation of our collection of STAT1 mutants in absence or presence of P. We expected three classes of mutants: first, those that translocate in the nucleus after IFN treatment, but remain in the cytoplasm in presence of P. Second, those that always stay in the cytoplasm. And third, those that translocate in the nucleus even in presence of P.
Figure 3 shows the analyses of the STAT1 nuclear translocation of all our mutants after IFN activation in absence or presence of MV P protein. For each mutant, the left panel documents the cellular localization of STAT1 (red staining) after IFN induction in the absence of P. The next panel is a nuclear DAPI staining (blue). The next three panels (P + IFN) document the cellular localization of STAT1 (red staining) after IFN induction in the presence of P (green staining); blue staining (DAPI) shows the nuclei. As expected, we observed that standard STAT1 translocates to the nucleus after IFN induction, but remains in the cytoplasm when P is expressed. Five mutants 542FC, 637KEL, 640SAV, 700GY and 702 IKT did not translocate to the nucleus after IFN treatment (Fig. 3 and Fig. 2B). Since these mutants are not phosphorylated (Fig. 2A, 542FC, 637KEL, 700GY and 702IKT) or poorly phosphorylated (Fig. 2A, 640SAV), impaired translocation was expected. Five mutants 531ASP, 534DG, 645PD, 694DGP, and 697KGT behaved like standard STAT1, implying that residues covered by these mutants may be irrelevant for the interaction with P (Fig. 3 and Fig. 2B). Finally, six mutants 540TR, 545ENI, 548NDK, 551NF, 622NGG, and 628FHA translocated to the nucleus after IFN treatment even in presence of P and thus cannot interact effectively with it (Fig. 3 and Fig. 2B). Four of them are localized in the linker domain (540TR to 551NF) and two in the SH2 domain (622NGG and 628FHA). Thus, both the linker and the SH2 domain are important for the interaction with MV-P. Interestingly, two of these mutants (551NF and 628FHA) are relevant for both the interaction with P and the phosphorylation of STAT1 by JAKs. Figure 2D shows the four mutants that cannot interact with P in yellow, and those two that are both P- and STAT1-phosphorylation deficient in green.
Fig. 3.

Confocal microscopy analysis of nuclear translocation of STAT1 mutants in presence or absence of P. U3A cells were transfected with the expression plasmid encoding the STAT1 mutants indicated on the left of the panels, in absence (IFN) or presence (P+IFN) of P, as indicated above the panels. After stimulation with IFN, cells were fixed, permeabilized and stained with an antibody to STAT1 (red), with DAPI (blue) or with an antibody to the P protein (green). STAT1 (top row) is the positive control. Left two columns (IFN): cells transfected with different mutants and treated with IFN. Right three columns (P+IFN): cells transfected with different mutants and with P, and treated with IFN.
Individual residues in the linker domain of STAT1 required for the interaction with P
We then focused on the four block mutants that cluster on one face of the linker domain (Fig. 2D, yellow residues 540–541, 545–550 and green residues 551–552). We generated 10 single amino acid mutants (Fig. 4A, listed above the immunoblots). Six mutants were expressed at similar levels as standard STAT1, three mutants (K550, N551 and F552) were expressed at a slightly lower levels (Fig. 4A, top gel) and T540A expression was strongly reduced (Fig. 4A and B). While one mutant, F552A showed reduced phosphorylation, all others were phosphorylated at similar or higher levels than standard STAT1 (Fig. 4A, P-STAT1). All mutants retained at least in part the ability to translocate to the nucleus after IFN treatment in absence or presence of P (data not shown). Thus, mutation of individual residues in this region is sufficient to impair the interaction with P.
Fig. 4.
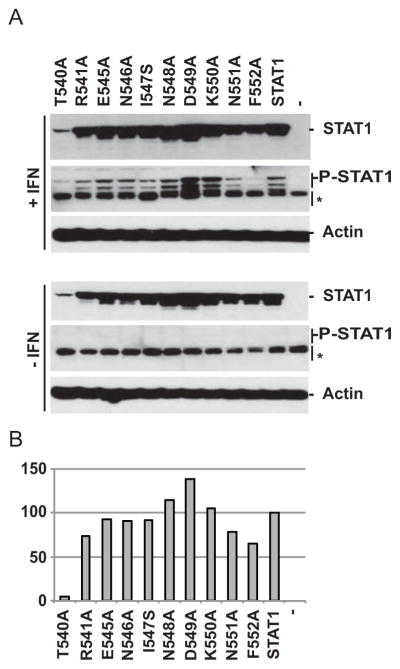
Phosphorylation of STAT1 single residue mutants. (A) Expression and phosphorylation of single residue STAT1 mutants in presence or absence of IFN. Immunoblot analysis of U3A cells transfected with the plasmids indicated above each lane. Thirty-six hours post transfection, cells were incubated (top three gels) or not (bottom three gels) with IFN for 1h, then cells extracts were processed for SDS-PAGE analysis. Membranes were blotted with antibodies against STAT1 or phospho-STAT1 (P-STAT1), as indicated on the right. Actin is a loading control. The * indicate non-specific bands. (B) Quantification of STAT1 mutant expression in presence of IFN. Protein bands were quantified using the Typhoon fluorimager and the Imagequant 5.0 software. Expression levels are reported as percentiles of control STAT1.
Additional residues in the linker domain of STAT1 are involved in the interaction with P
To assess whether neighboring residues not included in the first round of mutagenesis are important for the interaction, we then mutated all solvent-exposed residues which α-carbon atom is located within 10 Å of any residues previously identified. Figure 5A documents the expression of four additional mutants, E524A, K525A, K584A and E585A. While mutant K525A was not expressed, E524A, K584A and E585A were expressed at similar levels as STAT1 (Fig. 5A, top panels, STAT1). As expected, none of the mutants were phosphorylated in the absence of IFN treatment (Fig. 5A, bottom panels, P-STAT1). After treatment with IFN, mutants E524A and E585A were phosphorylated, but mutant K584A was not (Fig. 5A, top panels, P-STAT1). Finally, both phosphorylated mutants translocated to the nucleus in presence of IFN (Fig. 5B, left column, IFN), but only E585A translocated in presence of P (Fig. 5B, middle and right columns). Thus, we identified one additional residue, E585, important for the interaction with P. This residue is located between the cluster of residues influencing the P interaction (Fig. 5C, yellow) and the block of residues (628FHA) influencing both P and STAT1-phosphorylation interactions (Fig. 5C, green). Additionally, the adjacent residue K584 (Fig. 5C, blue) is important for the STAT1-phosphorylation by JAKs but since it is not phosphorylated, its interaction with P could not be assessed.
Fig. 5.
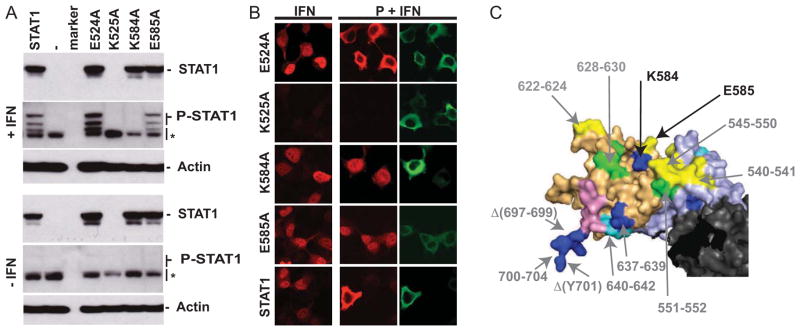
Phosphorylation and cellular localization of STAT1 mutants located in a 10Å radius of the previously identified residues. (A) Expression and phosphorylation of single residue STAT1 mutants in presence or absence of IFN. Immunoblot analysis of U3A cells transfected with the plasmids indicated above each lane and treated as described in Figure 4. (B) Confocal microscopy analysis of nuclear translocation of STAT1 mutants in absence or presence of P. U3A cells were transfected with an expression plasmid encoding the STAT1 single mutants, in absence (IFN) or presence (P+IFN) of P, as indicated above the panels. After stimulation with IFN, cells were fixed, permeabilized and stained with an antibody to STAT1 (red) or with an antibody to the P protein (green). (C) Localization of the STAT1 residues affecting the interaction with P and/or the phosphorylation of STAT1. Residues affecting STAT1 phosphorylation: blue and green. Residues affecting STAT1 nuclear translocation in presence of P: yellow and green. The linker, SH2 and TAD are in light blue, light orange and pink, respectively. Residues numbers are indicated.
Discussion
We have identified here several residues in the STAT1 linker domain, and others in the SH2 domain that sustain the STAT1 interaction with the MV-P protein. In the SH2 domain, the P-interaction may interfere with the phosphorylation of STAT1 by JAK1: both functional interactions are affected by mutations at STAT1 residues 551–552 and 628–630.
Two residues shown here to be relevant for STAT1-phosphorylation, K584 and H629 are also involved in STAT1 binding to the phosphotyrosine peptide of the IFNγ receptor as observed in a co-crystal (Mao et al., 2005). Remarkably, most of the residues which mutation caused decrease or complete inhibition of STAT phosphorylation, 637–642, K584, 697–701 (blue in Fig. 5C), 628–630 (green in Fig. 5C) are located in the same face of the SH2 domain. These data are consistent with previous structural and functional analyses indicating that the SH2 domain is important for STAT1-phosphorylation.
The MV-P protein functional footprint on STAT1 is centered on the linker domain with an extension in the SH2 domain (Fig. 5C). Residues 540–552 define one end of the footprint while two almost contiguous blocks of residues, 622–624 and 628–630 define the other end, and residue E585 is roughly in the middle (Fig. 5C). Interestingly, residues 544–550 of STAT1 were not resolved in the receptor-bound structure (Mao et al., 2005), suggesting that this region might form a flexible loop and be a potential interaction target. Altogether, these data suggest that MV-P interaction with STAT1 may interfere with its phosphorylation. However, other mechanisms cannot be excluded.
A 22-residue segment in the amino-terminal domain of the P protein (residues 110–131) sustains the interaction with STAT1 (Devaux et al., 2011; Devaux et al., 2007). Structural information on this P protein segment is not yet available, but it was reported that the N-terminal half of the P protein is unfolded and unstructured (Karlin et al., 2002). Remarkably, the 22-residue segment within this region is conserved among Morbilliviruses (Devaux et al., 2007), consistent with an interaction with a conserved cellular partner.
Inhibition of the JAK/STAT pathway by the concerted action of the MV-P and V proteins
While MV-P only interacts with STAT1, MV-V, which shares the amino-terminal half of P but has a different carboxyl-terminal domain, interacts with both STAT1 and STAT2 (Caignard et al., 2009; Ramachandran et al., 2008). Remarkably, the V-STAT2 interaction is much stronger than the P or V interactions with STAT1, and dominates the system (Caignard et al., 2009; Ramachandran et al., 2008). We know that P can block STAT1 phosphorylation (Caignard et al., 2007; Devaux et al., 2007), while V sequesters STAT1 and STAT2 in high molecular complexes (Palosaari et al., 2003). Both interactions may synergize to inhibit the JAK/STAT pathway. In a non-infected cell, after IFN binding to its receptor, JAK1 and TYK2 are phosphorylated, then STAT2 binds to one receptor subunit and induces the binding of STAT1 to the phosphotyrosine of the other receptor subunit, allowing its phosphorylation by JAK1. In P-expressing cells, P would encumber the linker and SH2 domains of STAT1, which might sterically hinder STAT1-phosphorylation by JAKs or binding of STAT1 to the IFN receptor. In MV infection, the V protein carboxyl-terminal domain interacts with STAT 2, in addition (Ramachandran et al., 2008). Altogether, these complementary strategies sustain efficient silencing of IFN signaling transduction.
Materials and methods
Cells
U3A, kindly provided by G. Stark (Cleveland, OH) (Leung et al., 1995; Muller et al., 1993), were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Mediatech Inc., Herndon, VA) supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin, 1mM sodium pyruvate (Mediatech Inc) and 250 μg/ml hygromycin (Invitrogen, Carlsbad, CA).
Expression plasmid construction
An expression plasmid coding for the P protein but not for the C protein, pCG-Pvac and the control plasmid P-inv were previously described (Devaux et al., 2011; Devaux et al., 2007). To produce pCG-STAT1, the P ORF of the pCG-Pvac vector was substituted by the open reading frame of STAT1, using the SalI and PacI restriction sites. The pCG-STAT1 plasmid was then mutagenized using the Quick-change system (Stratagene, La Jolla, CA). The integrity of the entire STAT1 coding region after mutagenesis was verified by sequencing.
Immunoblot analysis of cell extracts
U3A cells (2×105) were transfected with the mutant plasmids using Lipofectamine 2000 (Invitrogen). After 36h the cells were incubated with 1000U of IFNβ (PBL, Piscataway, NJ) for 60 minutes at 37°C and lysed in lysis buffer (Cell Signaling Technology, Danvers, MA) containing anti-protease and anti-phosphatase. The lysates were then cleared by centrifugation at 14000 rpm for 10 minutes at 4°C. After fractionation on 4–15% SDS-polyacrylamide gels (Biorad, Hercules, CA), and transfer to polyvinylidene difluoride membranes (Immobilon-P, [Millipore, Billerica, MA]), the samples were subjected to enhanced chemiluminescence detection (Fisher Scientific, Pittsburgh, PA) using a rabbit antibody against the phosphorylated Y701 of STAT1 (Millipore), a mouse antibody against human STAT1 (Santa Cruz Biotechnology, Santa Cruz, CA) and a mouse antibody against human β-actin conjugated with peroxidase (Sigma-Aldrich, St. Louis, MO).
Confocal microscopy
U3A cells (3×104) in chamber slides (Lab Teck II Chamber Slide system [Nalge Nunc International Corp, Naperville, IL]) were transfected with expression plasmids coding for the STAT1 mutants in presence or absence of an expression plasmid coding P protein using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. Twenty-four hours post-transfection, cells were treated with IFNβ (1000U/ml) for 60 minutes at 37°C, washed once with PBS and fixed for 15 minutes with PBS-2% paraformaldehyde (PFA). Cells were then permeabilized with PBS-2% PFA-0.1% Triton X-100 for 15 minutes, washed with PBS, incubated for 1 hour in blocking solution (PBS-2% FCS), and immuno-stained for STAT1 protein and for P protein. A mouse monoclonal anti-P antibody (kindly provided by D. Gerlier, Lyon, France), and rabbit antibodies directed against STAT1α p91 (C-24) (Santa Cruz Biotechnology) were used. Incubations were performed in PBS-2% FCS for 1h at room temperature. Five washes were performed after incubation with primary and secondary antibodies. After the last washes, cells were mounted with Vectashield containing DAPI (Vector Laboratories, Burlingame, CA) and analyzed with a Zeiss LSM 510 confocal microscope.
Residue in the linker and SH2 domains of STAT1 are important for MV-P interaction
Residue in the linker and SH2 domains of STAT1 are important for STAT1 phosphorylation
Residue interferring with both functions have similar location on STAT1
The viral P and V proteins may operate in concert to inhibit the JAK/STAT pathway
Acknowledgments
We thank George Stark for providing cell lines, Denis Gerlier for the anti-P antibody, Chanakha Navaratnarajah, Swapna Apte and Mathieu Mateo for reading the manuscript. This research was supported by grant R01AI63476 to RC, and by the Mayo Clinic Cancer Center. The Mayo Graduate School SURF program supported part of LP’s stipend.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Caignard G, Bourai M, Jacob Y, Tangy F, Vidalain PO. Inhibition of IFN-alpha/beta signaling by two discrete peptides within measles virus V protein that specifically bind STAT1 and STAT2. Virology. 2009;383:112–120. doi: 10.1016/j.virol.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Caignard G, Guerbois M, Labernardiere JL, Jacob Y, Jones LM, Wild F, Tangy F, Vidalain PO. Measles virus V protein blocks Jak1-mediated phosphorylation of STAT1 to escape IFN-alpha/beta signaling. Virology. 2007;368:351–362. doi: 10.1016/j.virol.2007.06.037. [DOI] [PubMed] [Google Scholar]
- Cattaneo R, Kaelin K, Baczko K, Billeter MA. Measles virus editing provides an additional cysteine-rich protein. Cell. 1989;56:759–764. doi: 10.1016/0092-8674(89)90679-x. [DOI] [PubMed] [Google Scholar]
- Ciancanelli MJ, Volchkova VA, Shaw ML, Volchkov VE, Basler CF. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J Virol. 2009;83:7828–7841. doi: 10.1128/JVI.02610-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux P, Hudacek AW, Hodge G, Reyes-Del Valle J, McChesney MB, Cattaneo R. STAT1-blind measles virus cannot control inflammation and is attenuated in rhesus monkeys. J Virol. 2011;85:348–356. doi: 10.1128/JVI.00802-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux P, von Messling V, Songsungthong W, Springfeld C, Cattaneo R. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 protein phosphorylation. Virology. 2007;360:72–83. doi: 10.1016/j.virol.2006.09.049. [DOI] [PubMed] [Google Scholar]
- Fontana JM, Bankamp B, Rota PA. Inhibition of interferon induction and signaling by paramyxoviruses. Immunol Rev. 2008;225:46–67. doi: 10.1111/j.1600-065X.2008.00669.x. [DOI] [PubMed] [Google Scholar]
- Horvath CM. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci. 2000;25:496–502. doi: 10.1016/s0968-0004(00)01624-8. [DOI] [PubMed] [Google Scholar]
- Horvath CM, Wen Z, Darnell JE., Jr A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 1995;9:984–994. doi: 10.1101/gad.9.8.984. [DOI] [PubMed] [Google Scholar]
- Karlin D, Longhi S, Receveur V, Canard B. The N-terminal domain of the phosphoprotein of Morbilliviruses belongs to the natively unfolded class of proteins. Virology. 2002;296:251–262. doi: 10.1006/viro.2001.1296. [DOI] [PubMed] [Google Scholar]
- Leung S, Qureshi SA, Kerr IM, Darnell JE, Jr, Stark GR. Role of STAT2 in the alpha interferon signaling pathway. Mol Cell Biol. 1995;15:1312–1317. doi: 10.1128/mcb.15.3.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, McMurray JS, Demeler B, Darnell JE, Jr, Chen X. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol Cell. 2005;17:761–771. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Muller M, Laxton C, Briscoe J, Schindler C, Improta T, Darnell JE, Jr, Stark GR, Kerr IM. Complementation of a mutant cell line: central role of the 91 kDa polypeptide of ISGF3 in the interferon-alpha and -gamma signal transduction pathways. EMBO J. 1993;12:4221–4228. doi: 10.1002/j.1460-2075.1993.tb06106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu Y, Takeda M, Ohno S, Koga R, Yanagi Y. Translational inhibition and increased interferon induction in cells infected with C protein-deficient measles virus. J Virol. 2006;80:11861–11867. doi: 10.1128/JVI.00751-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu Y, Takeda M, Ohno S, Shirogane Y, Iwasaki M, Yanagi Y. Measles virus circumvents the host interferon response by different actions of the C and V proteins. J Virol. 2008;82:8296–8306. doi: 10.1128/JVI.00108-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- Ohno S, Ono N, Takeda M, Takeuchi K, Yanagi Y. Dissection of measles virus V protein in relation to its ability to block alpha/beta interferon signal transduction. J Gen Virol. 2004;85:2991–2999. doi: 10.1099/vir.0.80308-0. [DOI] [PubMed] [Google Scholar]
- Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol. 2003;77:7635–7644. doi: 10.1128/JVI.77.13.7635-7644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien JP, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J Virol. 2002;76:4190–4198. doi: 10.1128/JVI.76.9.4190-4198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Parisien JP, Horvath CM. STAT2 is a primary target for measles virus V protein-mediated alpha/beta interferon signaling inhibition. J Virol. 2008;82:8330–8338. doi: 10.1128/JVI.00831-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JJ, Cruz CD, Horvath CM. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol. 2004;78:5358–5367. doi: 10.1128/JVI.78.10.5358-5367.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw ML, Garcia-Sastre A, Palese P, Basler CF. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J Virol. 2004;78:5633–5641. doi: 10.1128/JVI.78.11.5633-5641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai K, Stark GR, Kerr IM, Darnell JE., Jr A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science. 1993a;261:1744–1746. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- Shuai K, Ziemiecki A, Wilks AF, Harpur AG, Sadowski HB, Gilman MZ, Darnell JE. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature. 1993b;366:580–583. doi: 10.1038/366580a0. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Kadota SI, Takeda M, Miyajima N, Nagata K. Measles virus V protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett. 2003;545:177–182. doi: 10.1016/s0014-5793(03)00528-3. [DOI] [PubMed] [Google Scholar]
- Toth AM, Devaux P, Cattaneo R, Samuel CE. Protein kinase PKR mediates the apoptosis induction and growth restriction phenotypes of C protein-deficient measles virus. J Virol. 2009;83:961–968. doi: 10.1128/JVI.01669-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota S, Saito H, Kubota T, Yokosawa N, Amano K, Fujii N. Measles virus suppresses interferon-alpha signaling pathway: suppression of Jak1 phosphorylation and association of viral accessory proteins, C and V, with interferon-alpha receptor complex. Virology. 2003;306:135–146. doi: 10.1016/s0042-6822(02)00026-0. [DOI] [PubMed] [Google Scholar]
- Zhong M, Henriksen MA, Takeuchi K, Schaefer O, Liu B, ten Hoeve J, Ren Z, Mao X, Chen X, Shuai K, Darnell JE., Jr Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. PNAS. 2005;102:3966–3971. doi: 10.1073/pnas.0501063102. [DOI] [PMC free article] [PubMed] [Google Scholar]