

Abstract
While hypoglycemia occurs commonly among neonates, treatment can be challenging if hypoglycemia persists beyond the first few days of life. This review discusses the available treatment options for both transient and persistent neonatal hypoglycemia. These treatment options include dextrose infusions, glucagon, glucocorticoids, diazoxide, octreotide, and nifedipine. A stepwise, practical approach to the management of these patients is offered.
INDEX TERMS: diazoxide, glucagon, hyperinsulinism, hypoglycemia, nifedipine, octreotide
INTRODUCTION
Hypoglycemia continues to represent a common metabolic issue facing the neonatal population. Both healthy and ill-appearing neonates can be affected by hypoglycemia during the first days of life. Factors placing neonates at higher risk for developing hypoglycemia are prematurity, perinatal stress1 or asphyxia2, small size for gestational age,1,2 and being born to diabetic mothers.3 The goals of treating low-serum glucose concentrations are to prevent poor neurodevelopmental outcomes4 and to encourage normal feeding behaviors.5 Should the hypoglycemia persist, another goal for the medical team is to determine the underlying cause.
The definition of hypoglycemia remains controversial.6,7 Approaches to defining hypoglycemia have included a statistical approach with standard deviations, counterregulatory responses with a metabolic approach, neurophysiological changes, and neurodevelopmental outcomes of symptomatic versus asymptomatic hypoglycemia.8 Using any of these techniques alone has not provided a clear definition of hypoglycemia. Considering the lack of data to support a definitive breakpoint for serum glucose concentrations, general consensus appears to accept the definition of concentration less than 47 mg/dL as hypoglycemia in need of intervention, although for practical purposes less than 50 mg/dL is generally used.6,9
POSTNATAL GLUCOSE ADAPTATION
During fetal life, glucose passively diffuses across the placenta, using a concentration gradient. This process results in a fetal plasma glucose concentration approximately 70% to 80% of that of the maternal venous plasma glucose concentration.8 Insulin does not cross the placenta; therefore, the fetus must secrete insulin independently.
With the clamping of the umbilical cord, the neonate’s supply of glucose ceases while insulin secretion continues. The residual fetal insulin leads to a rapid decline in plasma glucose within the first hours of life.10 To overcome decreasing glucose concentrations, the release of counterregulatory hormones such as glucagon and cortisol in combination with the production of endogenous glucose through gluconeogenesis and glycogenolysis occurs. In healthy neonates, feeding is also initiated within approximately 12 hours of birth, further aiding in the increase of serum glucose concentrations. If feeding cannot be initiated, other metabolic substrates such as ketones will likely increase to offset the effects of lower glucose concentrations. Transient hypoglycemia can occur during the first hours of life because of a slow or immature fasting adaptation process.
GLUCOSE HOMEOSTASIS
Insulin and glucagon are released from pancreatic islet cells during the fed and fasting states. β-cells within islet cells contain ATP-sensitive potassium channels (KATP channels) and voltage- gated calcium channels (VGCC) that coordinate the secretion of insulin; while glucagon is secreted from α-cells.
Increasing glucose concentrations activate glucokinase, and β-cell glycolysis begins. This glycolytic pathway along with free fatty acids increases adenosine triphosphate (ATP) production within the β-cell. KATP channels consist of two subunits, the sulfonylurea (SUR) and the inward rectifier potassium channel (Kir6.2) subunits. The increase of ATP activates the SUR subunit, which closes the KATP channel. With the closing, depolarization of the cell occurs, leading to an influx of Ca2+ through the VCGG, stimulating the release of insulin.11 In contrast, decreasing glucose concentrations during the fasting state stimulate the release of glucagon. The exact mechanism is not completely understood; however, the mechanism may also be related to KATP channels within the α-cells.12 Glucagon helps regulate gluconeogenesis and glycogenolysis within the liver as a counterregulatory agent to insulin.
Exaggerated insulin secretion from β-cells can occur during the first few hours to days of life, resulting in hyperinsulinemia, which is the leading cause of hypoglycemia in neonates.5 Many mechanisms have been associated with this hypersecretion of insulin. The most prevalent cause of persistent hyperinsulinemia is the loss of function mutation of the KATP channel, which may lead to profound hypoglycemia. Other less common mechanisms include mutations of mitochondrial enzyme (GDH), activating mutation in glucokinase, and mutations of short-chain 3-hydroxyacyl-coenzyme A dehydrogenase.5 Hyperinsulinemia may be of a transient, prolonged, or persistent nature, leading to various degrees of severity of hypoglycemia.
TRANSIENT VERSUS PERSISTENT HYPOGLYCEMIA
Factors related to transient hypoglycemia are often related to conditions or events occurring during birth. Infants of mothers receiving intravenous dextrose during delivery13 and those treated with hypoglycemic agents during pregnancy14 have been linked to neonatal hypoglycemia. Infants born to diabetic mothers often secrete higher amounts of insulin to accommodate for excess fetal glucose concentrations. Preterm neonates and small-for-gestational-age infants have fewer glycogen and fat stores than full-term neonates.8 This lack of storage in combination with high insulin levels15 places them at risk for hypoglycemia. Birth asphyxia and perinatal stress increase the risk of hyperinsulinism in the neonatal period because of the use of anaerobic metabolism to maintain blood glucose concentrations.2 Transient hypoglycemia in these patients is a common occurrence during the first hours to days of life.
Congenital conditions such as Beckwith-Wiedemann, Mosaic Turner syndrome, and Costello syndromes have also been linked to hyperinsulinemia and subsequent hypoglycemia.16,17 These patients may experience a more prolonged hyperinsulinism lasting from several days to weeks. Persistent hyperinsulinemic hypoglycemia of infancy (PHHI), previously termed nesidioblastosis and congenital hyperinsulinism of infancy, is a rare condition that presents treatment challenges. It is associated with persistently elevated levels of circulating insulin caused by hypersecretion by pancreatic β-cells and an absence of ketone production. PHHI may present in the first few days of life or may present later in infancy.11 Many different metabolic disorders can also lead to persistent hypoglycemia, including glycogen storage disorders, disorders of gluconeogenesis, and fat oxidation.18
Understanding the glucose adaptation process that occurs shortly after birth, conditions leading to transient hypoglycemia, and the possible conditions affecting insulin secretion is necessary prior to selecting treatment. For infants experiencing transient hypoglycemia, an extensive diagnostic workup is not necessary. If persistent hyperinsulinism is suspected, the diagnostic workup will include plasma insulin, β-hydroxybutyrate, and free fatty acid levels. Additional tests may include plasma ammonia levels, plasma acyl-carnitine profile, and urine organic acids.5 The underlying cause of hypoglycemia and severity will also dictate the approach to management.
THERAPEUTIC MANAGEMENT
There are several treatment options available for the management of neonatal hypoglycemia (Table); however, selecting the appropriate intervention can be challenging as the underlying cause may take weeks to diagnose. Therefore, during the diagnostic process, it is important to prevent or minimize periods of hypoglycemia in an effort to mitigate potential adverse neurological outcomes caused by insufficient glucose availability for optimal brain function.
Table.
Pharmacologic Agents for Treatment of Neonatal Hypoglycemia
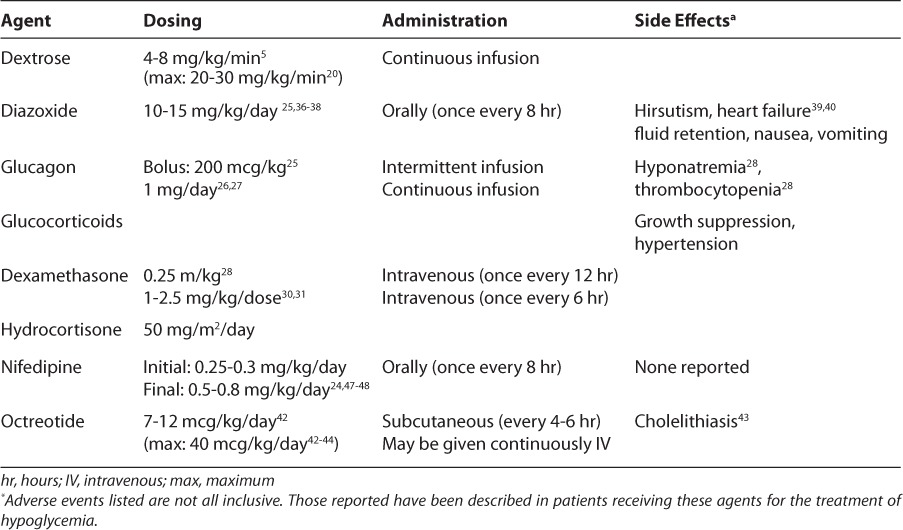
Dextrose
Hyperinsulinism is the most common cause of both transient and persistent/permanent forms of neonatal hypoglycemia. Administration of dextrose titrated to maintain euglycemia is the most practical and expedient initial approach.5 Dextrose infusions may be used regardless of the presence of enteral feeds. Conventionally, a 2 mL/kg to 3 mL/kg (200−300 mg/kg) intravenous bolus of 10% dextrose is given, followed by a continuous infusion.8,19 Initial glucose infusion rates generally used for full-term infants are 4 to 6 mg/kg/min, while rates for premature infants may be 6 to 8 mg/kg/min.6 An isotope tracer study noted that the glucose production rate of the liver in a single-term neonate was approximately 5 mg/kg/min.20 Glucose infusion rates should be titrated to achieve euglycemia, and hypoglycemic infants may require considerably higher rates.
Dextrose concentrations of up to 20% to 25% may be required in order to deliver glucose infusion rates in the 15 to 30 mg/kg/min range.5 Glucose infusions rates of up to 30 mg/kg/min have been used in patients unresponsive to lower rates.21 Most of these patients will require the placement of central lines in order to deliver a sufficient amount of dextrose in a volume that does not overload the patient with fluids. Peripheral access can be a concern not only from a volume perspective but also because of the hypertonicity of dextrose infusions. Concentrations greater than 12.5% must be administered using a central line.22 Because these patients require frequent serum glucose monitoring, it may be prudent to place an arterial line until stable euglycemia is established to minimize or prevent pain and tissue damage to the infants’ heels, toes, and fingers.
If patients continue to experience hypoglycemia episodes with a glucose infusion rate of approximately 20 mg/kg/min, additional therapeutic options may need to be explored, if the infant’s serum glucose cannot be maintained above 60 mg/dL.6 Unfortunately, the issue of when to progress to additional therapies for hypoglycemia is not well described. Many case reports begin to use additional medications after the glucose infusion rate remains at 12 mg/kg/min without the ability to wean therapy. The times of additional interventions in these case reports vary greatly from within the first 36 hours of life23 to 7 to 10 days of life.24 It may be prudent to delay additional therapies as long as possible (up to 14 days of life) to allow the transient forms of hypoglycemia to resolve in order not to expose infants to unnecessary drug therapy. Other than the infant who continues to experience consistent episodes of hypoglycemia or requires persistently high dextrose concentrations (>D25%), most infants should remain on dextrose infusions for at least 7 to 10 days before the addition of other therapeutic options is considered. However, infants who continue to experience profound hypoglycemia while receiving dextrose infusions may benefit from additional short-term therapy options such as glucagon and glucocorticoids. During this time, the focus should remain on the differential diagnosis.
Glucagon
Endogenous glucagon is secreted from α-cells in the pancreas as a counterregulatory hormone of insulin. Under normal physiologic conditions, hypoglycemia will induce the secretion of glucagon to raise serum glucose levels. Glucagon injections have been used for many years in neonatal intensive care units for the treatment of hypoglycemia. Glucagon has demonstrated less effectiveness in infants with PHHI or familial hyperinsulinism.25 However, glucagon is quite effective in elevating serum glucose concentrations in preterm and term neonates not exhibiting hyperinsulinemia.27,28
Intermittent glucagon doses have been variable. Doses have ranged from 200 mcg/kg to as low as 3 mcg/kg in a single patient.26 More commonly, glucagon is given as a continuous infusion over 24 hours. Doses range from 20 to 40 mcg/kg/hr in sick, preterm infants to a flat dose of 1 mg/day infused over 24 hours for infants regardless of gestational age or birth weight.27,28
Reports of severe hyponatremia with the use of glucagon have drawn attention to potential risks associated with this therapy.23 Hypertonic saline (3% sodium chloride) has been used to treat hyponatremia in infants receiving glucagon.25 While the use of glucagon may contribute to the decrease in serum sodium, another possible explanation is a dilutional effect decrease caused by the large volumes of intravenous dextrose solutions in addition to the volume required to administer the glucagon. There are reports of crystallization of glucagon when delivered in small volumes, resulting in occlusion of intravenous catheters. Further diluting the glucagon solution and or changing the solution more frequently than once a day may minimize the problem of crystallization. Monitoring of serum sodium should be conducted while receiving glucagon therapy. Other rare adverse effects described in case reports include thrombocytopenia and a rare cutaneous paraneoplastic phenomenon, erythema necrolyticum migrans.23,29
Glucocorticoids
The use of glucocorticoids, as an acute treatment of hypoglycemia, has been advocated by some.30 Data to support this use are limited to case reports; however, dosing is found in common tertiary references. Physiologically glucocorticoids reduce insulin secretion and increase insulin resistance as well as enhancing both gluconeogenesis and glycogenolysis. In theory these effects should induce an increase in serum glucose concentrations. Dexamethasone23 and hydrocortisone31,32 have been used to treat hypoglycemia. In case reports, patients received glucocorticoids at varying stages during the treatment process (initially versus salvage). Belik et al.23 gave a 35-week-gestation infant dexamethasone at 0.25 mg/kg every 12 hours; however, the infant later required glucagon therapy. Two other investigators, Bhownick et al.31 and Lindley et al.,32 gave hydrocortisone at 2.5 mg/kg/dose intravenously every 6 hours to a small-for-gestational-age term infant and 50 mg/m2/day intravenously divided every 6 hours to a 31-week gestation female, respectively. Both patients required additional therapies after the addition of steroids for prolonged glycemic control.
Systemic glucocorticoid therapy carries significant risks. Common side effects associated with glucocorticoid therapy include growth suppression, feed intolerance, and hypertension. Preterm, very-low-birth weight infants treated with hydrocortisone have an increased risk of spontaneous perforation of the gastrointestinal tract.33 In addition, glucocorticoids may unnecessarily elevate blood pressure in patients without hypotension and may increase the risk for adverse neurodevelopmental outcomes.34
Despite dextrose and glucocorticoid therapy, persistent hypoglycemia may be secondary to persistent hyperinsulinemia. Hyperinsulinism does not diagnose the cause of the excess secretion of insulin without further testing including genetic testing and acyl carnitine levels may take days to weeks for results. A trial of diazoxide may be indicated if hyperinsulinism is demonstrated.
Diazoxide
Diazoxide is a benzothiazine derivative that acts as a potent β-cell KATP channel opener. The stabilization of open KATP channels leads to the inhibition of insulin secretion. Patients with known or suspected genetic defect of the SUR and Kir 6.2 subunits may not fully respond to diazoxide therapy.35 These alterations have been demonstrated in families with hyperinsulinism who respond poorly to diazoxide therapy.36 Diazoxide has historically been a first line agent, however patients experiencing hypoglycemia during the neonatal period are less responsive than those presenting in infancy.37
Diazoxide is typically initiated at 10 to 15 mg/ kg/day orally in 2 to 3 divided doses. Some reports have initiated therapy at doses as small as 5 mg/kg/day.38 Doses may then be titrated upward or downward based on laboratory results. Diazoxide responsive patients tend to have lower dosage requirements than nonresponsive patients or those requiring surgical intervention.37,39 Maximum doses of up to 30 mg/kg/day have been reported; however, efficacy was not increased at doses above 15 mg/kg/day.39 When effective, hypoglycemia normalizes within 2 to 4 days of therapy initiation; however, because of variations in kinetic parameters, a trial of 5 to 8 days is required before judging therapy a failure.37 Onset of action is within 1 hour of administration, with a duration of action of 8 hours, assuming normal renal function. Diazoxide is available for oral use and, when successful, is one of only two therapies that provide an enteral option.
The most commonly reported side effect associated with diazoxide use is hypertrichosis, which is reported to some degree in nearly all patients.37 While clinically less significant, this can be quite concerning for parents. Another common and more clinically significant side effect is fluid retention. Diazoxide leads to an increase in sodium retention while limiting free water clearance. Patients without structural cardiac defects have developed cardiac failure while receiving diazoxide therapy. In these patients, the cardiac failure resolved upon discontinuation of therapy.40,41 Therefore, caution should be used in patients with known cardiac conditions susceptible to fluid overload.
Failure to respond to a trial of diazoxide therapy would suggest dysfunction associated with the KATP receptor, at which point a trial of octreotide is suggested as it works more distally in the insulin secretory pathway.5,7,39
Octreotide
Octreotide is a treatment option that is initiated in patients for whom diazoxide therapy failed. Octreotide is a long-acting somatostatin analogue. Somatostatin inhibits insulin secretion by hyperpolarization of β-cell and direct inhibition of VGCC. Endogenous somatostatin has a short half-life (1−3 minutes) making it a less desirable therapeutic option. The half-life of octreotide is approximately 1.5 hours, which allows for intermittent dosing in some patients. It may be administered as intravenous or subcutaneous intermittent doses or as a continuous infusion.
Initial dosing in the literature is limited to case reports and varies from standard dosing regimens to weight-based regimens.42–45 Doses of 5 to 25 mcg/dose given every 4 to 6 hours were initiated in two case series of 9 and 6 infants, respectively.42,43 Doses of 5 mcg/kg/dose every 4 to 6 hours were also used. Fifty-five percent of these patients (5 of 9 patients) were maintained with octreotide doses ranging from 7 to 12 mcg/ kg/day in combination with intermittent feedings.43 Thornton et al.45 began therapy at a dose of 5 mcg/kg/dose every 6 to 8 hours in 16 patients and titrated up to 40 mcg/kg/day. Others have also reported maximum doses of up to 40 mcg/kg/day.43,44
Octreotide demonstrated some efficacy in stabilizing glucose concentrations but not consistently avoiding subtotal pancreatectomy in nine patients.45 Nine patients received octreotide long term, up to 4.3 years with evidence of transient malabsorption and some compromise in linear growth. Resistance to octreotide occurred in all patients, even with escalated doses. This may be related to a possible ceiling effect at larger doses. Dose-related hypoglycemia may be seen with escalating doses. This phenomenon was described in an infant experiencing hypoglycemia once every 5 days while receiving 200 mcg/ day. As therapy was escalated to 400 mcg/day, the patients experienced increased frequency of hypoglycemic episodes (two−three per day). When therapy returned to the initial dose, hypoglycemia stabilized and occurred once every 4 to 5 days.42 Glaser et al.46 reported the use of octreotide in 8 patients with an initial starting dose of 4 to 5 mcg/kg/day divided in four doses. Doses required to maintain euglycemia ranged from 3 to 10 mcg/kg/day either IV or subcutaneously via an insulin infusion pump. For all patients, it was not necessary to increase the dose as the patient grew.
Intermittent dosing of octreotide should be administered 1 to 2 hours following selected feeds.42 Patient conditions may be controlled using doses with every other feed. If patients do not remain euglycemic with intermittent dosing, octreotide may be administered via continuous infusions.
Another important action of somatostatin is the inhibition of the release of growth hormone. The effects of long-term use of octreotide on normal growth are a concern. When growth hormone is measured following the administration of octreotide, a suppression of growth hormone is observed 2 hours following injections. However an increase in growth hormone is seen at 4 hours.47 Most side effects of octreotide are minor and require no interventions. One report of cholelithiasis in an infant required a reduction of the octreotide dose and initiation of ursodeoxycholic acid.44
Patients may have a good response to octreotide and can successfully wean off of dextrose infusions, while others may have no response or an incomplete response such that a continuous infusion of dextrose is still required to maintain euglycemia. For these patients, a combination of glucagon and octreotide may be necessary to maintain euglycemia and permit the successful discontinuation of a continuous dextrose infusion. This last subset of patients is most at risk for the necessity of a surgical intervention to treat the hyperinsulinism (Figure).
Figure.
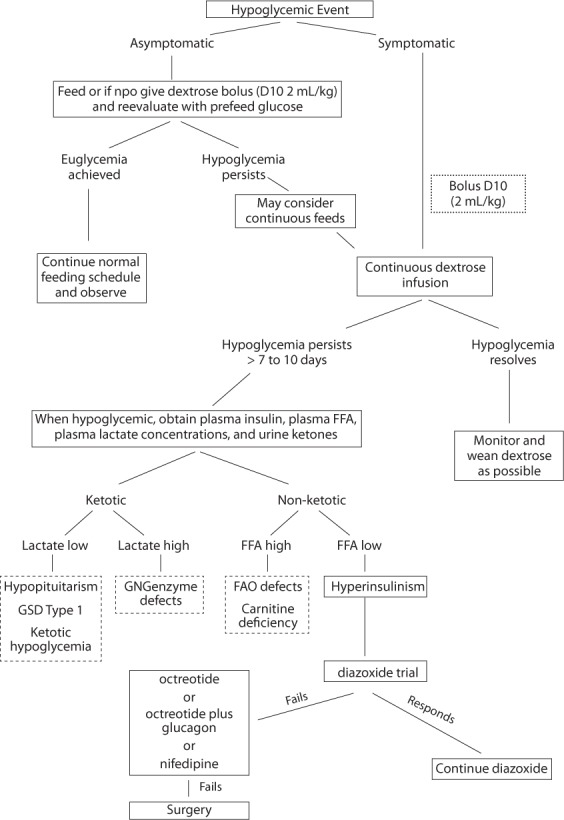
Treatment Strategies.
D10, dextrose 10%; FAO, fatty acid oxidation; FFA, free fatty acids; GN, genzymes (gluconeogenesis enzymes); GSD, glycogen storage disease.
Nifedipine
Nifedipine has been used by some investigators as a therapeutic option in patients for whom diazoxide therapy has failed with variable success.43,44,47 At this point, this therapy should be considered investigational because of the few patient case reports published.11 Calcium is required for adequate cardiac contractility in the infant population. The long-term safety of nifedipine has not been evaluated and may place some patients at risk for sudden cardiac death caused by calcium channel blockade.
Calcium channels located on pancreatic β-cell open to allow an influx of calcium leading to increased intracellular calcium which causes the secretion of insulin.11 Use of the calcium channel blocker nifedipine can inhibit insulin secretion. This treatment approach has been discussed more recently in the literature in patients fro whom other therapeutic options have failed; however, only 4 patients have been described in the published case series.24,48,49 Nifedipine was initiated at 0.25 to 0.3 mg/kg/day, orally, divided every 8 hours. Doses were increased by 0.1 mg/kg/day until patients were euglycemic and dextrose was weaned. Successful therapy was achieved with nifedipine at 0.5 to 0.8 mg/ kg/day.48,49
Safety of nifedipine in this population has not been studied; however, blood pressure was monitored routinely and was measured normal for sex and age.48 The case studies did not identify any adverse effects, nonetheless common side effects experienced with the use of calcium channel blockers include dizziness, flushing, headaches, and nausea.
DISCUSSION
Aggressive management of neonatal hypoglycemia is important as impaired neurodevelopmental outcomes are recognized in this patient population.11 Developmental delay has been reported in 30% of patients with congenital hyperinsulinism managed medically and is higher in those treated surgically.50 In another case series, the incidence of diabetes mellitus was 27% after pancreatectomy but reached 71% in those who had undergone more than one surgery. In addition, the incidence of neurodevelopmental delay was 44%.51 Adverse outcomes are presumed to be caused by hypoketotic hypoglycemia, but there may be other genetic abnormalities affecting neurodevelopment in this patient population as yet unidentified.39 Until further studies are available, it seems prudent to aggressively treat hypoglycemia in the neonate regardless of the infant’s age or underlying cause. Further studies in PHHI are still needed to develop a more consistent treatment approach; however, given its rarity, a multicenter strategy would best address the problem in a timely manner. Prospective multicenter trials are necessary to determine the kinds of treatment approaches that may maximize infants functional and neurodevelopmental outcomes.
For patients in whom a metabolic disorder has been diagnosed, appropriate disease-specific therapy should correct the hypoglycemia. There is continuing evolvement of the molecular and genetic causes of congenital hyperinsulinism which is beyond the scope of this review.5 The therapies previously described are palliative in that they are used until the specific diagnosis can be determined.
ABBREVIATIONS
- ATP
adenosine triphosphate
- D25
dextrose 25%
- KATP
ATP-sensitive potassium channels
- Kir6.2
inward rectifier potassium channel
- PHHI
persistent hyperinsulinemia hypoglycemia of infancy
- SUR
sulfonylurea subunit
- VGCC
voltage-gated calcium channels
Footnotes
DISCLOSURE The authors declare no conflicts or financial interest in any product or service mentioned in the manuscript, including grants, equipment, medications, employment, gifts, and honoraria.
REFERENCES
- 1.Cornblath M, Odell GB, Levin GY. Symptomatic neonatal hypoglycemia associated with toxemia of pregnancy. J Pediatr. 1959;55:545–562. doi: 10.1016/s0022-3476(59)80239-0. [DOI] [PubMed] [Google Scholar]
- 2.Collins JE, Leonard JV. Hyperinsulinism in asphyxiated and small for dates infant with hypoglycemia. Lancet. 1984;2(8398):311–313. doi: 10.1016/s0140-6736(84)92685-0. [DOI] [PubMed] [Google Scholar]
- 3.Persson B, Hanson U, Marcus C. Gestational diabetes mellitus and paradoxical macrosomia: a case report. Early Hum Dev. 1995;41(3):203–213. doi: 10.1016/0378-3782(95)01629-h. [DOI] [PubMed] [Google Scholar]
- 4.Zaffanello M, Zamboni G, Maffeis C. Neonatal hyperinsulinemic hypoglycemia: two case reports. Minerva Pediatr. 2002;54(4):325–333. et al. [PubMed] [Google Scholar]
- 5.De Leon DD, Stanley CA. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;3(1):57–68. doi: 10.1038/ncpendmet0368. [DOI] [PubMed] [Google Scholar]
- 6.Cornblath M, Hawdon JM, Williams AF. Controversies regarding the definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics. 2000;105(5):1141–1145. doi: 10.1542/peds.105.5.1141. et al. [DOI] [PubMed] [Google Scholar]
- 7.Hay WW, Cornblath M. Historical perspectives: transient symptomatic neonatal hypoglycemia. NeoReviews. 2003;4:e1–4. [Google Scholar]
- 8.Hypoglycemia of the newborn: review of the literature. Geneva, Switzerland: World Health Organization; 1997. World Health Organization. http://www.who.int/child_adolescent_health/documents/chd_97_1/en/index.html. Accessed April 24, 2013. [Google Scholar]
- 9.Lucas A, Morley R, Cole TJ. Adverse neurodevelopmental outcome of moderate neonatal hypoglycaemia. Br Med J. 1988;297(6659):1304–1308. doi: 10.1136/bmj.297.6659.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srinivasan G, Pildes RS, Cattamachi G. Plasma glucose values in normal neonates a new look. J Pediatr. 1986;109(1):114–117. doi: 10.1016/s0022-3476(86)80588-1. et al. [DOI] [PubMed] [Google Scholar]
- 11.Schwitzgebel VM, Gitelman SE. Neonatal hyperinsulinism. Clin Perinatol. 1998;25(4):1015–1038. [PubMed] [Google Scholar]
- 12.Munoz A, Hu M, Hussain K. Regulation of glucagon secretion at low glucose concentrations: evidence for adenosine triphosphate-sensitive potassium channel involvement. Endocrinology. 2005;146(12):5514–5521. doi: 10.1210/en.2005-0637. et al. [DOI] [PubMed] [Google Scholar]
- 13.Carmen S. Neonatal hypoglycemia in response to maternal glucose infusions before delivery. J Obstet Gynecol Neonatal Nurs. 1986;15(4):319–323. doi: 10.1111/j.1552-6909.1986.tb01403.x. [DOI] [PubMed] [Google Scholar]
- 14.Christesen HB, Melander A. Prolonged elimination of tolbutamide in a premature newborn with hyperinsulinaemic hypoglycaemia. Eur J Endocrinol. 1998;138(6):698–701. doi: 10.1530/eje.0.1380698. [DOI] [PubMed] [Google Scholar]
- 15.Hawdon JM, Hubbard M, Hales CN, Clark PM. The use of a specific radioimmunometric assay to determine preterm neonatal insulin-glucose relationship. Arch Dis Child. 1995;73(3):F166–169. doi: 10.1136/fn.73.3.f166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alkhayyat H, Christesen HB, Steer J. Mosaic turner syndrome and hyperinsulinaemic hypoglycaemia. J Pediatr Endocrinol Metab. 2006;19(12):1451–1457. doi: 10.1515/jpem.2006.19.12.1451. et al. [DOI] [PubMed] [Google Scholar]
- 17.Alexander S, Ramadan D, Alkhayyat H. Costello syndrome and hyperinsulinemic hypoglycemia. Am J Med Genet. 2005;139(3):227–230. doi: 10.1002/ajmg.a.31011. et al. [DOI] [PubMed] [Google Scholar]
- 18.Kahler SG. Metabolic disorders associated with neonatal hypoglycemia. NeoReviews. 2004;5:e377–e381. [Google Scholar]
- 19.Hawdon JM, Ward Platt MP, Aynsley-Green A. Prevention and management of neonatal hypoglycemia. Arch Dis Child Fetal Neonatal Ed. 1994;70(1):F60–F64. doi: 10.1136/fn.70.1.f60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bougneres PF. Stable isotope tracers and the determination of fuel fluxes in newborn infants. Biol Neonate. 1987;52(suppl 1):87–96. doi: 10.1159/000242742. [DOI] [PubMed] [Google Scholar]
- 21.Mohnike K, Blankenstein O, Pfuetzner A. Long-term non-surgical therapy of severe persistent congenital hyperinsulinism with glucagon. Horm Resp. 2008;70(1):59–64. doi: 10.1159/000129680. et al. [DOI] [PubMed] [Google Scholar]
- 22.Pereria-da-Silva L, Virella D, Henriques G. A simple equation to estimate the osmolarity of neonatal parenteral nutrition solutions. JPEN J Parenter Nutr. 2004;28(1):34–37. doi: 10.1177/014860710402800134. et al. [DOI] [PubMed] [Google Scholar]
- 23.Belik J, Musey J, Trussell RA. Continuous infusions of glucagon induces severe hyponatremia and thrombocytopenia in premature infants. Pediatrics. 2001;107(3):595–597. doi: 10.1542/peds.107.3.595. [DOI] [PubMed] [Google Scholar]
- 24.Kurtoglu S., Akcakus M, Keskin M. Severe hyperinsulinaemic hypoglycaemia in a baby born to a mother taking oral ritodrine therapy for preterm labour. Horm Resp. 2005;64(2):61–63. doi: 10.1159/000087471. et al. [DOI] [PubMed] [Google Scholar]
- 25.Horev Z, Ipp M, Levey P, Daneman D. Familial hyperinsulinism: successful conservative management. J Pediatr. 1991;119(5):717–720. doi: 10.1016/s0022-3476(05)80285-9. [DOI] [PubMed] [Google Scholar]
- 26.Mehta A, Wooten R, Cheng KN. Effect of diazoxide or glucagon on hepatic glucose production rate during extreme neonatal hypoglycaemia. Arch Dis Child. 1987;62(9):924–930. doi: 10.1136/adc.62.9.924. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charsha DS, McKinley PS, Whitfield JM. Glucagon Infusion for the treatment of hypoglycemia: efficacy and safety in sick, preterm infants. Pediatrics. 2003;111(1):220–221. doi: 10.1542/peds.111.1.220. [DOI] [PubMed] [Google Scholar]
- 28.Miralles RE, Lodha A, Perlman M, Moore AM. Experience with intravenous glucagon infusions as a treatment for resistant neonatal hypoglycemia. Arch Pediatr Adolesc Med. 2002;156(10):999–1004. doi: 10.1001/archpedi.156.10.999. [DOI] [PubMed] [Google Scholar]
- 29.Wald M, Lawrenz K, Luckner D. Glucagon therapy as a possible cause of erythema necrolyticum migrans in two neonates with persistent hyperinsulinaemic hypoglycemia. Eur J Pediatr. 2002;161(11):600–603. doi: 10.1007/s00431-002-1022-9. et al. [DOI] [PubMed] [Google Scholar]
- 30.Mehta A. Prevention and management of neonatal hypoglcaemia. Arch Dis Child. 1994;70(1):F54–F65. doi: 10.1136/fn.70.1.f54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhowmick SK, Lewandowski C. Prolonged hyperinsulinism and hypoglycemia in an asphyxiated, small for gestation infant: case management and literature review. Clin Pediatr (Phila) 1989;28(12):575–578. doi: 10.1177/000992288902801205. [DOI] [PubMed] [Google Scholar]
- 32.Lindley KJ, Dunne MJ, Kane C. Ionic control of β cell function in nesidioblastosis. A possible therapeutic role for calcium channel blockage. Arch Dis Child. 1996;74(5):373–378. doi: 10.1136/adc.74.5.373. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watterburg KL, Gerdes JS, Cole CH. Prophylaxis of early adrenal insufficiency to prevent bronchopulmonary dysplasia: a multicenter trial. Pediatrics. 2004;114(6):1649–1657. doi: 10.1542/peds.2004-1159. et al. [DOI] [PubMed] [Google Scholar]
- 34.American Academy of Pediatrics, Committee on Fetus and Newborn and Canadian Paediatric Society, Fetus and Newborn Committee. Post-natal corticosteroids to treat or prevent chronic lung disease in pre-term infants. Pediatrics. 2002;109:330–338. doi: 10.1542/peds.109.2.330. [DOI] [PubMed] [Google Scholar]
- 35.Peranteau WH, Ganguly A, Steinmuller L. Prenatal diagnosis and postnatal management of diffuse congenital hyper-insulinism: a case report. Fetal Diagn Ther. 2006;21(6):515–518. doi: 10.1159/000095664. et al. [DOI] [PubMed] [Google Scholar]
- 36.Kane C, Lidley KJ, Johnson PR. Therapy for persistent hyperinsulinemic hypoglycemia of infancy: understanding the responsiveness of β cells to diazoxide and somatostatin. J Clin Invest. 1997;100(7):1888–1893. doi: 10.1172/JCI119718. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Touati G, Poggi-Travert F, Ogier de Baulny H. Long-term treatment of persistent hyperinsulinaemic hypoglycemia of infancy with diazoxide: a retrospective review of 77 cases and analysis of efficacy-predicting criteria. Eur J Pediatr. 1998;157(8):628–633. doi: 10.1007/s004310050900. et al. [DOI] [PubMed] [Google Scholar]
- 38.Hussain K. Ketotic hypoglycemia in children with diazoxide responsive hyperinsulinism of infancy. Eur J Pediatr. 2005;164(6):387–390. doi: 10.1007/s00431-005-1654-7. [DOI] [PubMed] [Google Scholar]
- 39.Tyrrell VJ, Ambler GR, Yeow WH. Ten years‘ experience of persistent hyperinsulinaemic hypoglycemia of infancy. J Paediatr Child Health. 2001;37(5):483–488. doi: 10.1046/j.1440-1754.2001.00748.x. et al. [DOI] [PubMed] [Google Scholar]
- 40.McGraw ME, Price DA. Complications of diazoxide in the treatment of nesidioblastosis. Arch Dis Child. 1985;60(1):62–63. doi: 10.1136/adc.60.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gillies DR. Complications of diazoxide in the treatment of nesidioblastosis. Arch Dis Child. 1985;60(5):500–501. doi: 10.1136/adc.60.5.500-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glaser B, Landau H, Smilovici A, Nesher R. Persistent hyperinsulinaemic hypoglycaemia of infancy: long-term treatment with the somatostatin analogue sandostatin. Clin Endocrinol (Oxf) 1989;31(1):71–80. doi: 10.1111/j.1365-2265.1989.tb00455.x. [DOI] [PubMed] [Google Scholar]
- 43.Glaser B, Landaw H. Long-term treatment with the somatostatin analogue SMS 201–995: alternative to pancreatectomy in persistent hyperinsulinaemic hypoglycaemia of infancy. Digestion. 1990;45(suppl 1):27–35. doi: 10.1159/000200258. [DOI] [PubMed] [Google Scholar]
- 44.Radetti G, Gentili L, Paganini, Messner H. Cholelithiasis in a newborn following treatment with the somatostatin analogue octreotide. Eur J Pediatr. 2000;159(7):550. doi: 10.1007/s004310051335. [DOI] [PubMed] [Google Scholar]
- 45.Thornton PS, Alter CA, Katz LE. Short and long term use of octreotide in the treatment of congenital hyperinsulinism. J Pediatr. 1993;123(4):637–643. doi: 10.1016/s0022-3476(05)80969-2. et al. [DOI] [PubMed] [Google Scholar]
- 46.Glaser B, Hirsch HJ, Landau H. Persistent hyperinsulenemic hypoglycemia of infancy: longterm octreotide treatment without pancreatectomy. J Pediatr. 1993;123(4):644–650. doi: 10.1016/s0022-3476(05)80970-9. [DOI] [PubMed] [Google Scholar]
- 47.DeClue TJ, Malone JI, Bercu BB. Linear growth during long-term treatment with somatostatin analog (SMS 201–995) for persistent hyperinsulinemic hypoglycemia of infancy. J Pediatr. 1990;116(5):747–750. doi: 10.1016/s0022-3476(05)82665-4. [DOI] [PubMed] [Google Scholar]
- 48.Shanbag P, Pathak A, Vaidya M, Shahid SK. Persistent hyperinsulinemic hypoglycemia of infancy: successful therapy with nifedipine. Indian J Pediatr. 2002;69(3):271–272. doi: 10.1007/BF02734240. [DOI] [PubMed] [Google Scholar]
- 49.Bas F, Darendeliler F, Demirkol D. Successful therapy with calcium channel blocker (nifedipine) in persistent neonatal hyperinsulinemic hypoglycemia of infancy. J Pediatr Endocrinol Metab. 1999;12(6):873–878. doi: 10.1515/jpem.1999.12.6.873. et al. [DOI] [PubMed] [Google Scholar]
- 50.Steinkrauss L, Lipman TH, Hedell CD. Effects of hypoglycemia on developmental outcomes in children with congenital hyperinsulinism. J Pediatr Nurs. 2005;20(2):109–118. doi: 10.1016/j.pedn.2004.12.009. et al. [DOI] [PubMed] [Google Scholar]
- 51.Meissner T, Wendel U, Burgard P. Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol. 2003;149(1):43–51. doi: 10.1530/eje.0.1490043. et al. [DOI] [PubMed] [Google Scholar]