

Abstract
Caloric restriction (CR) is a dietary regimen known to promote lifespan by slowing down the occurrence of age-dependent diseases. The greatest risk factor for neurodegeneration in the brain is age, from which follows that CR might also attenuate the progressive loss of neurons that is often associated with impaired cognitive capacities. In this study, we used a transgenic mouse model that allows for a temporally and spatially controlled onset of neurodegeneration to test the potentially beneficial effects of CR. We found that in this model, CR significantly delayed the onset of neurodegeneration and synaptic loss and dysfunction, and thereby preserved cognitive capacities. Mechanistically, CR induced the expression of the known lifespan-regulating protein SIRT1, prompting us to test whether a pharmacological activation of SIRT1 might recapitulate CR. We found that oral administration of a SIRT1-activating compound essentially replicated the beneficial effects of CR. Thus, SIRT1-activating compounds might provide a pharmacological alternative to the regimen of CR against neurodegeneration and its associated ailments.
Introduction
Caloric restriction (CR), the reduction in the consumption of calories, is an effective regimen to slow aging in a variety of organisms ranging from yeast, nematodes, and fruitflies to mammals (Bishop and Guarente, 2007; Fontana et al., 2010). CR acts on a broad spectrum of tissues including liver, muscle, fat, pancreatic β-cells, and, interestingly, also the brain (Bordone and Guarente, 2005, 2007). There, CR has been shown to lead to an attenuation of age-associated ailments, in particular, Alzheimer's disease (AD)-related pathologies: CR reduced the load of amyloid-β, a pathological hallmark of AD, in different mouse models of AD (Patel et al., 2005; Wang et al., 2005; Qin et al., 2006b, Halagappa et al., 2007; Wu et al., 2008b; Mouton et al., 2009) and in aged squirrel monkeys (Qin et al., 2006a), and in the 3xTg AD mouse model, CR also reduced the phosphorylation of tau (Halagappa et al., 2007), another pathological aggregation in AD. Furthermore, CR was shown to rescue against memory deficits in several of these models (Halagappa et al., 2007; Wu et al., 2008b), and similar effects were also observed in wild-type mice (Fontán-Lozano et al., 2007) and in aged humans (Witte et al., 2009). Last, in an endpoint study with rhesus monkeys, CR was found to have significantly attenuated brain atrophy by preserving gray matter volume (Colman et al., 2009). However, whether CR can delay the onset of neurodegeneration and the mechanisms of this process currently remain unknown.
One potential mechanism that might mediate the beneficial effects of CR on brain health is the activation of the nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylase SIRT1 (Guarente and Picard, 2005; Bishop and Guarente, 2007; Bordone et al., 2007; Chen and Guarente, 2007; Lavu et al., 2008). SIRT1 was shown to at least partly mediate the beneficial effects of CR on AD-related pathologies (Qin et al., 2006b), on inflammatory responses following neurotoxic insults (Chen et al., 2008), and on enhancing learning and memory capacities (Fusco et al., 2012). Moreover, SIRT1 overexpression or activation in parallel to CR has been shown to reduce amyloid-β deposition (Chen et al., 2005; Donmez et al., 2010), tau phosphorylation (Min et al., 2010), and microglia and neurotoxicity-dependent inflammation (Chen et al., 2005; 2008), and to improve learning, memory, and synaptic plasticity (Gao et al., 2010; Michán et al., 2010). Furthermore, SIRT1 activation reduced axonal degeneration in the Wallerian degeneration mouse model (Araki et al., 2004) and protected against neuronal loss in mouse models of AD and amyotrophic lateral sclerosis (Kim et al., 2007). However, the potentially beneficial implication of SIRT1 activation following CR against neurodegeneration has not been investigated.
For this study, we hypothesized that CR delays the onset of neurodegeneration and neurodegeneration-associated pathologies such as brain atrophy, loss of synaptic plasticity, and cognitive deficits via the activation of SIRT1. Furthermore, we reasoned that if this was the case, SIRT1-activating compounds (STACs) should be able to recapitulate the beneficial effects of CR and provide a pharmacological alternative to this dietary regimen.
Materials and Methods
Animals.
Adult (3-month-old) double-transgenic CK-p25 mice of either sex (Cruz et al., 2003, 2006) and their single-transgenic or nontransgenic littermates were used for all experiments. All animal work was approved by the Committee for Animal Care of the Division of Comparative Medicine at the Massachusetts Institute of Technology.
Experimental manipulations.
CR treatment on CK-p25 mice consisted of 30% CR, representing 70% of the normal ad libitum caloric consumption. Mice were first accustomed to this diet during a 6 week period and then maintained on it for another 6 weeks, for a total duration of 3 months CR. During the last half of this treatment, p25 was induced by doxycycline removal from the diet. Control treatment consisted of ad libitum feeding of age- and sex-matched CK-p25 mice during the same time. Ad libitum feeding allowed for unrestricted continuous access to solid food and water. In total, the CR regimen was repeated two independent times.
Treatment of CK-p25 mice with SRT3657 [tertα-butyl 4-((2-(2-(6-(2-(tert-butoxycarbonyl(methyl)amino)ethylamino)-2-butylpyrimidine-4- carboxamido)phenyl)thiazolo[5,4-b]pyridin-6-yl)methoxy)piperidine-1-carboxylate][Sirtris-GSK; compound 10 in the study by Dai et al. (2010)] was performed by daily oral gavage with 30 mg/kg SRT3657 for 6 weeks. SRT3657 was diluted in Vitamin E TPGS (d-α-tocopheryl polyethylene glycol 1000 succinate; Cognis) as vehicle. Control treatment consisted of daily oral gavage with vehicle alone. Injection volume was 0.1 ml per animal. At this formulation, the brain/plasma ratio of SRT3657 at 3 mg/kg was 0.13.
Behavior.
Behavioral experiments were essentially conducted as described previously (Fischer et al., 2005; Gao et al., 2010). Open-field behavior was monitored using the VersaMax system (Accuscan) during 20 min. For fear conditioning, mice were put in the conditioning chamber (TSE Systems) for 3 min, after which a constant tone was played during 30 s, the last 2 s of which were paired with a one-time 2 s footshock (0.8 mA). Animals were then left in the box for another 30 s. Twenty-four hours later, mice were first tested for their contextual fear memory by putting them into the same training chamber for 3 min, during which their freezing behavior was scored. For assessing their cued fear memory, which took place at least 4 h after the contextual memory task, mice were put into a visually (gray instead of transparent) and contextually (flat instead of gridded surface) different box with a different smell (vanilla scented instead of ethanol scented) for a total of 3 min, the first half of which no tone was played, which was taken to measure baseline freezing levels, and the second half of which the same tone as during training was played, which served to measure their cued memory performance. Freezing was scored manually by an experimenter blind to the treatment group, by measuring freezing as the absence of movement except briefing every 10 s for 2 s. For the novel object recognition test, mice were habituated to the open testing arena (41.3 × 41.3 × 30.5 cm) over 2 d, for four trials per day, lasting 5 min each. During training, mice were exposed to two unknown objects that they were allowed to explore during four sessions of 5 min each. The objects had previously been put into a dirty cage to avoid olfactory preferences. Object memory was tested 24 h later by replacing one of the objects with a novel one and measuring the time with which the animals explored the novel object during a 10 min exploration session. The location of the object was counterbalanced between mice. Object exploration was scored by an automated software (Ethovision XT) and expressed as a discrimination ratio, DR = (tnovel − tfamiliar1)/ttotal with tnovel and tfamiliar being the time spent with the novel and the familiar objects, respectively, and ttotal being the total time spent with both objects.
Electrophysiology.
Mice were killed by cervical dislocation, and the brains were rapidly removed and transferred to ice-cold artificial CSF (ACSF) equilibrated with 95% O2/5% CO2. Transverse hippocampal slices (400 μm thick) were cut with a Leica VT1000S vibratome and recovered in a holding chamber at 32–34°C for 30 min, and were then allowed to return to room temperature for at least another 60 min before recording. The ACSF used for cutting contained the following (in mm): 75 sucrose, 87 NaCl, 2.5 KCL, 1.25 NaH2PO4, 21.4 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.3 ascorbic acid, and 20 d-glucose. The same ACSF was used for both incubation and recording, and contained the following (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1 NaH2PO4, 1.3 MgSO4 26.2 NaHCO3, and 11 d-glucose, saturated with 95% O2/5% CO2, pH 7.4. For extracellular recordings in the CA1 region of the hippocampus, a bipolar platinum–iridium stimulating electrode was placed in the Schaffer collateral axons to elicit field population responses. The field EPSPs (fEPSPs) were recorded via a glass micropipette filled with ACSF (1–3 MΩ) placed in the stratum radiatum region of the CA1. Stimuli (0.1 ms duration) were delivered every 30 s. Data were acquired using a HEKA EPC 10 amplifier and analyzed by Patchmaster software (HEKA). Peak stimulation-induced fEPSP amplitudes were required to be at least 0.7 mV, and stimulus intensity was set to produce 30–50% of the maximal response. Responses were allowed to stabilize, then a baseline was recorded for 30 min. Long-term potentiation (LTP) was induced by theta-burst stimulation (TBS), which was delivered as four trains of four pulses at 100 Hz separated by 200 ms. Recordings were made at baseline test intensity for an additional hour after LTP induction. In all experiments, the initial slope of the fEPSP was measured, and LTP for each time point was expressed as a percentage of the preinduction baseline. For analysis, measurements of evoked responses during the last 10 min of the recording session were compared with the last 5 min of the baseline period using Student's t tests.
Immunohistochemistry.
For immunohistochemistry, mice were perfused with 10% paraformaldehyde under deep anesthesia (ketamine, xylazine) and their brains sectioned at 40 μm thickness using a vibratome (Leica). Slices were permeabilized with 0.1% Triton X-100, blocked, and incubated overnight with 0.1% Triton X-100/10% fetal bovine serum in PBS containing primary antibodies: NeuN (Millipore), GFP (Aves Labs), synaptophysin (SVP; Sigma), SIRT1 (Abcam), SIRT3 (Santa Cruz Biotechnology), and histone 3 lysine 56 acetylation (AcH3K56; Epitomics). Primary antibodies were visualized with Alexa-Fluor 488, Cy3, and Cy5 antibodies (Invitrogen) in 1× PBS, and neuronal nuclei with Hoechst 33342 (Invitrogen). Note that the specificity of the SIRT1 antibody was confirmed in SIRT1F/F animals (Cheng et al., 2003) (data not shown). Slices were covered with Fluoromount G (Electron Microscopy Sciences). Images were acquired using a confocal microscope (LSM 510; Zeiss) and analyzed using ImageJ 1.42q. Three or four animals per group were used.
Electron microscopy.
Animals were perfused with 2.5% glutaraldehyde/2% paraformaldehyde in 0.1 m sodium cacodylate buffer, pH 7.4. The hippocampus was dissected out, sliced into 1 mm slices, washed in 0.1 m cacodylate buffer, and postfixed with 1% osmiumtetroxide (OsO4)/1.5% potassiumferrocyanide (KFeCN6) for 1 h, washed in water three times, and incubated in 1% aqueous uranyl acetate for 1 h followed by two washes in water and subsequent dehydration in grades of alcohol (10 min each at 50, 70, and 90%; two times for 10 min at 100%). The samples were then placed in propyleneoxide for 1 h and subsequently infiltrated overnight in a 1:1 mixture of propyleneoxide and TAAB Epon (Marivac Canada). The following day, the samples were embedded in TAAB Epon and polymerized at 60 nm for 48 h. Ultrathin sections (about 60 nm) were cut on a Reichert Ultracut-S microtome, placed onto copper grids, stained with uranyl acetate and lead citrate and examined in a TecnaiG2 Spirit BioTWIN. Images were recorded with an AMT 2k CCD camera. Fifteen distinct apical regions (stratum radiatum) of CA1, ∼50–100 μm from the cell body layer of the hippocampus were imaged per animal. A synapse was defined as an electron dense postsynaptic density area juxtaposed to a presynaptic terminal filled with synaptic vesicles. The number of synapses was determined by an experimenter blind to treatment and genotype.
Golgi staining.
Mice were perfused with 10% paraformaldehyde, and their entire brains Golgi-Cox stained using the Rapid Golgistain Kit (FD NeuroTechnologies) as per the manufacturer's instructions. Brains were cut at 60 μm thickness using a Leica vibratome. The number of apical and basal spines on hippocampal CA1 pyramidal neurons was counted by an experimenter blind to treatment group and genotype. Only mature spines with a clear mushroom-like body and a distinctive shaft were included. For each experimental group, a minimum of 10 cells from three or four animals each was analyzed.
Immunoprecipitation and Western blot.
Immunoprecipitation (IP) was performed by homogenizing whole-tissue hippocampal lysates in 500 μl of sterile-filtered 50 mm Tris, 120 mm NaCl, and 0.5% NP-40 containing proteinase inhibitors (Roche), followed by 15 min centrifugation at 14,000 rpm at 4°C and collection of the supernatant, which was incubated 60–90 min with 1–2 μg of p53 (Cell Signaling Technologies). After incubation, BSA-precleared protein G beads (GE Healthcare) were added for 45 min at 4°C, and the immune complexes were collected at 8000 rpm for 3 min, by one wash each in high-salt homogenization buffer (containing 50 mm Tris, 500 mm NaCl, and 1% NP-40) and regular homogenization buffer (see above). For Western blot (WB), whole-tissue lysates were homogenized in 400 μl 1× RIPA buffer containing proteinase inhibitors (Roche). IP or WB samples were loaded on 10–12% SDS gels, transferred onto PVDF membranes, and incubated with primary antibodies against acetylated lysine, p53 (Cell Signaling Technologies), IgG, synaptophysin (Sigma), or SIRT1 (Millipore) in 1× TBST, and visualized with HRP-conjugated secondary antibodies (GE Healthcare) and Western Lightning ECL solutions (PerkinElmer). Band intensities were quantified using ImageJ 1.42q.
SIRT1 activity assay.
SIRT1 activity was measured by a Fluor-de-Lys enzymatic assay (Enzo Life Sciences) with the following specifications. In vitro, 1.5 μl of the Fluor-de-Lys substrate was incubated with 1 μl of recombinant human SIRT1 protein in 30 μl 1× assay buffer (50 mm Tris-Cl, pH8.0, 1 mm DTT, 137 mm NaCl, 2.7 mm KCl, 4 mm MgCl2, 0.1 mm EDTA, 1 mm NAD+, and 10% glycerol in ddH2O) in the presence or absence of 1 μl 50 mm nicotinamide or 1 μl 0.5 μg/100 μl suramin sodium. Fluorescence was measured in transparent polystyrol plates (Corning) by excitation wavelength 355 nm and emission wavelength 460 nm using an infinite 200Pro microplate reader (Tecan). In vivo, the protein content of whole-tissue lysates was extracted as described under immunoprecipitation (see above) and incubated overnight with 1–2 μg of SIRT1 antibody (Millipore). After incubation, BSA-precleared protein A beads (GE Healthcare) were added for 45 min at 4°C, and the immune complexes were collected by centrifugation at 8000 rpm for 3 min and washed twice with high-salt homogenization buffer (see Immunoprecipitation and Western blot, above). The immune complexes were then redissolved in 30 μl 1× assay buffer together with 1.5 μl of the Fluor-de-Lys substrate for 30 min at 37° shaking, and the supernatant was used for the fluorescent measurement. SIRT1 activity was normalized to SIRT1 protein levels.
Statistics.
Statistical analyses were performed using GraphPad Prism 5. One-way ANOVAs followed by Tukey's post hoc analyses or one-tailed Student's t tests were used. All data are represented as mean ± SEM. Statistical significance was set at p ≤ 0.05.
Results
CR rescues against neuronal loss and brain atrophy
To examine whether CR can protect against neuronal loss, we used the CK-p25 mouse model of neurodegeneration (Cruz et al., 2003, 2006; Fischer et al., 2005). These mice allow for a time-controlled onset of neurodegeneration in the forebrain by doxycycline-mediated inducible activation of the CdK5 coactivator p25 (coupled to GFP) under the CamKIIα promoter. At 6 weeks of p25 induction, these mice display substantial neuronal loss, reduced dendritic arborization, reduced synaptic density and plasticity, as well as learning and memory deficits. CK-p25 mice were subjected to 3 months of 30% calorie restriction (CK-p25,CR) (Fig. 1A), the typical regimen of food reduction found to increase rodent lifespan (Bishop and Guarente, 2007). After 6 weeks of CR, when the animals' reduction in body weight reached asymptotic levels (Fig. 1B), we induced transgene production by removing doxycycline from the diet (Fig. 1A). CK-p25,CR animals were compared to CK-p25 mice fed ad libitum (CK-p25,AL) and to control mice fed ad libitum (CON,AL) (see Materials and Methods) (Fig. 1A,B).
Figure 1.
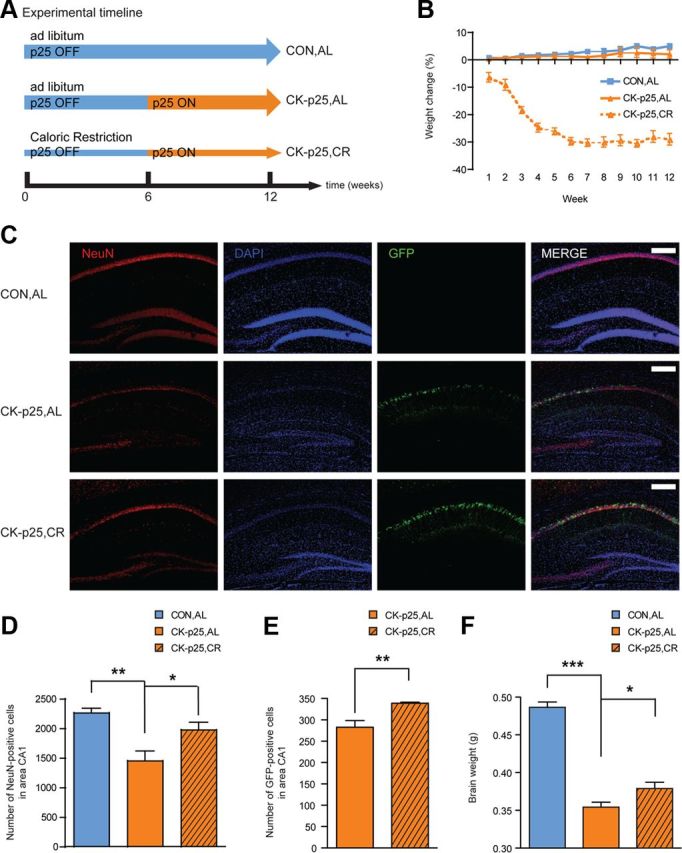
A regimen of 3 month 30% CR reduces neurodegeneration. A, Schematic of the experimental timeline. Three groups of mice were used: CON,AL, CK-p25,AL, and CK-p25,CR. B, Weight development over the experimental timeline of all three groups of mice used in this study. C, CR-mediated preservation of neuronal integrity in CK-p25,CR animals. Immunohistochemical images depicting the number of neurons as evidenced by NeuN-staining and the number of p25-expressing cells by GFP-staining. Scale bars, 200 μm. D, Quantification of NeuN-positive cells under C. One-way ANOVA (F(2,9) = 10.01, p ≤ 0.01), followed by Tukey's post hoc tests; n = 4 mice each. E, Quantification of GFP-positive cells under C (one-tailed t test, t(4) = 3.56; n = 3 each). F, CR-mediated preservation of overall brain mass. One-way ANOVA F(2,30) = 102.6, p ≤ 0.0001, followed by Tukey's post hoc tests; n = 12 for CON,AL and CK-p25,AL; n = 9 for CK-p25,CR. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001 for Tukey's post hoc comparisons. All values are mean ± SEM.
Following the regimen of CR, we found that CK-p25,CR animals had a similar number of neurons in hippocampus area CA1 compared to CON,AL animals as evidenced by NeuN immunohistochemistry, despite the profound neuronal loss displayed by CK-p25,AL animals (Fig. 1C,D) (Fischer et al., 2007). The number of neurons in CK-p25,CR animals was significantly higher than in CK-p25,AL animals (Fig. 1C,D), an effect that was also detectable by measuring the amount of GFP-positive p25-expressing cells in this brain area (Fig. 1E). This suggests that CR not only rescues against neuronal death in p25-positive neurons, but in a non cell-autonomous way throughout this brain area. These neuroprotective effects of CR were also observed by an attenuated loss of brain mass in CK-p25,CR animals compared to CK-p25,AL mice (Fig. 1F). Therefore, a regimen of 30% CR effectively rescued against p25-driven neurodegeneration.
CR preserves synaptic density
To assess whether CR also preserves structural synaptic integrity in CK-p25 mice, we measured synaptic density by immunohistochemical labeling of SVP, a marker of presynaptic terminals. SVP labeling was markedly reduced in the stratum radiatum of CK-p25,AL animals compared to CON,AL mice (Fig. 2A,B) (Fischer et al., 2007). In contrast, in CR-treated CK-p25 mice the levels of SVP were comparable to CON,AL mice, and significantly elevated over those in CK-p25,AL animals (Fig. 2A,B). Similar results were obtained by Western blot analysis of SVP levels (data not shown). Thus, CR rescued against the loss of presynaptic terminals.
Figure 2.
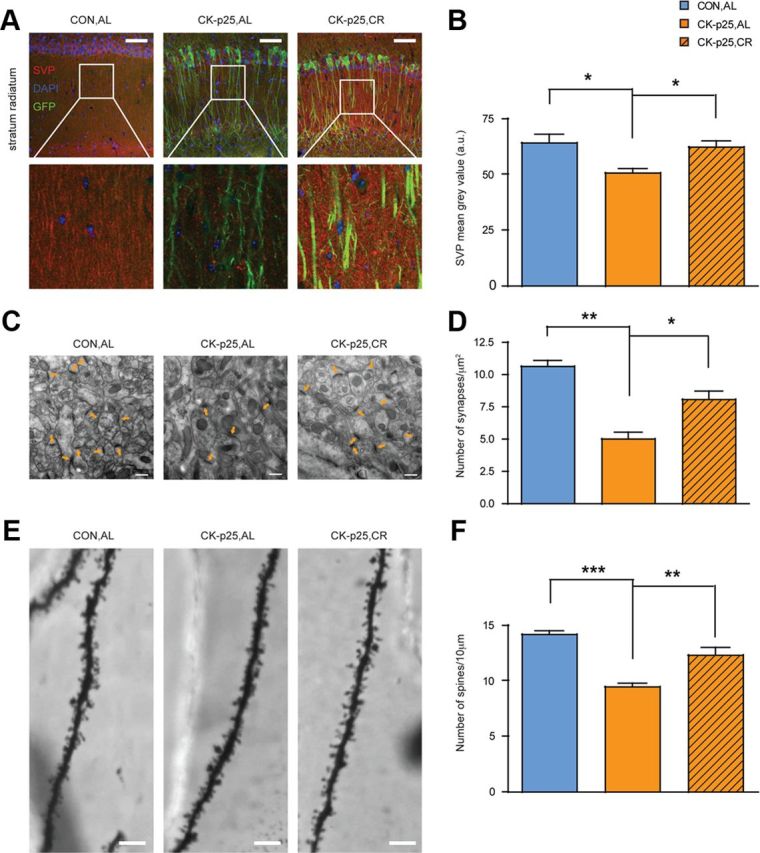
CR protects structural synaptic and dendritic integrity. A, CR-mediated preservation of presynaptic terminals in CK-p25,CR animals as evidenced by SVP immunohistochemistry in the stratum radiatum of hippocampal area CA1. B, Quantification of A [one-way ANOVA (F(2,8) = 8.99, p ≤ 0.01) followed by Tukey's post hoc tests; n = 3 for CON,AL and CK-p25,CR; n = 5 for CK-p25,AL]. C, CR-mediated preservation of the number of synapses in the hippocampus of CK-p25,CR animals as evidenced by transmission electron microscopy. D, Quantification of C. One-way ANOVA (F(2, 6) = 24.84, p ≤ 0.01) followed by Tukey's post hoc tests; n = 3 mice each. E, CR-mediated preservation of the number of spines per given dendritic length in the hippocampus of CK-p25,CR animals as evidenced by Golgi impregnation. F, Quantification of E [one-way ANOVA (F(2,22) = 23.31, p ≤ 0.01) followed by Tukey's post hoc tests; n = 7–10 dendrites per animal, 3 animals each]. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001 for Tukey's post hoc comparisons. All values are mean ± SEM. Scale bars: A, 100 μm; C, 0.5 μm; E, 2 μm.
Next, to more precisely determine the effect of CR on synaptic integrity, we examined the number of functional synapses by transmission electron microscopy. We defined a synapse as an electron-rich postsynaptic density area juxtaposed to a presynaptic terminal filled with synaptic vesicles (see Materials and Methods). We found that in the stratum radiatum of CK-p25,CR animals, the number of functional synapses was markedly elevated over CK-p25,AL mice, and not significantly different from that in CON,AL animals (Fig. 2C,D). In addition to preserving presynaptic terminals, CR was thus also effective to prevent the loss of functional synapses that normally occurs during p25-mediated neurodegeneration (Fischer et al., 2005).
Another aspect of synaptic integrity is the number of dendritic spines, which reflects the degree of connectivity between neurons and thereby synaptic strength (Fiala et al., 2002). To explore whether CR affects dendritic spine density, we used Golgi impregnation of CA1 pyramidal neurons. We found that whereas CK-p25,AL mice had lower spine density than their control littermates (CON,AL), CK-p25 mice that underwent CR had a similar number of spines per given length to CON,AL animals, a number that was significantly higher when compared to CK-p25,AL mice (Fig. 2E,F). Together, these data indicate that, in addition to rescuing against neurodegeneration, CR is also effective in maintaining synaptic density.
CR preserves synaptic plasticity and memory capacities
As structural synaptic integrity was rescued by the CR regimen, we next questioned if such beneficial effects would also translate into ameliorated synaptic plasticity and enhanced memory capacities. Six-week induced CK-p25 mice show impairments in hippocampal LTP and associative and spatial memory tasks (Fischer et al., 2005). We therefore asked whether CR had a positive effect on LTP and these types of memory. We first measured fEPSPs following theta-burst stimulation of Schaffer collaterals. While this stimulation was not capable of evoking LTP in CK-p25,AL animals, it did so in CK-p25,CR animals to the extent that fEPSPs of CK-p25,CR mice were undistinguishable from CON,AL animals (Fig. 3A). Importantly, basal synaptic transmission as measured by the synaptic input–output relationship was similar between all three groups, albeit slightly reduced in CK-p25,AL and CK-p25,CR animals (Fig. 3B). Thus, CR rescued against neurodegeneration-mediated synaptic plasticity deficits.
Figure 3.
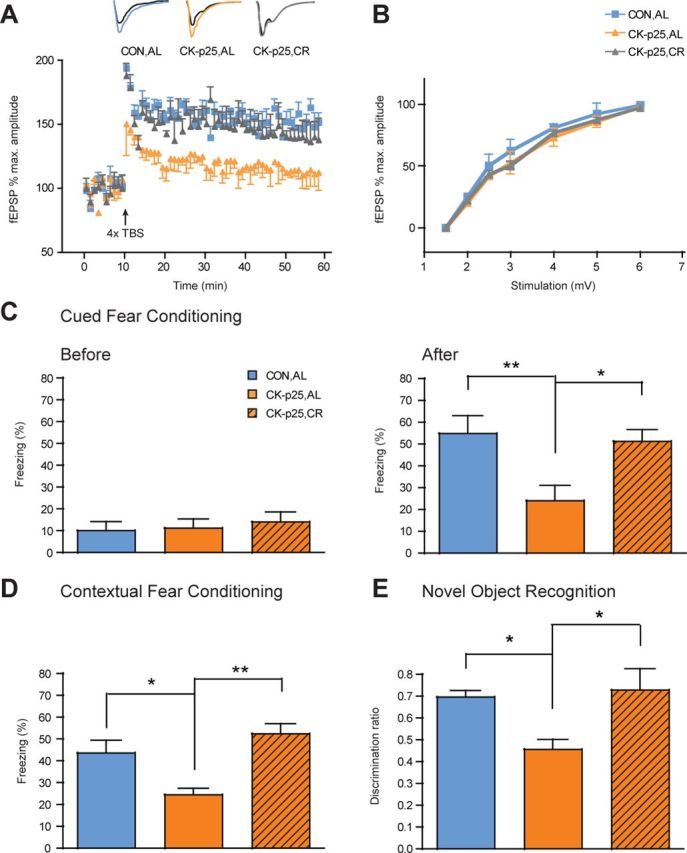
CR maintains synaptic plasticity and memory. A, Preservation of long-term potentiation by CR. fEPSPs (expressed in percentage of maximal amplitude) in hippocampal area CA1 of CON,AL, CK-p25,AL and CK-p25,CR animals (n = 3–4 slices from 3–4 mice each) before and after TBS. *p ≤ 0.05 by ANOVA for the effect of treatment. Sample traces above the line chart represent fEPSPs at 1 min before (black) and 1 h after (colored) TBS. B, Input–output relationship of comparable baseline synaptic transmission between CON,AL, CK-p25,AL, and CK-p25,CR animals. C, Cued fear conditioning. Left, Freezing responses before the onset of the tone were comparable between CON,AL, CK-p25,AL and CK-p25,CR animals 24 h after training. Right, Freezing responses after the onset of the tone show comparable memory retention between CON,AL and CK-p25,CR animals, but reduced memory for CK-p25,AL mice. One-way ANOVA (F(2,30) = 6.29, p ≤ 0.01) followed by Tukey's post hoc tests; n = 10–12 mice each. D, Contextual fear conditioning. Freezing responses 24 h after training on a contextual fear conditioning task show comparable memory retention between CON,AL and CK-p25,CR animals, but reduced memory for CK-p25,AL mice [one-way ANOVA (F(2,30) = 7.52, p ≤ 0.01) followed by Tukey's post hoc tests; n = 10–13 mice each]. E, Novel object recognition. Discrimination ratio 24 h after training revealing a significantly enhanced object memory in CK-p25,CR compared to CK-p25,AL animals [one-way ANOVA (F(2,19) = 7.16, p ≤ 0.01) followed by Tukey's post hoc tests; n = 7–8 mice each]. *p ≤ 0.05; **p ≤ 0.01 for Tukey's post hoc comparisons. All values are mean ± SEM.
Then, we measured the animals' memory performance on a cued and contextual version of fear conditioning and on an object recognition paradigm to assess associative and spatial memories for objects, respectively. On the cued fear conditioning task, CK-p25,CR animals remembered the tone significantly better than CK-p25,AL animals 24 h after training, at levels indistinguishable from those of CON,AL animals (Fig. 3C, After). Importantly, the fear response of CK-p25,CR mice upon reexposure to the conditioning chamber alone, i.e., before the onset of the tone, was comparable between all three groups (Fig. 3C, Before), suggesting a selective enhancement of the trained memory, and not an overall increase of the fear reaction. A similar rescue in memory performance was observed in the contextual fear conditioning paradigm 24 h after training: CK-p25,CR animals performed significantly better than CK-p25,AL mice, at levels comparable to CON,AL animals (Fig. 3D). Last, on the novel object recognition task, CK-p25,AL mice spent significantly less time exploring the novel object than CON,AL mice 24 h after training, indicating impaired object memory (Fig. 3E). CK-p25,CR mice, however, spent significantly more time with the novel object than CK-p25,AL mice, at levels indistinguishable from CON,AL animals (Fig. 3E), suggesting that CR was capable of preserving object memory. There was no difference in object preference during training (data not shown).
Importantly, CR treatment per se did not alter overall locomotor activity as assessed by total distance moved and time spent moving in an open-field test, and CR did further not affect the animals' anxiety as measured by the time spent in the center of an open arena, nor their exploratory behavior, indicated by their time spent rearing (data not shown). Therefore, without affecting the animals' general behavior, the regimen of CR specifically improved synaptic plasticity as well as associative and object recognition memories.
CR activates SIRT1 and SIRT1-mediated neuroprotective mechanisms
One potential mechanism that might mediate the beneficial effects of CR against neurodegeneration and its associated structural and behavioral impairments involves the activation of SIRT1, since SIRT1 overexpressing mice were shown to recapitulate the majority of the systemic effects of CR (Bordone et al., 2007), and because SIRT1 expression was found to be increased in the brains of CR-treated animals (Qin et al., 2006b; Chen et al., 2008). Based on this, we first measured the levels of hippocampal SIRT1 by immunohistochemistry. In agreement with previous reports (Qin et al., 2006b; Chen et al., 2008), we found SIRT1 protein expression to be markedly increased in this brain area in CK-p25,CR animals compared to CK-p25,AL mice (Fig. 4A,B). Concomitant to the increase of SIRT1, we observed a significant reduction in acetylation of H3K56, a target of SIRT1's deacetylase activity (Kong et al., 2011), in CK-p25,CR compared to CK-p25,AL mice (Fig. 4C,D). As SIRT1 levels in CK-p25 animals have been found previously to be elevated over those in control animals, and as this upregulation confers protection against neurotoxicity (Kim et al., 2007), CR is likely to further potentiate the neuroprotective effects of SIRT1. In contrast, the levels of SIRT3, a sirtuin with antioxidant characteristics that is also known to be responsive to CR (Someya et al., 2010), remained unchanged (data not shown).
Figure 4.
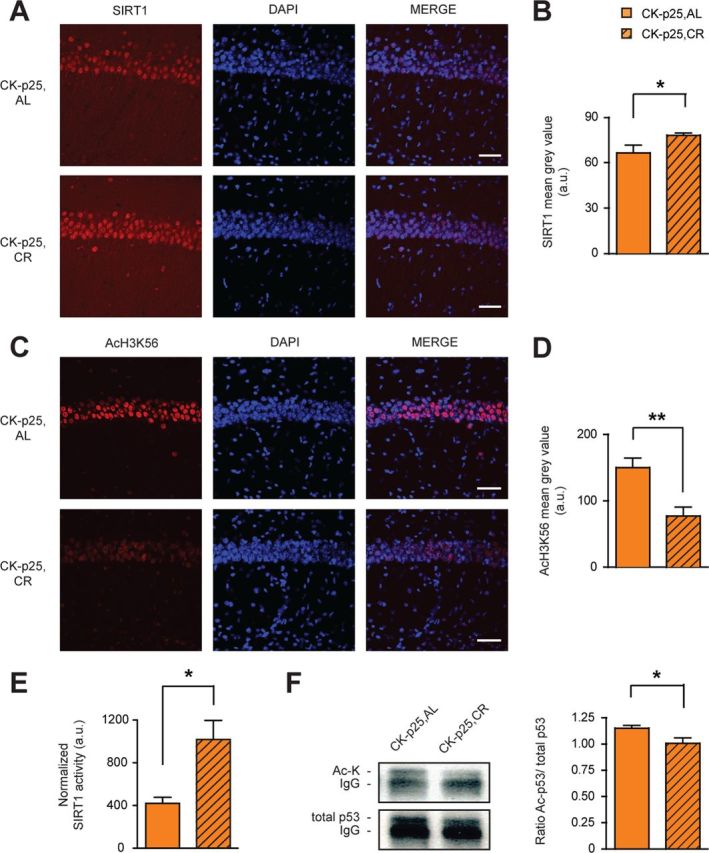
SIRT1 activation by the CR regimen. A, Increased SIRT1 expression following CR in hippocampal area CA1 as evidenced by SIRT1 immunohistochemistry. B, Quantification of A (one-tailed t test, t(4) = 2.68; n = 3 each). C, Decreased AcH3K56 following CR in hippocampal area CA1 as evidenced by AcH3K56 immunohistochemistry. D, Quantification of C (one-tailed t test, t(4) = 3.84; n = 3 each). E, Increased SIRT1 deacetylase activity following CR. SIRT1 deacetylase activity in CK-p25,AL and CK-p25,CR hippocampi. One-tailed t test, t(5) = 2.03; n = 3 for CK-p25,AL; n = 4 for CK-p25,CR. F, Decreased acetylation of p53 following CR. Left, Representative images of Western blot analysis following immunoprecipitation of acetylated (top) and total (bottom) p53 levels in CK-p25,AL and CK-p25,CR hippocampi. Right, Quantification of the ratio of acetylated/total p53 protein levels (one-tailed t test, t(13) = 2.04; n = 6 for CK-p25,AL; n = 9 for CK-p25,CR). *p ≤ 0.05; **p ≤ 0.01 for one-tailed t tests. All values are mean ± SEM. Scale bars: 100 μm.
Next, we measured the activity of SIRT1 in CK-p25,CR and CK-p25,AL animals. We immunoprecipitated SIRT1 out of hippocampal lysates and then fluorometrically assessed its enzymatic activity (see Materials and Methods). To assure that this method does indeed measure the deacetylase activity of SIRT1, we first performed an in vitro test reaction, in which we combined recombinant SIRT1 together with a deacetylase substrate and the known SIRT1-specific inhibitors nicotinamide or suramin sodium. The fluorometric assay reflected a complete in vitro inhibition of recombinant SIRT1 activity [recombinant SIRT1 plus substrate, 42426 fluorescent activity in arbitrary units (f.a.a.u.); recombinant SIRT1 plus substrate plus nicotinamide, 7327 f.a.a.u.; recombinant SIRT1 plus substrate plus suramin sodium, 7547 f.a.a.u.; substrate alone, 4649 f.a.a.u.]. Using this assay in vivo, we observed that CR significantly increased the activity of SIRT1 in CK-p25,CR compared to CK-p25,AL animals (Fig. 4E). Therefore, the CR regimen in CK-p25 mice not only increased SIRT1 protein levels, but also its activity.
One of the downstream mechanisms by which SIRT1 increases cellular lifespan is p53 (Chen and Guarente, 2007), a proapoptotic transcription factor that promotes cellular senescence (Oren, 2003). Upon SIRT1-mediated deacetylation, p53 becomes inactivated (Luo et al., 2001; Vaziri et al., 2001). Based on this knowledge, we measured p53 acetylation in hippocampal extracts of CK-p25 animals following CR or AL treatment by immunoprecipitation of p53 coupled to Western blot analysis. We observed that the acetylation of p53 was significantly reduced following CR treatment (Fig. 4F), suggesting that p53 becomes inactivated following CR. Together, these results indicate that CR upregulates SIRT1 and activates its deacetylase function, which co-occurs with a deacetylation of p53 and likely p53 inactivation. As a reduction of p53 has been shown to be neuroprotective (Kim et al., 2007), this SIRT1-p53 pathway constitutes a potential mechanism by which CR might rescue against neurodegeneration.
SRT3657, a small-molecule activator of SIRT1
Since SIRT1 expression and activity were increased by CR, we reasoned that by using small-molecule STACs we might be able to obtain the same beneficial effects against neurodegeneration as under the CR regimen. To test this, we administered a STAC to CK-p25 mice during the 6 weeks of p25 transgene induction (Fig. 5A). This timeline was chosen to most closely recapitulate the asymptotic phase of CR during p25-transgene induction (Fig. 1B). CK-p25 mice were given a known SIRT1-specific STAC, SRT3657 (CK-p25,SRT3657) (Dai et al., 2010) by oral gavage, or its vehicle alone (CK-p25,VEH) (Fig. 5A). As shown in Figure 5B, SRT3657 treatment significantly upregulated the activity of SIRT1 in hippocampal extracts of CK-p25 animals. Such increased SIRT1 activity was also reflected by decreased H3K56 acetylation (Fig. 5C,D). Thus, SRT3657 treatment effectively activated SIRT1 in vivo.
Figure 5.
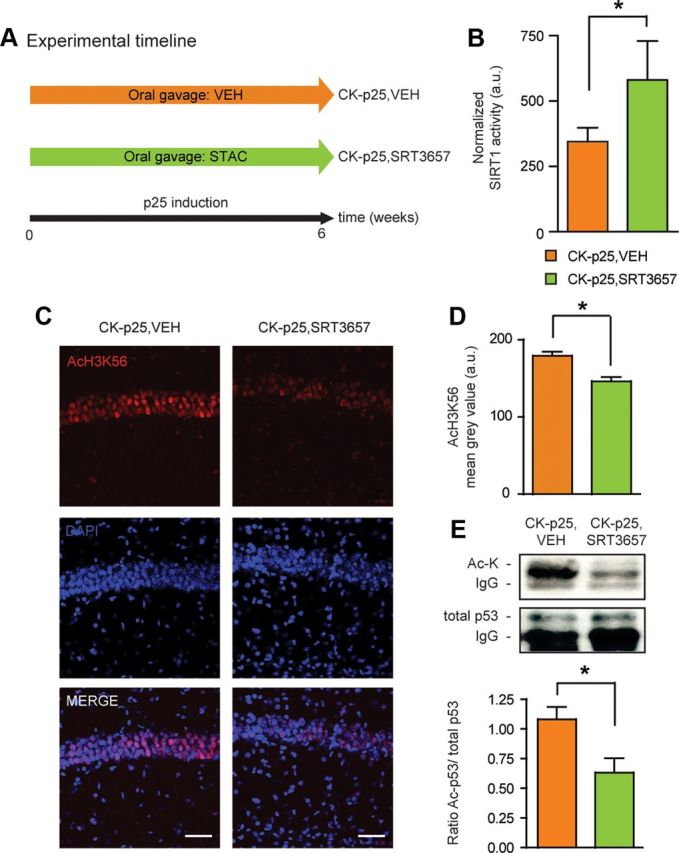
SRT3657 activates SIRT1 in CK-p25 mice. A, Schematic of the experimental timeline. Two groups of mice were used: CK-p25,VEH and CK-p25,SRT3657. B, Increased SIRT1 activity following SRT3657 treatment. SIRT1 deacetylase activity in CK-p25,VEH and CK-p25,SRT3657 hippocampi (one-tailed t test, t(8) = 2.65; n = 4 for CK-p25,VEH; n = 6 for CK-p25,SRT3657). C, Decreased AcH3K56 following SRT3657 treatment in hippocampal area CA1 as evidenced by AcH3K56 immunohistochemistry. Scale bar, 100 μm. D, Quantification of C. One-tailed t test, t(8) = 2.61; n = 4 for CK-p25,VEH; n = 6 for CK-p25,SRT3657. E, Decreased acetylation of p53 following SRT3657 treatment. Top, Representative images of Western blot analysis following immunoprecipitation of acetylated (top) and total (bottom) p53 levels in CK-p25,VEH and CK-p25,SRT3657 hippocampi. Bottom, Quantification of the ratio of acetylated/total p53 protein levels (one-tailed t test, t(8) = 2.61; n = 4 for CK-p25,VEH; n = 6 for CK-p25,SRT3657). *p ≤ 0.05 for one-tailed t tests. All values are mean ± SEM.
If a STAC is to recapitulate CR in vivo, it should activate SIRT1 downstream mechanisms similar to the CR treatment. Therefore, we next tested whether the acetylation of the proapoptotic protein p53 might be altered by SRT3657 treatment. Similar to the CR regimen, we found the acetylation of p53 to be significantly reduced in the hippocampus of CK-p25,SRT3657 mice compared to CK-p25,VEH animals (Fig. 5E). This suggests that similar neuroprotective pathways might be activated by the SRT3657 treatment.
SRT3657 recapitulates the neuroprotective potential of CR
To further assess a potential neuroprotective role of SRT3657, we examined synaptic density by SVP immunohistochemistry. Compared to CK-p25,VEH animals, CK-p25,SRT3657 mice displayed higher synaptic density in the stratum radiatum of the hippocampus at levels indistinguishable from CON, VEH animals (Fig. 6A,B), indicating preserved synaptic integrity. Then, to account for the extent of neurodegeneration, we examined the number of neurons by NeuN-immunohistochemistry in hippocampal area CA1. We found that the number of NeuN-positive cells was significantly increased in CK-p25,SRT3657 animals compared to CK-p25,VEH mice, to a number comparable to CON,VEH animals (Fig. 6C,D), demonstrating that neurodegeneration can be at least partially prevented by the STAC SRT3657. Intriguingly, this was further evidenced by a partial rescue in overall brain loss (Fig. 6E), similar to the effect observed under the CR regimen (Fig. 1F).
Figure 6.
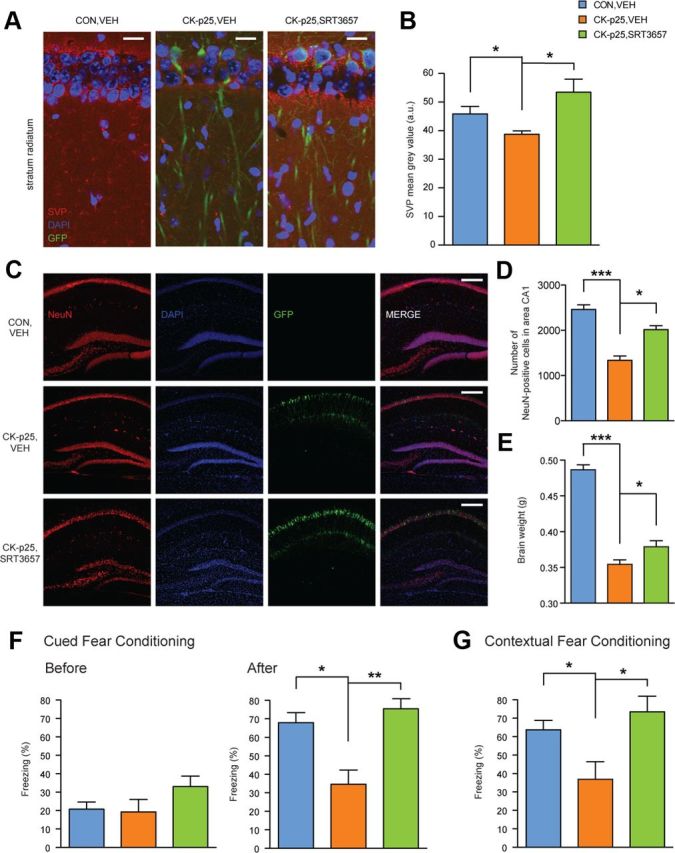
SRT3657 recapitulates the beneficial effects of CR. A, SRT3657-mediated increase in presynaptic terminals in the stratum radiatum of hippocampal area CA1 in CK-p25,SRT3657 animals compared to CK-p25,VEH mice as evidenced by SVP immunohistochemistry. B, Quantification of A [one-way ANOVA (F(2,11) = 5.77, p ≤ 0.05) followed by Tukey's post hoc tests; n = 4–5 mice each]. C, SRT3657-mediated increase in the number of neurons in hippocampal area CA1 in CK-p25,SRT3657 animals compared to CK-p25,VEH mice as revealed by NeuN-immunohistochemistry. D, Quantification of C [one-way ANOVA (F(2,12) = 29.99, p ≤ 0.0001) followed by Tukey's post hoc tests; n = 3–6 mice each]. E, SRT3657-mediated preservation of overall brain mass. One-way ANOVA (F(2,11) = 19.68, p ≤ 0.001) followed by Tukey's post hoc tests; n = 4–7 mice each. F, Cued fear conditioning. Left, Freezing responses before the onset of the tone were comparable between CK-p25,VEH and CK-p25,SRT3657 animals 24 h after training. Right, Freezing responses after the onset of the tone show improved memory retention in CK-p25,SRT3657 mice [one-way ANOVA (F(2,23) = 7.47, p ≤ 0.01) followed by Tukey's post hoc tests; n = 8–10 mice each]. G, Contextual fear conditioning. Freezing responses 24 h after training on a contextual fear conditioning task show improved memory retention in CK-p25,SRT3657 mice [one-way ANOVA (F(2,22) = 5.28, p ≤ 0.05) followed by Tukey's post hoc tests; n = 8–10 mice each]. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001 for Tukey's post hoc comparisons. All values are mean ± SEM. Scale bars: A, 50 μm; C, 20 μm.
Last, to evaluate whether the so-maintained neuronal and synaptic integrity might translate into behavioral improvements, we tested CK-p25,SRT3657 and control littermates for fear memory. Twenty-four hours after training, CK-p25,SRT3657 mice froze significantly more than CK-p25,VEH mice on the cued fear conditioning task after the onset of the tone, but not before (Fig. 6F), indicating enhanced memory retention. Likewise, CK-p25,SRT3657 mice outperformed their VEH-treated counterparts on the contextual fear memory test (Fig. 6G). Importantly, both cued and contextual fear memory retention were comparable between CK-p25,SRT3657 and CON,VEH animals. Furthermore, SRT3657 treatment per se did not affect overall locomotor and exploratory activity, nor did it alter the animals' anxiety levels (data not shown). Together, these data indicate that SRT3657 treatment prevented synaptic and neuronal loss and effectively rescued against neurodegeneration-driven memory impairments. For that reason, SRT3657 treatment might constitute a pharmacological alternative to CR.
Discussion
In the current study, we have shown that a regimen of CR effectively delays the onset of neurodegeneration, preserves structural and functional synaptic plasticity as well as memory capacities, and activates the expression and activity of SIRT1, a known promoter of neuronal lifespan (Tang, 2009; Zhang et al., 2011). Importantly, mimicking the dietary regimen by oral administration of the STAC SRT3657 recapitulated the beneficial effects of CR against neurodegeneration-associated pathologies and thus might constitute a pharmacological alternative to CR against neurodegeneration.
Expanding the favorable effects of CR on memory performance (Halagappa et al., 2007; Fontán-Lozano et al., 2008; Wu et al., 2008a; Witte et al., 2009) and against AD-related pathologies such as amyloid-β accumulation (Patel et al., 2005; Wang et al., 2005; Qin et al., 2006a,b; Halagappa et al., 2007; Wu et al., 2008b; Mouton et al., 2009) and tau hyperphosphorylation (Halagappa et al., 2007), this study newly shows that a regimen of CR can also delay the onset of neurodegeneration per se. It thereby complements results in rhesus monkeys, where CR was shown to preserve gray matter volume (Colman et al., 2009), and in Caenorhabditis elegans, where it was found to prevent the destruction of dopaminergic neurons in a Parkinson's disease model (Jadiya et al., 2011). One question that remains to be answered is whether the regimen of CR would also prove effective against neurodegeneration when started later, i.e., after the onset of neurodegeneration. Likewise, future studies will need to assess whether the beneficial effects of CR are permanent or whether neurodegeneration regains the upper hand once the regimen of CR is stopped.
Mechanistically, the CR regimen used here was found to activate SIRT1. This is in line with previous reports showing that CR increases hippocampal SIRT1 expression (Qin et al., 2006b; Chen et al., 2008) and that, conversely, a fat-rich diet reduces hippocampal SIRT1 expression (Heyward et al., 2012). Although we cannot determine that the beneficial effects of CR are solely mediated by SIRT1, nor that SIRT1 acts in parallel to CR, it is nevertheless likely that such activation of SIRT1 does contribute to the positive effects of CR: overexpression of SIRT1 has been shown previously to not only improve memory performance (Gao et al., 2010; Michán et al., 2010), but also to protect against neurodegeneration (Araki et al., 2004; Kim et al., 2007), whereas RNAi-mediated knockdown of SIRT1 levels precluded the neuroprotective effects of CR in C. elegans (Jadiya et al., 2011).
This possibility is further supported by our findings that, using the STAC SRT3657, the beneficial effects of CR could be essentially recapitulated. Such treatment represents the first direct evidence of a STAC as a pharmacological alternative to a dietary regimen against neurodegeneration. Since dietary changes do not always produce the anticipated beneficial outcomes (Mattison et al., 2012), STAC treatments might be preferable over CR. While we did not observe any adverse effects following SRT3657 treatment, more refined studies are still needed to determine the overall safety and tolerability of this STAC. Moreover, similar questions as raised by the results obtained with the CR treatment concerning the timing of administration as well as the persistence of the beneficial effects are also applicable here. In addition, it would be interesting to assess the combined effects of pharmacological SIRT1 activation and CR on the progression of neurodegeneration. Such experiments would also help to disentangle the question as to whether SIRT1 activation acts via the same pathway as CR.
Last, although we identified p53 deactivation as a potential target of the SRT3657 and CR regimen, other neuroprotective pathways such as the Forkhead box protein O transcription factors, insulin/IGF-signaling, PGC1α, or NfκB might also be activated following both STAC and CR treatments (Gan and Mucke, 2008; Lavu et al., 2008). These limitations notwithstanding, the present study proposes the STAC SRT3657 as an attractive alternative route to convey the beneficial effects of CR against neurodegeneration, without the need for CR itself.
Footnotes
This work was supported by a Swiss National Science Foundation Grant for Prospective Researchers (J. Gräff), the Theodor and Ida Herzog-Egli Foundation (M.K.), and the Glenn Foundation for Medical Research and NIH Grant PO1 AG027916 (L.-H.T.). L.-H.T. is an HHMI investigator. We thank Vipin X. Suri and Jim Ellis from Sirtris-GSK for the SRT3657 compound, Maria Ericsson for the electron microscopy experiments, Katie Schlieper and Mali Taylor for mouse colony maintenance and the oral gavage, Alison Mungenast for text editing, and Matthew M. Dobbin for fruitful scientific discussions. Sirtris Pharmaceuticals contributed to this study in providing and characterizing SRT3657 under a material transfer agreement.
L.-H.T. is a scientific advisor of Sirtris-GSK.
References
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Bishop NA, Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007;8:835–844. doi: 10.1038/nrg2188. [DOI] [PubMed] [Google Scholar]
- Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- Chen D, Guarente L. SIR2: a potential target for calorie restriction mimetics. Trends Mol Med. 2007;13:64–71. doi: 10.1016/j.molmed.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Chen D, Steele AD, Hutter G, Bruno J, Govindarajan A, Easlon E, Lin SJ, Aguzzi A, Lindquist S, Guarente L. The role of calorie restriction and SIRT1 in prion-mediated neurodegeneration. Exp Gerontol. 2008;43:1086–1093. doi: 10.1016/j.exger.2008.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280:40364–40374. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/S0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci. 2006;26:10536–10541. doi: 10.1523/JNEUROSCI.3133-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Kustigian L, Carney D, Case A, Considine T, Hubbard BP, Perni RB, Riera TV, Szczepankiewicz B, Vlasuk GP, Stein RL. SIRT1 activation by small molecules: kinetic and biophysical evidence for direct interaction of enzyme and activator. J Biol Chem. 2010;285:32695–32703. doi: 10.1074/jbc.M110.133892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142:320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/S0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron. 2005;48:825–838. doi: 10.1016/j.neuron.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Fontana L, Partridge L, Longo VD. Extending healthy life span–from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontán-Lozano A, Sáez-Cassanelli JL, Inda MC, de los Santos-Arteaga M, Sierra-Domínguez SA, López-Lluch G, Delgado-García JM, Carrión AM. Caloric restriction increases learning consolidation and facilitates synaptic plasticity through mechanisms dependent on NR2B subunits of the NMDA receptor. J Neurosci. 2007;27:10185–10195. doi: 10.1523/JNEUROSCI.2757-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontán-Lozano A, Romero-Granados R, Troncoso J, Múnera A, Delgado-García JM, Carrión AM. Histone deacetylase inhibitors improve learning consolidation in young and in KA-induced-neurodegeneration and SAMP-8-mutant mice. Mol Cell Neurosci. 2008;39:193–201. doi: 10.1016/j.mcn.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Fusco S, Ripoli C, Podda MV, Ranieri SC, Leone L, Toietta G, McBurney MW, Schutz G, Riccio A, Grassi C, Galeotti T, Pani G. A role for neuronal cAMP responsive-element binding (CREB)-1 in brain responses to calorie restriction. Proc Natl Acad Sci U S A. 2012;109:621–626. doi: 10.1073/pnas.1109237109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Mucke L. Paths of convergence: sirtuins in aging and neurodegeneration. Neuron. 2008;58:10–14. doi: 10.1016/j.neuron.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wang WY, Mao YW, Gräff J, Guan JS, Pan L, Mak G, Kim D, Su SC, Tsai LH. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466:1105–1109. doi: 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L, Picard F. Calorie restriction—the SIR2 connection. Cell. 2005;120:473–482. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, Mattson MP. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Heyward FD, Walton RG, Carle MS, Coleman MA, Garvey WT, Sweatt JD. Adult mice maintained on a high-fat diet exhibit object location memory deficits and reduced hippocampal SIRT1 gene expression. Neurobiol Learn Mem. 2012;98:25–32. doi: 10.1016/j.nlm.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadiya P, Chatterjee M, Sammi SR, Kaur S, Palit G, Nazir A. Sir-2.1 modulates ‘calorie-restriction-mediated’ prevention of neurodegeneration in Caenorhabditis elegans: implications for Parkinson's disease. Biochem Biophys Res Commun. 2011;413:306–310. doi: 10.1016/j.bbrc.2011.08.092. [DOI] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong S, Kim SJ, Sandal B, Lee SM, Gao B, Zhang DD, Fang D. The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. J Biol Chem. 2011;286:16967–16975. doi: 10.1074/jbc.M111.218206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins—novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841–853. doi: 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/S0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, Barnard D, Ward WF, Qi W, Ingram DK, de Cabo R. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–321. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michán S, Li Y, Chou MM, Parrella E, Ge H, Long JM, Allard JS, Lewis K, Miller M, Xu W, Mervis RF, Chen J, Guerin KI, Smith LE, McBurney MW, Sinclair DA, Baudry M, de Cabo R, Longo VD. SIRT1 is essential for normal cognitive function and synaptic plasticity. J Neurosci. 2010;30:9695–9707. doi: 10.1523/JNEUROSCI.0027-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton PR, Chachich ME, Quigley C, Spangler E, Ingram DK. Caloric restriction attenuates amyloid deposition in middle-aged dtg APP/PS1 mice. Neurosci Lett. 2009;464:184–187. doi: 10.1016/j.neulet.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- Patel NV, Gordon MN, Connor KE, Good RA, Engelman RW, Mason J, Morgan DG, Morgan TE, Finch CE. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol Aging. 2005;26:995–1000. doi: 10.1016/j.neurobiolaging.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Qin W, Chachich M, Lane M, Roth G, Bryant M, de Cabo R, Ottinger MA, Mattison J, Ingram D, Gandy S, Pasinetti GM. Calorie restriction attenuates Alzheimer's disease type brain amyloidosis in Squirrel monkeys (Saimiri sciureus) J Alzheimer's Dis. 2006a;10:417–422. doi: 10.3233/jad-2006-10411. [DOI] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006b;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang BL. Sirt1's complex roles in neuroprotection. Cell Mol Neurobiol. 2009;29:1093–1103. doi: 10.1007/s10571-009-9414-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/S0092-8674(01)00527-X. [DOI] [PubMed] [Google Scholar]
- Wang J, Ho L, Qin W, Rocher AB, Seror I, Humala N, Maniar K, Dolios G, Wang R, Hof PR, Pasinetti GM. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer's disease. Faseb J. 2005;19:659–661. doi: 10.1096/fj.04-3182fje. [DOI] [PubMed] [Google Scholar]
- Witte AV, Fobker M, Gellner R, Knecht S, Floel A. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106:1255–1260. doi: 10.1073/pnas.0808587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, Lahiri DK, Zawia NH. Alzheimer's disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008a;28:3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Shen Q, Dong S, Xu Z, Tsien JZ, Hu Y. Calorie restriction ameliorates neurodegenerative phenotypes in forebrain-specific presenilin-1 and presenilin-2 double knockout mice. Neurobiol Aging. 2008b;29:1502–1511. doi: 10.1016/j.neurobiolaging.2007.03.028. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang S, Gan L, Vosler PS, Gao Y, Zigmond MJ, Chen J. Protective effects and mechanisms of sirtuins in the nervous system. Prog Neurobiol. 2011;95:373–395. doi: 10.1016/j.pneurobio.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]