

Mitogen-activated protein kinases (MAPKs) are important components of signaling cascades that regulate many normal cellular events, such as embryogenesis, cell differentiation, cell proliferation and cell death. MAPK pathways can also play an important role in cancer cell survival.[1–3] The vast majority of MAPK inhibitors reported to date have targeted the highly conserved ATP binding sites. A few inhibitors have been found to bind to the “DFG-out” site.[4,5] Although these compounds show good inhibition of their target kinases, many also inhibit other protein kinases with similar or greater potency.[6]
Recruitment sites on MAPKs recognize their protein substrates’ and regulators’ docking domains. These docking interactions may account for the pathway specificity among MAPK modules, and recruitment-site interactions can also induce conformational changes leading to allosteric effects.[7–9] There is a growing interest in developing small molecule MAPK inhibitors that target these recruitment sites. Such inhibitors could overcome limitations to selective and durable inhibition by ATP-competitive inhibitors.[10] Recently, inhibitors targeting the D-recruitment site (DRS) of JNK and ERK have been reported.[11–15] Inhibitors that bind to an allosteric site in the MAPK insert region of p38α, [16,17] and a substrate-selective inhibitor that binds in regions adjacent to the ATP-binding site[18,19] have been described, but no DRS-targeting p38α inhibitors have been reported. ATP-competitive and “DFG-out” type p38α inhibitors have been evaluated in the clinic for treatment of inflammatory diseases[20–23] and p38α may also be a target for cancer treatment.[24–27] The discovery of p38α recruitment site inhibitors could provide additional selectivity for these and other therapeutic applications.[28] Here we report a new class of small molecules that can serve as probes of the DRS of p38α. One such probe has been used to identify the natural product rooperol as an inhibitor that targets the DRS of p38α.
In our prior work, we showed that the dialkynylimidazole (DAIm) 1, designed to undergo aza-Bergman rearrangement in the ATP binding site (Scheme 1), forms a covalent adduct with p38α at 37 °C (25 % adduct after 12 h).[29] In an effort to study the site and mechanism of this covalent modification, we synthesized the N-alkynylimidazole 2 (Figure 1C). Interestingly, 2 is even more efficient that 1 in adducting p38α. When 2 (100 μM) was incubated with non-phosphorylated p38α (5 μM) under the same conditions (12 h, room temperature, 50 mM HEPES, 10 mM MgCl2, 2 mM DTT, 1 mM EGTA, pH 7.5) followed by extensive dialysis, ESI-MS analysis revealed a single peak in the mass spectrum at m/z = 41824, which corresponds to addition of a one molecule of 2 (MW = 263 Da) to p38α (Figure 1B).
Scheme 1.
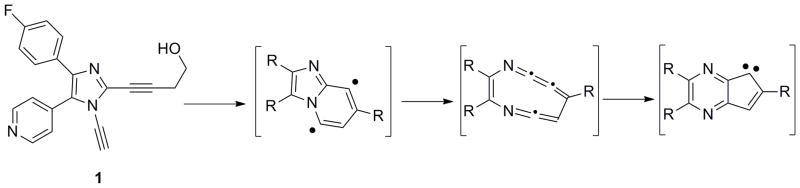
Structure of the p38α inhibiting alkynylimidazole 1, which forms covalently adduct p38α, and the proposed aza-Bergman rearrangement of 1 to afford diradical and carbene intermediates
Figure 1.

(A) ESI-MS spectrum of unphosphorylated p38α. (B) ESI-MS spectrum of p38α incubated in the presence of 2 for 12 h at 37 °C. (C) structure of compound 2.
In order to identify the site of adduction, the sample was digested with chymotrypsin and further analyzed by MALDI-MS/MS. De novo sequencing analysis indicated 2 selectively modifies Cys-119, with a mass addition of 263 Da (see Supporting Information). Although we cannot exclude the possibility that the covalent adduction of p38α by 1 involves binding-induced cyclization, we were intrigued by the result that the N-alkynylimidazole 2 selectively modifies Cys-119, which is directly adjacent to the (DRS) of this kinase (Figure 2), despite the presence of three other Cys residues in p38α.
Figure 2.

Structure of p38α (from PDB 1DI9) illustrating the ATP binding pocket (orange), DFG loop (purple), MAPK insert (green), and the D-site recognition docking site (red surface) which includes Cys119 (sulfur atom in yellow). The location of the other Cys residues are shown in yellow.
While 2 forms an adduct with p38α at the recruitment site, it also possesses the 5-pyridylmidazole scaffold common to many ATP-competitive p38α inhibitors.[30] In order to avoid complications from non-covalent interactions at the ATP-binding site, we prepared the DAIm 3[31] which lacks the 5-pyridyl imidazole substituent. The second terminal alkyne at the 2-position of the imidazole 3 can serve as a handle for further functionalization such as azide-alkyne Huisgen cycloaddition “click” chemistry (Figure 3). After incubation of 3 with p38α at 25 °C for 18 h, we carried out a Cu-catalyzed cycloaddition with the azide labeled fluorescent dye Alexa594-azide. Then the reaction mixture was subjected to denaturing gel electrophoresis and analyzed by both in-gel fluorescence scanning and Coomassie blue staining (Figure 4). In-gel fluorescence analysis of samples of p38α treated with 3 followed by “click” reaction showed a strong fluorescent band with a loading of 0.4 μg of the protein (Figure 4A, lane 3). No fluorescence signal was observed for p38α alone or p38α treated directly with the “click” reaction reagents (Figure 4A, lane 1,2). The approximate detection limit for the adducted p38α after click reaction was determined to be 1 ng (Supporting Information)
Figure 3. Design of DAim.

3 as an In vitro and in-cell recruitment site probe for p38α
Figure 4.

SDS-PAGE analysis of p38α treated with 3 followed by “click” addition of Alexa594-azide: lane 1, p38α (2 μg) alone; lane 2. p38α (2 μg) subjected to “click” reagents; lane 3. p38α (0.4 μg) incubated with 3 followed by “click” reaction. (A): gel visualized by fluorescence. (B): Coomassie blue staining of the same gel.
To determine the rate of this covalent adduction, 3 (25 μM) was incubated with p38α (5 μM) and aliquots were removed at various time points followed by “click” chemistry and in-gel fluorescence SDS-PAGE analysis. Quantification of the fluorescent bands’ intensity as a function of time was fit to a first-order rate equation [P = Pmax(1-e−k’t)] to give a pseudo-first order rate for covalent adduction k′ = 0.11 h−1 (Figure 5). At higher concentrations of 3 (up to 200 μM), no rate saturation was observed (see Supporting Information). Thus, the non-covalent association of 3 with p38α is rather weak. Despite this, 3 is selective for adduction of Cys119, as demonstrated by competition experiments with 2 (see Supporting Information). A combination of weak binding at the docking site and kinetic preference for Cys119 adduction by the N-alkynylimidazole moiety may explain the selectivity of 3.
Figure 5.

Determination of the pseudo-first-order rate of the covalent adduction of p38α by 3.
We next investigated the ability of this probe to detect p38α in cells. By transient transfection, p38α bearing an N-terminal Flag-tag was expressed in HEK 293T cells. The cells were treated with 3 (1 μM, 5 μM, 50 μM) for 4 hours at 37 °C. Subsequently, the cells were pooled, washed, and harvested. After lysis, the cell lysate was immunoprecipitated with anti-FLAG antibody. The resulting supernatant was incubated with Alexa594-azide under click reaction conditions followed by SDS-PAGE and in-gel fluorescence analysis (Figure 6). There was no variation in the total amount of p38α in each lane as determined by anti-Flag Western blot analysis (Supporting Information). In contrast, the observation of a dose-dependent fluorescent band corresponding to the labelled p38α•3 adduct (Figure 6, lanes 2,3,4) demonstrates that 3 is both cell-permeable and able to form adducts with p38α within a cellular context. However, 3 is only modestly selective for p38α. When 3 was incubated with the lysate of the p38α-expressing HEK293 cells followed by click reaction and in-gel fluorescence SDS-PAGE analysis, a number of bands were observed in addition to the band corresponding to p38α (Supporting Information).
Figure 6.

In-gel fluorescence SDS-Page analysis of HEK 293T cells over-expressing Flag-tagged p38α treated with 3 and then subjected to lysis, immunoprecipitation, and click reaction with Alexa594-azide. Lane 1. Cells treated with vehicle (0.05 % DMSO); 2. Cells treated with 1 μM 3. Lane 3. Cells treated with 5 μM 3. Lane 4. Cells treated with 50 μM 3. Lane 5. Isolated flag-tagged p38α (~0.6 μg) treated with 50 μM 3 in vitro followed by click reaction.
The adduction of p38α by 3 is selectively blocked by ligands that bind to the recruitment site of the kinase. p38α was incubated in the presence of 3 (100 μM) and an equimolar concentration of a peptide derived from the D-site of MKK3, which is a remarkably potent inhibitor of p38α (IC50 <10 nM).[32] After click reaction, in-gel fluorescence SDS-PAGE analysis demonstrates near complete inhibition of adduction by the MKK3 peptide (Figure 7A, lane MKK3). In contrast, incubation of p38α in the presence of 3 and a Ste7 D-site peptide, [33] which is not a p38α cognate D-site peptide, results in minimal inhibition of adduct formation (Figure 7A, lane Ste7). Interestingly, when these competition experiments are carried out in the presence of ATP-binding site inhibitors SB 203580 and BIRB-796, a slight increase in the adduct formation by 3 is observed (Figure 7B). Presumably, there is some cross-talk between the ATP binding site and the recruitment site such that occupancy of the former alters the conformation of the recruitment site to facilitate adduct formation.
Figure 7.

Adduction of p38α by 3 is selectively blocked by recruitment site ligands. p38α was incubated in the presence of 3 (100 μM) alone (Control) or (A): with an equimolar concentration of the MKK3 D-site peptide (MKK3) or the Ste7 D-site peptide (Ste7) or (B) with an equimolar concentration of the ATP-binding site inhibitors SB 203580 or BIRB-796 for 16 h followed by click reaction and by in-gel fluorescence SDS-PAGE analysis.
Polyphenolic natural products have emerged as a promising class of inhibitors of kinase pathways including the MAPK pathway.[34–36] Although the mode of kinase inhibition by these natural products is often not known, recently the lignan (−)-zuonin has been found to inhibit JNK by targeting its DRS.[13,14] We employed a fluorescent-based assay using probe 3 to screen for natural products that similarly recognize the DRS of p38α. Incubation of p38α (5μM) with 3 (100 μM) was carried out in the presence of individual members of a small library of plant polyphenol natural products and synthetic analogs, each at 200 μM, for 16 h. The reaction mixtures were subjected to click reaction conditions and in-gel fluorescence analysis, and the inhibition of adduct formation by 3 was determined by quantification of the fluorescence bands (Figure 8). Within this limited number of compounds, the African potato (Hypoxis rooperi)-derived rooperol[37] shows the strongest inhibition, followed closely by the honey bee propolis-derived caffeic acid phenethyl ester (CAPE[38]) and its double-bond reduced analog DHC. Other synthetic analogs of CAPE (e.g., the corresponding amide, CAPA) and other H. rooperi-derived natural products (e.g, nyasol) are much less effective inhibitors (Figure 8).
Figure 8.

Inhibition of p38α adduct formation with 3 by a collection of natural products and analogs. Inhibitors were assayed at 200 μM concentration (54 μg/ml in the case of African potato extract (APE)).
To confirm the ability of rooperol to inhibit p38α through its DRS interaction, its effect on the rate of p38α phosphorylation of ATF2(1–115) was determined. Rooperol inhibits p38α in a dose-dependent fashion, with maximal inhibition of 60% and an IC50 of 18.6 μM (Figure 9). Similar rate profiles have been obtained for small molecule DRS inhibitors of JNK.[14] African potato is used in traditional medicine to treat wide a variety of aliments, and pharmacological investigations have demonstrated antinociceptive and anti-inflammatory effects in rodents.[37] Rooperol, the aglycone of the major phenolic constituent of H. rooperi corms, has been shown to inhibit cytokine mRNA levels in LPS-stimulated U937 cells.[38] DRS binding and inhibition of p38α by rooperol demonstrated here may play a role in the anti-inflammatory effects of this natural product.
Figure 9.

Inhibition of p38α phosphorylation of ATF2 by rooperol. The rate of phosphorylation of ATF2 (12.5 μM) by active p38α (10 nM) in the presence of 500 μM ATP was determined in the presence of 0 to 432 μM rooperol.
In summary, a novel class of N-alkynylimidazole small molecules have been identified as DRS probes for p38α through covalent interaction with Cys-119. Using alkyne-azide “click” chemistry, one such probe can be used to fluorescently label p38α in vitro and in cells. The adduction of p38α by this probe is specifically blocked by the p38α-cognate MKK3 D site peptide. Using this probe to identify small molecules that recognize the p38α DRS, the anti-inflammatory natural product rooperol was identified as a novel p38α inhibitor. This work demonstrates the utility of targeting specific cysteine residues associated with kinase recruitment sites for the discovery of novel inhibitors. The identification of natural products that bind to the p38α recruitment site not only provides scaffolds for the design of novel inhibitors, but also indicates that interaction with these kinase recruitment sites may be one means by which these pharmacologically active agents exert their effects.
Experimental Section
Adduct formation with DAIm 3, click reaction, and in-gel fluorescence PAGE analysis
Reaction mixtures (100 μL) containing 5 μM p38α, 100 μM 3 in 50 mM HEPES, pH 7.5, 1 mM EGTA, 2 mM DTT, 10 mM MgCl2 were incubated at 25 °C for 16 h. Without further purification, aliquots of this reaction mixture were subjected to click reaction. To reaction mixtures containing 25 ng of p38α in 50 mM potassium phosphate buffer were added stock solutions of CuSO4 (0.5 μL, 50 mM), tris(2-carboxyethyl)phosphine (0.5 μL, 50 mM), Alexa-594 azide (0.5 μL, 2.5 mM), and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (1.65 μL, 1.5 mM). The click reaction was allowed to proceed at 25 °C for 16 h, quenched by the addition of 2X SDS loading buffer and heat inactivation at 95 °C for 10 min. The samples were analyzed by 10 % SDS PAGE. The gel was scanned by Typhoon Trio from GE healthcare and the data were analyzed by Image J software.
In cell labeling of p38
HEK 293T cells (5 × 105 cells) were seeded on a 6 well polystyrene plate in DMEM supplemented with 10 % FBS (Invitrogen) and 1 % L-glutamine. Cells were grown to 90–95 % confluency in an atmosphere of 5 % CO2. pCDNA3 Flag p38α (Addgene) was transfected into HEK 293T cells using polyethyleneimine (PEI). After 48 h incubation, old medium was removed. Compound 3 was added in new growth medium. The cells were incubated at 37 °C for 4 h. After the treatment, cells were pooled, spinned down at 1200 rpm and washed twice with cold PBS pH 7.4. Cell pellets were lysed in lysis buffer containing protease inhibitors (Thermo Scientific). The resulting solution was incubated at 4 °C for 30 min. The cell lysates were centrifuged at 14,000 rpm at 4 °C for 10 min. Supernatant was collected and incubated with ANTI-FLAG M2 affinity gel overnight at 4 °C. The resin was centrifuged and washed with TBS three times. Flag-p38α was eluted by with FLAG peptide. The click reaction and in-gel fluorescence PAGE analysis was carried out following the procedures described above.
Competition Assay
Reaction mixtures (100 μL) containing 5 μM p38α, 100 μM 3, and 100 μM test compound in 50 mM HEPES, pH 7.5, 1 mM EGTA, 2 mM DTT, 10 mM MgCl2 were incubated at 25 °C for 16 h. Without further purification, aliquots of this reaction mixture were subjected to click reaction and the samples were analyzed as shown above. The inhibition of adduct formation by 3 was determined by quantification of the fluorescence bands
Inhibition Assay
Full length p38MAPKα (GenBank accession number NM_011951) and GST-ATF2 (1–115) were expressed, and purified and p38MAPKα was activated with MKK6 as described previously..[6] Briefly, non-activated p38α protein (0.5 μg) in 20 mM Tris-HCl buffer, pH 7.5 containing 10 %v/v glycerol, 10 mM MgCl2, and 1 mM DTT was incubated with 0.05 μg MKK6 and 10 μM ATP for 30 min and the reaction was stopped by addition of 2 μL of 0.5 M EDTA. Phosphorylation of p38α was confirmed by ESI-MS. Kinase inhibition assays were conducted at 30 °C in assay buffer (25 mM HEPES buffer-pH 7.5, 50 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 2 mM DTT and 10 μg mL-1 BSA), containing 500 μM [-32P] ATP (100–1000 c.p.m. pmol−1), 11 mM MgCl2 and different concentrations of rooperol in a final total volume of 70 μL, containing 5 % DMSO. 10 nM active full length p38MAPKα was assayed with 12.5 μM GST-ATF2 (1–115) protein substrate. Activity was assessed at different compound concentrations by the measurement of initial rates, where the total product formation represented less than 10% of the initial substrate concentrations. Different concentrations of rooperol were incubated with the enzyme for 90 minutes before initiation of the reaction by the addition of ATF2 and ATP. At set time points (0.5, 1, 1.5, 2, 4 min), 10 μL aliquots were taken from every reaction and spotted onto 2×2 cm2 squares of P81 cellulose paper; the papers were washed 3 times for 15 minutes each in 50 mM phosphoric acid (H3PO4), followed by a pure acetone wash, then dried. The amount of labeled protein was determined by counting the associated c.p.m. on a Packard 1500 scintillation counter at a sigma value of 2.
Supplementary Material
Acknowledgments
This research was supported by grants from the Robert Welch Foundation (F-1298 to SMK, F-1390 to KND), the Cancer Prevention Institute of Texas (to JL), the Texas Istitute for Drug and Diagnostic Development (to SMK), and the NIH (GM59802 to KND).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 2.Johnson GL, Lapadat R. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 3.Dhillon AS, Hagan S, Rath O, Kolch W. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 4.Regan J, Breitfelder S, Cirillo P, Gilmore T, Graham AG, Hickey E, Klaus B, Madwed J, Moriak M, Moss N, Pargellis C, Pav S, Proto A, Swinamer A, Tong L, Torcellini C. J Med Chem. 2002;45:2994–3008. doi: 10.1021/jm020057r. [DOI] [PubMed] [Google Scholar]
- 5.Wrobleski ST, Doweyko AM. Curr Top Med Chem. 2005;5:1005–1016. doi: 10.2174/1568026054985894. [DOI] [PubMed] [Google Scholar]
- 6.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akella R, Moon TM, Goldsmith EJ. Biochim Biophys Acta. 2008;1784:48–55. doi: 10.1016/j.bbapap.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bardwell AJ, Frankson E, Bardwell L. J Biol Chem. 2009;284:13165–13173. doi: 10.1074/jbc.M900080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharrocks AD, Yang SH, Galanis A. Trends Biochem Sci. 2000;25:448–453. doi: 10.1016/s0968-0004(00)01627-3. [DOI] [PubMed] [Google Scholar]
- 10.Hancock CN, Macias AT, MacKerell AD, Shapiro P. Med Chem. 2006;2:213–222. doi: 10.2174/157340606776056151. [DOI] [PubMed] [Google Scholar]
- 11.Hancock CN, Macias A, Lee EK, Yu SY, MacKerell AD, Shapiro P. J Med Chem. 2005;48:4586–4595. doi: 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- 12.Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen LH, Cellitti JF, Riel-Mehan M, Emdadi A, Solinas G, Karin M, Pellecchia M. Proc Natl Acad Sci U S A. 2008;105:16809–16813. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaoud TS, Yan C, Mitra S, Tseng C-C, Jose J, Taliaferro JM, Tuohetahuntila M, Devkota A, Sammons R, Park J, Park H, Shi Y, Hong J, Ren P, Dalby KN. doi: 10.1021/ml300129b. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaoud TS, Park J, Mitra S, Yan C, Tseng C-C, Shi Y, Jose J, Taliaferro JM, Ren P, Hong J, Dalby KN. submitted. [Google Scholar]
- 15.(a) De SK, Stebbins JL, Chen LH, Riel-Mehan M, Machleidt T, Dahl R, Yuan HB, Embadi A, Barile E, Chen V, Murphy R, Pellecchia M. J Med Chem. 2009;52:1943–1952. doi: 10.1021/jm801503n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) De SK, Chen V, Stebbins JL, Chen LH, Cellitti JF, Machleidt T, Barile E, Riel-Mehan M, Dahl R, Yang L, Emdadi A, Murphy R, Pellecchia M. Bioorg Med Chem. 2010;18:590–596. doi: 10.1016/j.bmc.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) De SK, Barile E, Chen V, Stebbins JL, Cellitti JF, Machleidt T, Carlson CB, Yang L, Dahl R, Pellecchia M. Bioorg Med Chem. 2011;19:2582–2588. doi: 10.1016/j.bmc.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comess KM, Sun CH, Abad-Zapatero C, Goedken ER, Gum RJ, Borhani DW, Argiriadi M, Groebe DR, Jia Y, Clampit JE, Haasch DL, Smith HT, Wang SY, Song DY, Coen ML, Cloutier TE, Tang H, Cheng XH, Quinn C, Liu B, Xin ZL, Liu G, Fry EH, Stoll V, Ng TI, Banach D, Marcotte D, Burns DJ, Calderwood D, Hajduk PJ. ACS Chem Biol. 2011;6:234–244. doi: 10.1021/cb1002619. [DOI] [PubMed] [Google Scholar]
- 17.Pollack SJ, Beyer KS, Lock C, Muller I, Sheppard D, Lipkin M, Hardick D, Blurton P, Leonard P, Hubbard PA, Todd D, Richardson CM, Ahrens T, Baader M, Hafenbradl DO, Hilyard K, Burli RW. J Comp-Aided Mol Des. 2011;25:677–687. doi: 10.1007/s10822-011-9454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davidson W, Frego L, Peet GW, Kroe RR, Labadia ME, Lukas SM, Snow RJ, Jakes S, Grygon CA, Pargellis C, Werneburg BG. Biochemistry. 2004;43:11658–11671. doi: 10.1021/bi0495073. [DOI] [PubMed] [Google Scholar]
- 19.Davis D, Bagley MC, Dix M, Murziani PGS, Rokicki MJ, Woddowson CS, Zayed JM, Bachler MA, Kipling D. Bioorg Med Chem Lett. 2007;17:6832–6835. doi: 10.1016/j.bmcl.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 20.Pettus LH, Wurz RP. Curr Top Med Chem. 2008;8:1452–1467. doi: 10.2174/156802608786264245. [DOI] [PubMed] [Google Scholar]
- 21.Schultz RM. Prog Drug Res. 2003;60:59–92. doi: 10.1007/978-3-0348-8012-1_2. [DOI] [PubMed] [Google Scholar]
- 22.English JM, Cobb MH. Trends Pharmacol Sci. 2002;23:40–45. doi: 10.1016/s0165-6147(00)01865-4. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, Boehm J, Lee JC. Nat Rev Drug Discov. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 24.Wagner EF, Nebreda AR. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 25.Noel JK, Crean S, Claflin JE, Ranganathan G, Linz H, Lahn M. Med Oncol. 2008;25:323–330. doi: 10.1007/s12032-008-9039-1. [DOI] [PubMed] [Google Scholar]
- 26.Mantovani A, Allavena P, Sica A, Balkwill F. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 27.Engelberg D. Sem Cancer Biol. 2004;14:271–282. doi: 10.1016/j.semcancer.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 28.Mayor F, Jr, Jurado-Pueyo M, Campos PM, Murga C. Cell Cycle. 2007;6:528–533. doi: 10.4161/cc.6.5.3920. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Kaoud TS, Laroche C, Dalby KN, Kerwin SM. Bioorg Med Chem Lett. 2009;19:6293–6297. doi: 10.1016/j.bmcl.2009.09.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson KP, McCaffrey PG, Hsiao K, Pazhanisamy S, Galullo V, Bemis GW, Fitzgibbon MJ, Garon PR, Murcko MA, Su MS. Chem Biol. 1997;4:423–431. doi: 10.1016/s1074-5521(97)90194-0. [DOI] [PubMed] [Google Scholar]
- 31.Laroche C, Li J, Kerwin SM. J Med Chem. 2011;54:5059–5069. doi: 10.1021/jm200289j. [DOI] [PubMed] [Google Scholar]
- 32.Bardwell AJ, Frankson E, Bardwell L. J Biol Chem. 2009;284:13165–13173. doi: 10.1074/jbc.M900080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SB, Warthaka M, Yan CL, Kaoud TS, Piserchio A, Ghose R, Ren PY, Dalby KN. Plos One. 2011;6(4) doi: 10.1371/journal.pone.0018594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crozier A, Jaganath IB, Clifford MN. Nat Prod Rep. 2009;26:1001–1043. doi: 10.1039/b802662a. [DOI] [PubMed] [Google Scholar]
- 35.Li W, Mei X, Tu YY. Mini-Rev Med Chem. 2012;12:120–126. doi: 10.2174/138955712798995011. [DOI] [PubMed] [Google Scholar]
- 36.Lamoral-Theys D, Pottier L, Dufrasne F, Neve J, Dubois J, Kornienko A, Kiss R, Ingrassia L. Curr Med Chem. 2010;17:812–825. doi: 10.2174/092986710790712183. [DOI] [PubMed] [Google Scholar]
- 37.Owira PMO, Ojewole JAO. Phytother Res. 2009;23:147–152. doi: 10.1002/ptr.2595. [DOI] [PubMed] [Google Scholar]
- 38.Guzdek A, Rokita H, Cichy J, Allison AC, Koj A. Med Inflamm. 1998;7:13–18. doi: 10.1080/09629359891324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.