

Abstract
The drug efflux pump P-glycoprotein possesses two common and often linked polymorphisms that result in variable drug action. G2677T results in A893S, whereas C3435T is synonymous and has been reported to alter protein folding. We tested the effect of these MDR1 variants on Human Ether-Related A Go-Go (HERG) block by ibutilide in CHO cells 48 h following transient transfection with an IRES-dsRed vector containing MDR1, G2677T MDR1, G2677T/C3435T MDR1 or an empty bicistronic site and an IRES-GFP vector containing HERG (KCNH2). Cotransfection of MDR1 variants had no effect on IKr amplitude at baseline. Cells cotransfected with MDR1-G2677T showed resistance to ibutilide vs HERG alone (IC50: 105.3±1.42 nM vs 27.4±2.5 nM; P<0.0001), consistent with the idea that A893S attenuates IKr block by enhancing drug efflux and thus reducing the drug available to interact with the channel binding site. However, G2677T/C3435T cells showed ibutilide sensitivity similar to cells expressing HERG alone (IC50: 22.2±0.9 nM). Immunostaining showed that the C3435T variant did not traffic to the cell surface. Coculture with fexofenadine(1 μM), an MDR1 substrate known to rescue misfolding in other membrane proteins, restored cell surface expression of MDR1 G2677T/C3435T and restored resistance to block HERG by ibutilide 200 nM (98.5±0.98% vs 42.3±2.2%, P<0.001). The non-synonymous MDR1 variant G2677 T (A893S) confers resistance to ibutilide block of IKr, which is mitigated by the C3435T polymorphism through reduced protein expression, an effect that can be restored by coculture with fexofenadine. These data identify ibutilide as an MDR1 substrate and further support the concept that variable drug transport function can modulate the action of HERG blockers.
Keywords: pharmacology, long QT syndrome, P-glycoprotein, ATP-binding cassette transporters, antiarrhythmia agents
Introduction
Administration of ion channel-blocking antiarrhythmics is associated with considerable and unpredictable variability in clinical response ranging from suppression of arrhythmia to life-threatening proarrhythmia.1–4 Pharmacological antagonism of the Human Ether-Related A Go-Go (HERG, KCNH2) potassium channel, which encodes the pore-forming subunit of the protein complex responsible for carrying the delayed rectifier potassium current, IKr, represents one well-studied mechanism of drug-induced proarrhythmia. The reduction of IKr with drug block can lead to impaired cardiac repolarization, exaggerated QT interval prolongation and the development of torsade de pointes.1,2,5–8 Although numerous risk factors, including gender, hypokalemia, hypomagnesemia, bradycardia and mutations in cardiac ion channels, have been identified, the degree of QT interval prolongation and incidence of torsade de pointes remains unpredictable among patients with the same risk profile and equivalent QT intervals at baseline. 1,3,4,9–13
The site of drug block on the HERG potassium channel complex has been identified at the intracellular face of the channel protein.5,6,14 Increasing evidence from other organ systems, including the blood–brain barrier, placenta, kidney and liver, has shown that access to such intracellular sites of action can be a highly regulated process determined by xenobiotic transporters that control drug uptake and efflux.15–22
Efflux transporters generally move drugs from the intracellular to the extracellular space. They are encoded by genes belonging to the ABC (ATP-binding cassette) superfamily, which includes MDR1 (formally known as ABCB1), encoding P-glycoprotein.23,24 A common nonsynonymous polymorphism in MDR1, G2677T, leading to A893S, has been associated with evidence of an altered response to substrate drugs, including a blunted response to antineoplastic therapy, reduced neurotoxicity following treatment with the immunosuppressant tacrolimus and lower systemic exposure to the antihypertensive calcium channel blocker, amlodipine.25–30 In addition, a second synonymous polymorphism, C3435T, has been associated with altered drug response. The explanation for the latter findings is unclear, although one study reported implicated altered protein folding during translation. Evidence for this included reduced inhibition of P-glycoprotein by cyclosporine and verapamil.31 We present here studies testing the role of wild-type and variant MDR1 on modulation of drug-induced HERG block.
Results
Coexpression of MDR1*7 but not MDR1*1 modulates IKr block by ibutilide
We first examined the impact of coexpressing MDR1 constructs on the characteristics of IKr. Figure 1 and Table 1 show that baseline current–voltage relationships and tail currents were not significantly different between groups.
Figure 1.

MDR1 coexpression does not impact current–voltage relationships. (a) Current–voltage relationship of activating currents. (b) Steady-state current–voltage relationship. (c–f) Representative trace for cells expressing HERG alone (c), MDR1*1 (d), MDR1*3 9 (e) and MDR1*7 (f).
Table 1.
MDR1 coexpression does not affect IKr
| Control | MDR*1 | MDR1*3 | MDR1*7 | |
|---|---|---|---|---|
| V1/2 activation (mV) | 19.8±3.0 | 19.6±0.6 | 20.5±4.8 | 22.6±3.42 |
| Tau activation @40 mV | 135±19.7 | 136±18.2 | 137±8.5 | 133±10.9 |
| Tau deactivation (ms) @40 mV | 591±31 | 597±49 | 576±53 | 569±67 |
| V1/2 deactivation | −0.4±0.2 | −0.4±0.3 | −0.4±0.1 | −0.3±0.6 |
| Peak tail current (pA/pF) | 8.7±0.6 | 8.7±0.8 | 8.7±1.7 | 8.9±1.0 |
n = 7 each; all P values >0.9. Peak tail current: pA/pF at −40 mV after a depolarizing pulse to +60 mV. Time constant of (Tau) activation (ms): monoexponential fit to activating current during a pulse to +40 mV. Time constant of (Tau) deactivation: (ms): monoexponential fit to deactivating current at −40 mV after a depolarizing pulse to +40 mV. V1/2 is defined as the voltage at which maximum current amplitude occurred in mV.
Ibutilide blocked IKr in cells expressing HERG+MDR1*1 to the same extent as cells expressing HERG alone (IC50: 22.5±0.9 vs 27.4±2.5 nM; P>0.05). This nanomolar block indicates that CHO cells take up ibutilide. However, cells expressing MDR1*7 showed a marked resistance to ibutilide (IC50: 105.3±1.42 nM vs 27.4±2.5 nM; P<0.0001). These data (Figures 2 and 3) suggest that the alanine-to-serine change generates a protein with the ability to enhance efflux of ibutilide.
Figure 2.
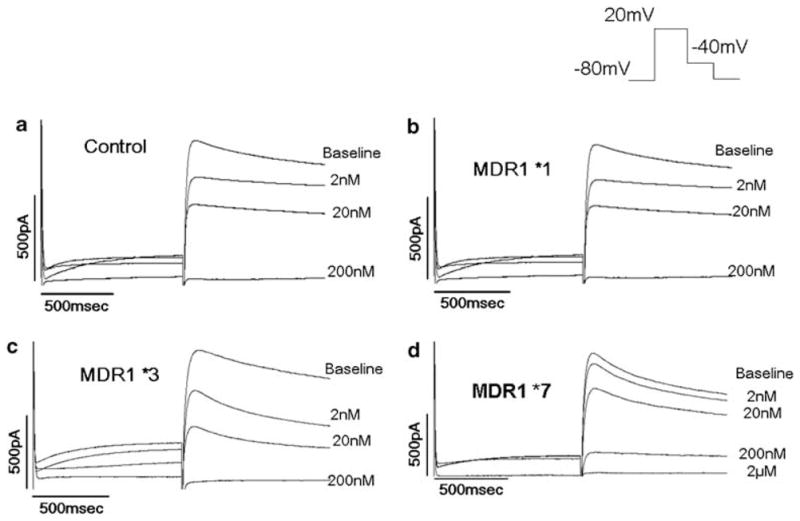
Effect of MDR1 variants of HERG block vs HERG alone. (a) HERG alone, (b) MDR1*1, (c) MDR1*3, (d) MDR1*7.
Figure 3.
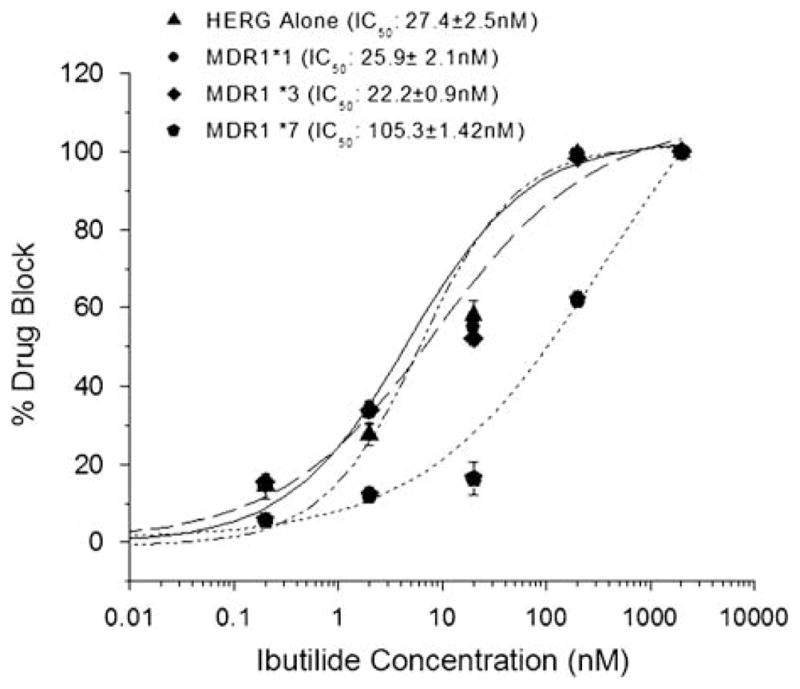
Dose–response relationship for MDR1 variants vs HERG alone. Coexpression of MDR1*7 increased the concentration of ibutilide needed to suppress IKr by 50% fivefold.
Influence of MDR1*3 on IKr block
Drug block in cells expressing the linked haplotype MDR1*3 unexpectedly did not show the same resistance to ibutilide block as the cells expressing MDR1*7, with an IC50 similar to that for MDR1*1 (IC50: 25.9±2.1). As discussed above, one possible explanation for this finding is that the reported altered protein folding and cell surface expression caused by C3435T might influence drug block by ibutilide. We reasoned that rescuing cell surface expression might therefore restore the effect of the A893S change. We therefore examined the effect of fexofenadine on HERG block by ibutilide and surface expression of MDR1*3. We selected fexofenadine because it is an MDR1 substrate shown to rescue misfolded HERG mutants, but does not suppress IKr.32
To test the effect of fexofenadine, we evaluated a high concentration of ibutilide, 200 nM, which suppressed IKr by 98.5±2% in the absence of MDR1. Culture of HERG+MDR1*3 cells with fexofenadine-supplemented media produced a marked resistance to ibutilide-induced IKr suppression relative to cells cultured in the absence of fexofenadine (98.5±0.98% vs 42.3±2.2%, P<0.001). Furthermore, in HERG+ MDR1*3 cells cocultured with fexofenadine, we were unable to completely suppress peak tail current using concentrations up to 2 μM. (Figure 4).
Figure 4.
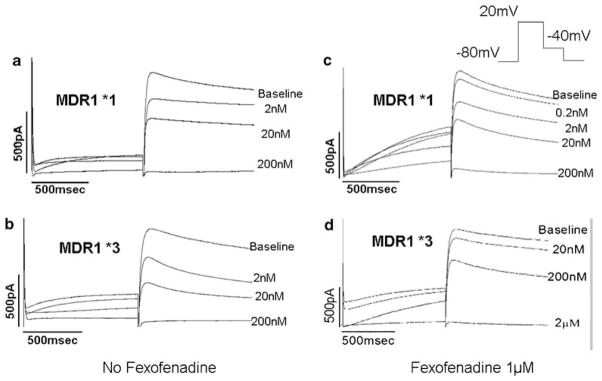
Impact of fexofenadine-supplemented media on ibutilide resistance caused by MDR1*3. (a) Ibutilide-induced IKr block in MDR1*1 cells not exposed to fexofenadine. (b) Ibutilide-induced IKr block in MDR1*3 cells not exposed to fexofenadine. (c) Ibutilide-induced IKr block in MDR1*1 cells exposed to fexofenadine does not alter degree of IKr block relative to untreated cells. (d) Fexofenadine coculture induces marked resistance to IKr block by ibutilide.
Immuofluoresence studies shown (Figure 5a–c) demonstrated that MDR1*3 surface expression was increased by fexofenadine. In the absence of fexofenadine, cells transfected with MDR1*7 showed surface staining, whereas those cells transfected with MDR1*1 or MDR1*3 did not. By contrast, culture of MDR1*3-transfected cells showed a marked increase in cell surface staining following culture with fexofenadine-supplemented media relative to untreated cells (18/20 vs 1/20 cells per field; P<0.05). Notably, exposure to fexofenadine during culture did not alter MDR1*1 cell surface expression (Figure 5a) or block by ibutilide (Figure 4). This indicates that the combined G2677/C3435 haplotype does not traffic to the cell surface in CHO cells and thus does not actively export ibutilide.
Figure 5.

Impact of fexofenadine supplementation on cell-surface expression of MDR1 variants. In each panel, the expression of P-glycoprotein variants cultured in the absence of fexofenadine is shown on the top row, and expression of P-glycoprotein variants cultured in the presence of fexofenadine is shown on the bottom row. In each row, di-8-ANEPPS, anti-MDR1 antibody and a combination overlay are presented from left to right. (a) MDR1*1, (b) MDR1*3 showing increased surface expression in the presence of fexofenadine and (c) MDR1*7.
Discussion
In this study, we showed that ibutilide-induced block of HERG and the ensuing suppression of IKr are blunted by coexpression of the variant, MDR1*7, but not wild type. Interestingly, subsequent immunostaining shows that the MDR1*7 variant was expressed at the cell surface, whereas MDR1*1 was not.
When we evaluated the functional effect of the linked haplotype, G2677T/C3435T (that is, MDR1*3), the IC50 for ibutilide-induced HERG block was similar to control cells expressing HERG alone. This raised the unanticipated possibility that the synonymous C3435T polymorphism contributed to this effect. As the C3435T polymorphism has been reported to alter folding of the MDR1 protein during translation, we supplemented our culture media with fexofenadine. Fexofenadine was selected because of data that the drug, an MDR1 substrate and not a HERG blocker, restored cell surface expression of certain misprocessed HERG mutants.31 Fexofenadine-supplemented media restored MDR1*3-mediated resistance to HERG block to the extent that we were unable to completely suppress IKr at concentrations exceeding 2 μM.
Immuofluoresence studies support this idea, in that protein from the combined G2677T/C3435T haplotype (that is, MDR1*3) was shown at the cell surface only in the presence of fexofenadine coculture. This finding has two important implications. First, our finding lends further support to the notion that this ‘silent’ coding-region polymorphism exerts a functional effect. Second, our data also contribute to the idea that concomitant medications that are thought to be innocuous, such as fexofenadine, may obscure the ability to determine the influence of a particular genotype or haplotype on pharmacokinetic parameters in human association studies.
Influence of the C3435T polymorphism on drug transport is variable
Two association studies report functional differences between MDR1*3 and MDR1*7 and so appear to support our results. In a cohort of patients with schizophrenia treated with olanzapine, a dopamine receptor antagonist, patients with the G2677T/C3435T haplotype (that is, MDR1*3) showed a marked improvement in objective measures of treatment response compared with MDR1*7.33 The influence of the G2677T/C3435T haplotype was also assessed in a cohort of 89 patients receiving the antimalarial agent mefloquine. In this study, presence of G2677T/C3435T resulted a higher incidence of neurological adverse effects relative to those without this haplotype, suggesting that the linked haplotype led to an increased drug concentration in the central nervous system.34 P-glycoprotein is expressed on the vascular endothelium of the blood–brain barrier, so these results are in general agreement with our data that the G2677T/C3435T polymorphism leads to reduced drug efflux.
Several association studies do not support our results. Two cancer studies associated the MDR1*3 haplotype with a higher rate of treatment failure relative to MDR1*1. However, it is not known whether the drugs used (idarubicin/cytaribine or etoposide) can alter MDR1 trafficking or if these compounds are transported by a different MDR1 haplotype.31,35,36 A similar result was observed in a cohort of Japanese patients: MDR1*3 was associated with resistance to antiepileptic therapy with carbamazepine (a sodium channel blocker and inducer of CYP3A4) relative to MDR1*1.37 As CYP3A4 and P-glycoprotein share common elements of transcriptional regulation, it may be possible that carbamazepine induced surface expression of P-glycoprotein (similar to our results with fexofenadine) or that carbamazepine is transported by a different MDR1 haplotype.
Implications for ion channel antagonists
Medically intractable epilepsy is a disorder in which ion channel blocker therapy is used to suppress abnormal ion channel activity. Using a model of medically intractable epilepsy in rodents, Clinckers et al.38 showed that the sodium channel blocker oxcarbazepine produced only an antiepileptic effect in the presence of the P-glycoprotein inhibitor verapamil and the nonspecific multidrug resistance protein inhibitor probenecid. In another study, brain concentrations of the sodium channel blocker phenytoin were inversely proportional to MDR1 mRNA expression.39 Although the mechanisms underlying variable MDR1 expression are not known, one possibility is that the disease modulates gene expression. The recognized electrical remodeling in atrial fibrillation40–42 raises the possibility that disease-mediated alterations in protein expression could be an additional mechanism leading to drug resistance in this arrhythmia.
Limitations
The cells used express neither gene studied, but it is possible that they express other related transporters (although this would not explain our results). We assessed transport function in a heterologous expression system that limits the interaction to one drug target (that is, HERG) and one transporter (that is, P-glycoprotein). Future studies will be needed to compare this model with similar experiments in cardiomyocytes. We did not directly assess protein folding, thus the effect of fexofenadine on protein folding is inferred. Although we did not directly measure intracellular ibutilide concentration, the effects shown here identify ibutilide as a substrate for P-glycoprotein, which may have important implications for the management of patients with atrial fibrillation. In fact, our results validate the approach of assessing a drug effect, not by measuring concentration, but by a direct evaluation of pharmacological action mediated by a drug interacting with an intracellular target. Rare mutations in KCNH2 are one cause of the congenital long QT syndrome; it is possible that transporter coexpression could modulate drug sensitivity of mutant channels.
Conclusions
In a heterologous system, we showed that the functional variant in MDR1, G2677A, leading to Ala893Ser, confers resistance to ibutilide-mediated HERG block. However, when a plasmid encoding MDR1*3 was studied, we unexpectedly found that the protein does not traffic to the cell surface and thus failed to confer resistance to ibutilide. Immunofluorescence data suggested that the effect of the silent C3435T polymorphism in MDR1*3 can be rescued by fexofenadine. The effect of MDR1 variants on the pharmacodynamic effects of ibutilide strongly suggests that variable function of drug transporters is a potential mechanism for resistance to antiarrhythmic pharmacotherapy.
Materials and methods
Cell preparation
Chinese hamster ovary cells (CHO-K1, ATCC) were grown at 37 °C in 5% CO2 in F-12 nutrient mixture medium supplemented with 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA, USA), 2mmoll−1 L-glutamine and penicillin (50Uml−1)–streptomycin (50 μgml−1). CHO cells were selected because they do not have endogenous potassium currents and do not express MDR1 as assessed by reverse transcriptase-PCR and western blot. In each experiment, cells were cotransfected (Fugene HD, Roche Laboratories, Nutley, NJ, USA) with two separate bicistronic plasmids. One encoded a red-fluorophore (IRES-dsRed) with HERG and the second contained a green fluorophore protein (IRES-GFP) with either an empty bicistronic site, wild-type MDR1 (HERG+G2677/C3435; MDR1*1), the G2677 T variant alone (HERG+G2677T/C3435; MDR1*7) or the linked haplotype (HERG+G2677T/C3435T; MDR1*3). The G2677T and C3435T variants were introduced by site-directed mutagenesis of the wild-type MDR1 clone. The ratio of wild-type or variant MDR1 and HERG plasmids relative to Fugene HD was 2 μg/2 μg per 20 μl. In some experiments, the effects of media supplementation with fexofenadine (1 μM), a drug that does not suppress IKr and has been reported to rescue misfolded HERG mutants, were assessed.32
Whole cell electrophysiology protocol
Cells showing yellow (that is, red plus green) fluorescence were selected for study 48 h after transfection. Patch pipettes were fabricated from thick-wall borosilicate glass (World Precision Instruments Inc., Sarasota, FL, USA) with a multistage P-97 Flaming-Brown micropipette puller (Sutter Instruments Co., Novato, CA, USA) and subsequently polished. Pipette resistance was 1–4 molΩ−1, and as reference electrode, a 1–2% agar bridge with composition similar to the bath solution was used. The intracellular pipette filling solution contained (in mM): KCl, 110; HEPES, 10; K4 BAPTA, 5; K2 ATP, 5; and MgCl2, adjusted to pH 7.2 with KOH yielding a final intracellular K + concentration of 145mM. The bath (Normal Tyrode’s) solution contained (in mM): NaCl, 145; KCl, 4; CaCl2, 1.8; MgCl2, 1; HEPES, 10; and glucose, 10, adjusted to pH 7.35 with NaOH. Experiments took place at room temperature (22 °C). Data were acquired using the whole cell patch configuration through an Axopatch 200B amplifier (Axon Instruments Inc., Foster City, CA, USA) at 1–10 kHz after anti-alias filtering at less than 50% of the sampling frequency (5 kHz).43 The holding potential was −80mV. The standard protocol to obtain current–voltage relations and activation curves consisted of 1-s steps that were imposed in 10-mV increments between −80mV and +60mV, followed by 1 s at −40mV to measure deactivating tail currents. The cycle time for protocols was 15 s or slower. Under these conditions, IKr is stable for >60 min in the absence of a drug intervention.44 After collection of baseline data, the IKr blocker ibutilide was added to the perfusate while pulsing to +20mV was applied every 15 s. IKr was monitored, and when a new steady state was reached (generally in 5 min), data in the presence of drug were collected. Ibutilide concentration was increased in stepwise increments from 200pM to 2 μM in the same cell. The IC50 and 95% confidence intervals for each group was calculated using the Hill equation (I=I[/IC50/Dn]). In experiments using fexofenadine (see below), 1 μM of drug was added to the medium, transfected cells were then cultured for 48 h, drug was washed out and IKr was subsequently assessed.
Immunofluorescence and confocal microscopy
To evaluate cell surface expression of P-glycoprotein, CHO cells were cultured in two-chamber tissue culture slides (Becton-Dickinson Falcon, Franklin Lakes, NJ, USA) and transfected with MDR1*1, MDR1*3 or MDR1*7 using Fugene HD at a ratio of 1 μg/5 μl. On each slide, one well contained cells cultured in untreated media, whereas the other well contained cells cultured in fexofenadine (1 μM)-supplemented media. After 48 h, media were removed and cells were washed in 1× phosphate-buffered saline (PBS) three times for 5 min each, fixed with 4% paraformaldehyde for 15 min and washed with 1× PBS for 5 min. Protein block was established using 1× PBS with 10% bovine serum albumin for 4 h. Primary anti-P-glycoprotein antibody (Santa Cruz BioTechnology, Santa Cruz, CA, USA) was introduced in blocking solution at a 1:250 dilution and subsequently incubated at 4 °C overnight (18 h). Primary antibody was removed and slides were successively washed with protein block solution three times each for 30, 15 and 5 min. Protein block was continued for an additional 30 min. GFP-conjugated anti-mouse secondary antibody (Sigma-Aldrich, St Louis, MO, USA) was subsequently introduced at a 1:5000 dilution and incubated at 22 °C for 4 h. Cells were then washed with 1× PBS three times each for 30, 15 and 5 min. To visualize the cell membrane, di-8-ANEPPS (0.05 μgml−1) was introduced after the last rinse for 60 s, removed and the cells rinsed with 1× PBS for 5 min. Experiments with all three constructs were carried out simultaneously, in triplicate, and repeated three times.
All cell groups were visualized on an LSM 510 META confocal microscope with a ×63/1.4 (oil immersion) magnification in differential interference contrast (DIC) mode, a pinhole diameter of 204 μM and a wavelength intensity of 10% at 633 and 488 nM in a plane, multitrack 8-bit configuration. Surface expression was assessed in the overlay mode and prospectively defined as the presence of yellow fluorescence on the entire circumference of the cell. The proportion of cells per field showing yellow fluorescence about the entire cell circumference was compared between groups in the presence and absence of fexofenadine.
Data analysis
To determine the influence of MDR1 variants on drug block, intergroup comparisons were made between cells expressing HERG alone, HERG+MDR1*1, HERG+MDR1*3 and HERG+MDR1*7 using one-way analysis of variance with a post hoc Bonferroni correction for individual pairwise comparisons. Individual pairwise comparisons using the Student’s t-test were used to assess the impact of fexofenadine on trafficking of P-glycoprotein between treated and untreated cells transfected with the same construct. Membrane surface expression of P-glycoprotein was determined by a single operator with pairwise comparisons between fexofenadine-treated and -untreated cells performed using the χ2 test for categorical data.
Acknowledgments
This work was supported by grants (GM07569; HL65962) from the National Heart Lung and Blood Institute.
Footnotes
Duality of interest
None declared.
References
- 1.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 2.Roden DM. Genetic polymorphisms, drugs, and proarrhythmia. J Intervent Cardiac Electrophysiol. 2003;9:131–135. doi: 10.1023/a:1026267903800. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 4.Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, et al. The potential for QT prolongation and proarrhythmia by non-antiarrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Eur Heart J. 2000;21:1216–1231. doi: 10.1053/euhj.2000.2249. [DOI] [PubMed] [Google Scholar]
- 5.Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. PNAS. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeola SW, Rich TC, Uebele VN, Tamkun MM, Snyders DJ. Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel : role of S6 in antiarrhythmic drug binding. Circ Res. 1996;78:1105–1114. doi: 10.1161/01.res.78.6.1105. [DOI] [PubMed] [Google Scholar]
- 7.Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 8.Napolitano C, Schwartz PJ, Brown AM, Ronchetti E, Bianchi L, Pinnavaia A, et al. Evidence for a cardiac ion channel mutation underlying drug-induced QT prolongation and life-threatening arrhythmias. J Cardiovasc Electrophysiol. 2000;11:691–696. doi: 10.1111/j.1540-8167.2000.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 9.Lasser KE, Allen PD, Woolhandler SJ, Himmelstein DU, Wolfe SM, Bor DH. Timing of new black box warnings and withdrawals for prescription medications. JAMA. 2002;287:2215–2220. doi: 10.1001/jama.287.17.2215. [DOI] [PubMed] [Google Scholar]
- 10.Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA. 1993;270:2590–2597. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- 11.Houltz B, Darpo B, Edvardsson N, Blomstrom P, Brachmann J, Crijns HJ, et al. Electrocardiographic and clinical predictors of torsades de pointes induced by almolanat infusion in patients with chronic atrial fibrillation or flutter: a prospective study. PACE. 1998;21:1044–1057. doi: 10.1111/j.1540-8159.1998.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 12.Gbadebo TD, Trimble RW, Khoo MSC, Temple J, Roden DM, Anderson ME. Calmodulin inhibitor W-7 unmasks a novel electrocardiographic parameter that predicts initiation of torsade de pointes. Circulation. 2002;105:770–774. doi: 10.1161/hc0602.103724. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Amer College Cardiol. 2000;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- 14.Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- 15.Jazwinska-Tarnawska E, Orzechowska-Juzwenko K, Niewinski P, Rzemislawska Z, Loboz-Grudzien K, Dmochowska-Perz M, et al. The influence of CYP2D6 polymorphism on the antiarrhythmic efficacy of propafenone in patients with paroxysmal atrial fibrillation during 3 months propafenone prophylactic treatment. Int J Clin Pharmacol Ther. 2001;39:288–292. doi: 10.5414/cpp39288. [DOI] [PubMed] [Google Scholar]
- 16.Woodahl EL, Yang Z, Bui T, Shen DD, Ho RJY. Multidrug resistance gene G1199A polymorphism alters efflux transport activity of P-glycoprotein. J Pharmacol Exp Ther. 2004;310:1199–1207. doi: 10.1124/jpet.104.065383. [DOI] [PubMed] [Google Scholar]
- 17.Schinkel AH, Wagenaar E, Mol CAAM, van Deemter L. P-glycoprotein in the blood–brain barrier of mice influences the brain penetration and pharmacological activity ofmany drugs. J Clin Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marzolini C, Kim RB. Placental transfer of antiretroviral drugs. Clin Pharmacol Ther. 2005;78:118–122. doi: 10.1016/j.clpt.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Chan LMS, Lowes S, Hirst BH. The ABCs of drug transport in intestine and liver: efflux proteins limiting drug absorption and bioavailability. Eur J Pharm Sci. 2004;21:25–51. doi: 10.1016/j.ejps.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Niemi M, Backman JT, Kajosaari LI, Leathart JB, Neuvonen M, Daly AK, et al. Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin Pharmacol Ther. 2005;77:468–478. doi: 10.1016/j.clpt.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Potschka H, Loscher W. Blood–brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kusuhara H, Sugiyama Y. Active efflux across the blood brain barrier: role of the solute carrier family. NeuroRx. 22005(2):73–85. doi: 10.1602/neurorx.2.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho RH, Kim RB. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther. 2005;78:260–277. doi: 10.1016/j.clpt.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 24.Grube M, Meyer zu Schwabedissen HEU, Prager D, Haney J, Moritz KU, Meissner K, et al. Uptake of cardiovascular drugs into the human heart: expression, regulation, and function of the carnitine transporter OCTN2 (SLC22A5) Circulation. 2006;113:1114–1122. doi: 10.1161/CIRCULATIONAHA.105.586107. [DOI] [PubMed] [Google Scholar]
- 25.Kim RB, Leake BF, Choo EF, Dresser GK, Kubba SV, Schwarz UI, et al. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther. 2001;70:189–199. doi: 10.1067/mcp.2001.117412. [DOI] [PubMed] [Google Scholar]
- 26.Kurata Y, Ieiri I, Kimura M, Morita T, Irie S, Urae A, et al. Role of human MDR1 gene polymorphism in bioavailability and interaction of digoxin, a substrate of P-glycoprotein. Clin Pharmacol Ther. 2002;72:209–219. doi: 10.1067/mcp.2002.126177. [DOI] [PubMed] [Google Scholar]
- 27.Kim KA, Park PW, Park JY. Effect of ABCB1 (MDR1) haplotypes derived from G2677T/C3435T on the pharmacokinetics of amlodipine in healthy subjects. Brit J Clin Pharmacol. 2007;63:53–58. doi: 10.1111/j.1365-2125.2006.02733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Illmer T, Schuler US, Thiede C, Schwarz UI, Kim RB, Gotthard S, et al. MDR1 gene polymorphisms affect therapy outcome in acute myeloid leukemia patients. Cancer Res. 2002;62:4955–4962. [PubMed] [Google Scholar]
- 29.Yamauchi A, Ieiri I, Kataoka Y, Tanabe M, Nishizaki T, Oishi R, et al. Neurotoxicity induced by tacrolimus after liver transplantation: relation to genetic polymorphisms of the ABCB1 (MDR1) gene. Transplantation 2002. 2002 Aug 27;74:571–572. doi: 10.1097/00007890-200208270-00024. [DOI] [PubMed] [Google Scholar]
- 30.Elens L, Capron A, Kerckhove VV, Lerut J, Mourad M, Lison D, et al. 1199G>A and 2677G>T/A polymorphisms of ABCB1 independently affect tacrolimus concentration in hepatic tissue after liver transplantation. Pharmacogenet Pharmacogenomics 2007. 2007 Oct 17;17:873–883. doi: 10.1097/FPC.0b013e3282e9a533. [DOI] [PubMed] [Google Scholar]
- 31.Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, et al. A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 32.Rajamani S, Anderson CL, Anson BD, January CT. Pharmacological rescue of human K+ channel long-QT2 mutations. Circulation. 2002;105:2830–2835. doi: 10.1161/01.cir.0000019513.50928.74. [DOI] [PubMed] [Google Scholar]
- 33.Bozina N, Kuzman MR, Medved V, Jovanovic N, Sertic J, Hotujac L. Associations between MDR1 gene polymorphisms and schizophrenia and therapeutic response to olanzapine in female schizophrenic patients. J Psychiatr Res. 2008;42:89–97. doi: 10.1016/j.jpsychires.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Aarnoudse ALHJ, van Schaik RHN, Dieleman J, Molokhia M, van Riemsdijk MM, Ligthelm RJ, et al. MDR1 gene polymorphisms are associated with neuropsychiatric adverse effects of mefloquine. Clin Pharmacol Ther. 2006;80:367–374. doi: 10.1016/j.clpt.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 35.Sohn JW, Lee SY, Lee SJ, Kim EJ, Cha SI, Kim CH, et al. MDR1 polymorphisms predict the response to etoposide–cisplatin combination chemotherapy in small cell lung cancer. Jpn J Clin Oncol. 2006;36:137–141. doi: 10.1093/jjco/hyi231. [DOI] [PubMed] [Google Scholar]
- 36.Kim DH, Park JY, Sohn SK, Lee NY, Baek JH, Jeon SB, et al. Multidrug resistance-1 gene polymorphisms associated with treatment outcomes in de novo acute myeloid leukemia. Int J Cancer. 2006;118:2195–2201. doi: 10.1002/ijc.21666. [DOI] [PubMed] [Google Scholar]
- 37.Seo T, Ishitsu T, Ueda N, Nakada N, Yurube K, Ueda K, et al. ABCB1 polymorphisms influence the response to antiepileptic drugs in Japanese epilepsy patients. Pharmacogenomics. 2006;7:551–561. doi: 10.2217/14622416.7.4.551. [DOI] [PubMed] [Google Scholar]
- 38.Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y. Quantitative in vivo microdialysis study on the influence of multidrug transporters on the blood–brain barrier passage of oxcarbazepine: concomitant use of hippocampal monoamines as pharmacodynamic markers for the anticonvulsant activity. J Pharmacol Exp Ther. 2005;314:725–731. doi: 10.1124/jpet.105.085514. [DOI] [PubMed] [Google Scholar]
- 39.Tishler D, Weingberg K, Hinton D, Barbaro N, Annett G, Raffel C. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia. 1995;36:1–6. doi: 10.1111/j.1528-1157.1995.tb01657.x. [DOI] [PubMed] [Google Scholar]
- 40.Cha TJ, Ehrlich JR, Zhang L, Shi YF, Tardif JC, Leung TK, et al. Dissociation between ionic remodeling and ability to sustain atrial fibrillation during recovery from experimental congestive heart failure. Circulation. 2004;109:412–418. doi: 10.1161/01.CIR.0000109501.47603.0C. [DOI] [PubMed] [Google Scholar]
- 41.Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, et al. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101:2631–2638. doi: 10.1161/01.cir.101.22.2631. [DOI] [PubMed] [Google Scholar]
- 42.Schoonderwoerd BA, Van Gelder IC, Van Veldhuisen DJ, Van den Berg MP, Crijns HJGM. Electrical and structural remodeling: role in the genesis and maintenance of atrial fibrillation. Progress Cardiovasc Dis. 2005;48:153–168. doi: 10.1016/j.pcad.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 43.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Archive: Eur J Physiol 1981. 1981 Aug 1;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 44.Yang T, Snyders D, Roden DM. Drug block of Izs: model systems and relevance to human arrhythmias. J Cardiovasc Pharmacol. 2001;38:737–744. doi: 10.1097/00005344-200111000-00010. [DOI] [PubMed] [Google Scholar]