

Abstract
Anxiety-like responses to stress are accompanied by elevation of brain cytokine-mRNAs. Because cytokines microinjected into central-amygdala (CeA) substitute for stress in a behavioral paradigm, the possibility was raised that cytokines increased by stress influence behavior by affecting CeA-neural activity. Previously, cytokines increased firing-rate of CeA-neurons comparable to that induced by corticotropin-releasing factor (CRF). In this investigation, tumor-necrosis-factor-α (TNFα) increased amplitude, but not frequency of mEPSCs from CeA-neurons. Additionally, TNFα decreased the threshold for triggering action potentials from CeA-neurons without altering membrane-properties during current-clamp recording. Glutamate-receptor-antagonist blockade of mEPSCs and the TNFα-induced reduction in firing threshold implicated glutamate in these changes. A phosphatidyl-inositol-3-kinase-antagonist prevented the TNFα-induced increased in amplitude of mEPSCs, documenting a TNFα intracellular influence. Additionally, TNFα increased frequency, but not amplitude of mIPSCs. CRF-receptor-antagonists were found to prevent the TNFα-induced increase in mIPSC-frequency, without altering the TNFα-induced amplitude increase in mEPSCs or the reduced threshold for action-potentials by TNFα. To clarify how TNFα was increasing CRF-release in the presence of tetrodotoxin, the possibility tested was whether preventing glial-activation would prevent this elevated mIPSC-frequency blocked by CRF-receptor antagonists. Minocycline, which blocks glial activation, prevented the TNFα-induced increase in mIPSC-frequency—a finding consistent with glia contributing to the CRF-involvement in this TNFα action. To fully understand the means by which a CRF1-receptor-antagonist and minocycline prevent TNFα from increasing mIPSC-frequency will require further clarification. Nonetheless, these data provide convincing evidence that release of TNFα by stress could influence GABA- and glutamate-function to alter neural activity of CeA-neurons.
Keywords: Central amygdala, CRF-receptor antagonists, Glia, mIPSCs, mEPSCs, Neural function, Minocycline, PI-3-kinase-antagonist
1. INTRODUCTION
Cytokines are elevated in brain following restraint stress (Porterfield et al., 2011), social defeat stress (Wohleb et al., 2011), withdrawal from chronic alcohol exposure (Whitman et al. 2013,) and following systemic lipopolysaccharide (LPS) administration (Qin et al., 2007; Whitman et al. 2013). Stress and withdrawal from chronic alcohol are known to be accompanied by anxiety- and depressive-like behaviors (Dunn et al., 1999; Breese et al., 2004, 2005a,b, 2011; Hammen, 2005; Lindqvist et al., 2009). In addition to the role of cytokines in immune function, there is a contention that cytokines may produce functional changes by being neural mediators in the CNS (Adler et al., 2006; Rostène et al., 2007; Vitkovic L et al., 2000). Evidence supporting cytokines having neural action came in part from finding the presence of cytokine receptors on neurons (Boka et al., 1994; Hermann et al., 2004; Holmes et al., 2004). Furthermore, various studies in brain have demonstrated that cytokines can influence neural function (Belkouch et al., 2011; Churchill et al., 2008; Callewaere et al., 2006; Emch et al., 2000, 2001; Gruol and Nelson, 2005; Kawasaki et al., 2008; Knapp et al., 2011; Lukats et al., 2005; Oh et al., 2003; Santello and Volterra, 2012; Stellwagen et al., 2005, 2006; Tabarean et al., 2006; Wilkinson et al., 1993).
To explore if brain cytokines released by stress could be linked to function, intracerebroventrical (ICV) administration of cytokines was established to substitute for the initial two withdrawals of a chronic-intermittent alcohol (CIA) exposure protocol that facilitated ethanol-withdrawal-induced anxiety (Breese et al., 2008, 2011), just as observed previously with stress (Breese et al., 2004). The central nucleus of the amygdala (CeA) has a known association with stress-induced negative affect (Huang et al. 2010). Therefore, to explore if the CeA could be involved in the ICV cytokine facilitation of ethanol-withdrawal anxiety (Breese et al., 2008), cytokine microinjection into the CeA prior to ethanol at weekly intervals was also found to facilitate ethanol withdrawal-induced anxiety-like behavior (Knapp et al., 2011). Importantly, other investigations have provided indirect evidence for cytokine action in the CeA being capable of influencing neural activity. Inagaki et al. (2012) found that increasing cytokines in individuals with an LPS challenge induced an fMRI change in the amygdala. Prager et al. (2012) recently noted changes in electrophysiological measures as well as altered cFos levels in amygdala being induced by systemic LPS. More directly relevant, the proinflammatory cytokine, tumor-necrosis-factor-α (TNFα), applied to CeA neurons was found to increase firing rate and facilitate GABA release (Knapp et al., 2011). In spite of these published observations, evaluations of other neural actions of cytokines in the CeA have not been provided.
Therefore, based upon stress increasing TNFα and other cytokines in brain, the present investigation defines whether TNFα application to CeA neurons influences selected electrophysiological determinations, including measures of membrane properties, sensitivity to current application, mEPSCs, and mIPSCs. First, the possible influence of TNFα on glutamate-related changes from CeA neurons was tested. Subsequently, the increased frequency of mIPSCs from TNFα-responsive neurons was confirmed (Knapp et al., 2011). Then, studies explored the potential mechanisms by which TNFα-induced alterations in CeA-neural activity. These investigations included exploring if TNFα-induced alterations depended upon CRF (Knapp et al., 2011), selected kinases (Stellwagen et al., 2005) and/or glial elements (Behan et al., 1995; Giuliani et al. 2005; Nutile-McMenemy et al., 2007; Tian and Bishop, 2003; Yan et al. 2008). Thus, the present efforts not only will provide further evidence that cytokines affect CeA neural function, but also will explore the means by which CeA-neural function can be associated with TNFα-induction of glutamate and GABA activity. Such extended information concerning actions of cytokines on CeA neurons is expected to further our understanding of the relationship stress-induction of cytokines in this brain site has to facilitation of anxiety-like behavior that follows stress (Breese et al., 2004, 2008, 2011; Knapp et al., 2011).
1. Materials and Methods
Slice Preparation
Coronal brain slices, 350–400 μm thick, containing the right or left amygdala, were obtained from 18–24 day-old Sprague-Dawley rats. Animals were lightly anesthetized by isoflurane inhalation and killed by decapitation. Brains were rapidly removed and placed in ice-cold sucrose buffer with the following composition (in mM): sucrose 112.5, NaCl 63, KCl 3, NaH2PO4 1.25, CaCl2 0.5, NaHCO3 24, MgSO4 6, and glucose 10. The solution was aerated with 95% O2 and 5% CO2. The brain was blocked using a stainless steel blade and maintained in the ice-cold sucrose buffer until tissue slices of 350–400 μM thickness were cut from the tissue blocks with a vibrating tissue slicer (Leica VT 1000S). The slices were stored in a beaker containing artificial cerebrospinal fluid (ACSF) gassed with 95% O2/5% CO2. ACSF contained (in mM): NaCl 124, KCl 3.25, KH2PO4 1.25, CaCl2 2, NaHCO3 20, MgSO4 2, and glucose 10. The brain slices were equilibrated at least 1 hour at room temperature (21–24°) before initiating experiments.
Electrophysiological Recording
Whole-cell patch clamp recording was obtained with an Axopatch 1D amplifier (Axon Instruments) at room temperature (21–24°). A slice was placed at the bottom of a chamber attached to a microscope (BX51WI, Olympus; Tokyo, Japan). The slice was superfused with ACSF gassed with 95%O2/5%CO2 at a flow rate of 1.5 ml/minute Recording electrodes were pulled from borosilicate glass (Drummond Scientific Company, Broomall, PA) that had a resistance of 2.5–3 MΩ. The electrodes were filled with internal solution with the following composition (in mM): KCl 150, HEPES 15, K-ATP 2, EGTA 5, and phosphocreatine 15 (pH 7.4, adjusted with KOH). For voltage clamp recording the holding potential was −60 mV. Data were digitized at 5 kHz, and collected with pClamp 10 (Axon Instruments). Various drugs were placed in sealed syringes containing either a control (ACSF) or a ACSF solution that were delivered through Teflon tubing connected to a multibarrel perfusion pencil (250 μM in diameter) positioned 150 to 250 μm from the cell tested. In both the mIPSC and the mEPSC recordings, 1 μM tetrodotoxin (TTX, Sigma) was included in the perfusion solution (ACSF) to block action-potential-dependent currents. To record mIPSCs, 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, Sigma-Aldrich) and 50 μM 2-amino-5- phosphonovaleric acid (AP-5, Sigma-Aldrich) were applied to neurons prior to recording to block glutamate mediated transmission. For mEPSCs recording, 20 μM bicuculline methiodide (Tocris) was applied prior to recording to block GABAA receptor mediated transmission.
The central-amygdala (CeA) was visualized using infrared illumination under differential interference contrast optics with a water-immersion lens (40x). The image was displayed on a monitor via video camera to permit the location of neurons in the CeA. After breaking the neural membrane of a cell, 5- to 7-minutes were required for membrane stabilization, at which time recordings were immediately initiated. When control recordings were complete, TNFα or other experimental combinations were administered and recordings made after 5 minutes of drug delivery. Then, a 5–8 minute wash out was followed by a final minute of recording. In circumstances in which recovery from TNFα did not occur within the washout period, sham experiments were performed by replacing TNFα with ACSF or ACSF with bovine serum albumin (BSA; the carrier for TNFα).
For the current clamp recordings, pre-test potential was maintained at −60 mV by injecting current that was altered in some cases just before application of current steps to insure that all measures were made at −60 mV. When injected current was altered, the change in current required to maintain membrane potential at −60 mV was determined for drug and control groups. To evaluate whether drug application altered membrane properties, resting potentials and input resistance before and during drug application were determined. To measure input resistance, 4 steps of hyperpolarizing current pulses (500-ms, 1 Hz with 0.1 nA increments) from pre-test membrane potential of −60 mV were applied. Input resistance was calculated as the slope of the current-voltage plot. To evaluate sensitivity of cells to cytokines, a series of 600-ms current injections (10 steps, 1 Hz) of increasing currents were applied at given increments to induce action potentials. Current increments were adjusted for each neuron to insure reaching the threshold for the initiation of action potentials. Once established, the current increments remained constant for a given neuron. The firing threshold was defined as the shift of potentials from pre-test membrane potential to the potential which initiated the action potentials (Figs. 2A & 2B). In the current-clamp experiments, unless indicated in text, inhibitory receptor antagonists, blockers of excitatory transmission, and tetrodotoxin (TTX) were not included.
Figure 2. TNFα Reduced the Threshold for current injection to elicit Action-Potentials in CeA.

Shown in the upper of figures, are (A) potential traces recorded under control conditions and (B) traces obtained from the same neuron after a 5-min treatment with 60-ng/ml TNFα. The injected current was increased by 100 pA with each presentation and traces for the highest subthreshold current and the threshold current are shown. Threshold current was 600 pA in (A) and 400pA in (B). (C) TNFα 60-ng/ml lowered the current required to trigger an action potential [t (6)=3.59, n=7, p<0.05]. (D) When TNFα (60-ng/ml) was applied in the presence of 10 μM CNQX and 50 μM AP-5, the current threshold for eliciting an action potential did not change [t(5)=0.93; n=6, p>0.05].
Drugs
NBI 27914, astressin 2B and α-helical CRF 9–41 were supplied by Tocris. The second messenger inhibitors for PI-3-kinase and P38-MAP-kinase were supplied by Tocris. TNFα (Recombinant rat TNFα, R&D Systems) was dissolved in 0.1% bovine serum albumin (BSA) in PBS (10 μg/ml) as a stock solution. In respect to the relevance of the doses of TNFα (30 to 60-ng/ml) utilized compared to the concentration at the receptor site is an unknown. TNFα levels in brain have been shown to be in the general range of 0.1- to 1.0-pg/ml. However, TNFα is released from glia by vesicular transport just as neurotransmitters are released from neurons. Consequently, the concentration of TNFα at the release site that acts on the TNFα receptor would be higher than in tissue. Regardless, the TNFα concentrations used will be shown to influence neural function.
Data Analysis
The data recorded with patch-clamp were analyzed with either Clampfit (pClamp 10, Axon Instruments) or mini Analysis (Synaptosoft, version 6.0.7). Numerical data were given as mean ± SEM, and n represents the number of cells tested. Data were submitted to either analysis of variance (ANOVA) followed by an LSD test or a Student’s t-test as appropriate. Linear trend was analyzed using a Pierson product moment correlation. Calculated p-values of less than 0.05 were accepted as evidence of significance.
3. Results
3.1. Effect of TNFα on mEPSCs from CeA-Neurons
This first investigation determined whether TNFα would alter mEPSC characteristics from CeA neurons. In Fig. 1, the TNFα was found to increase the amplitude of mEPSCs. In Fig. 1A, representative current traces of mEPSCs were recorded from the same CeA neuron prior to (control) and during 60-ng/ml TNFα application (TNFα) to demonstrate that TNFα increased the amplitude of mEPSCs. Additionally included in Fig. 1A is a representative trace that the combination of CNQX and AP-5 blocked the mEPSCs that were recorded from a CeA neuron in the absence of TNFα (see Fig. 1 legend), an indication that the recorded mEPSCs resulted from glutamate release. Fig. 1B shows an averaged trace of mEPSCs from the representative neuron in Fig. 1A in the presence and absence of TNFα to demonstrate the increase in mEPSC-amplitude by TNFα (60-ng/ml). Further, Fig. 1C provides statistical confirmation that the mEPSC amplitude increase by TNFα is dose-related with a significant linear trend [r (29)=0.74; p<0.05] [F (4,24)=12.30, n=5–8; p<0.0001]. A Post hoc LSD test demonstrated that the percent increase in mEPSC amplitude induced by 30 and 60-ng/ml TNFα was significantly greater than the change induced by 0, 6 and 18-ng/ml TNFα (Fig. 1C). Importantly, TNFα (Fig. 1D) did not change the rate of the mEPSCs [F (4,26) = 0.22, p>0.05; n=5–8]. Because the effect of TNFα on the amplitude of mEPSCs did not recover during wash out, sham experiments were performed with ACSF, data which are shown as a “0” TNFα concentration in Figs. 1C and 1D. Adding BSA, the carrier protein for TNFα, to ACSF (BSA+ACSF) had no effect on percent change in the amplitude or frequency of mEPSCs compared to ACSF alone (2.4 ± 2.4 t=0,072; p>0.05 and 1 ± 7.4; t=0.078, p>0.05, respectively; n=5). In summary, these results are consistent with TNFα inducing a change in post-synaptic sensitivity to glutamate without a change in presynaptic release probability.
Figure 1. TNFα increased the Amplitude of mEPSCs in CeA.

(A) Representative mEPSC recordings from the same neuron in the presence of 20 μM bicuculline (Control), and after TNFα (TNFα). Application of glutamate antagonists to that neuron (CNQX + AP-5) blocked the mEPSCs. The glutamate antagonist treatment completely blocked the mEPSCs in other neurons (n=5). (B) Averaged traces of mEPSCs from the same neuron in Fig. 1A prior (control) and in the presence of TNFα (60 ng/ml) showing the increase in mEPSC amplitude induced by TNFα (TNFα). (C) TNFα concentration enhanced the amplitude of mEPSCs concentration-dependently (n=5–8 for individual concentrations; F (4,24) = 12.30, p<0.0001) with a significant linear trend; r(29)=0.74, p<0.05. (D) TNFα did not to influence the frequency of mEPSCs [F (4,26)=0.22; p>0.05; n=5–8].
3.2 Effect of TNFα on CeA-Neural Membrane Properties and Sensitivity to Current Application
Because TNFα can increase firing rate (Knapp et al., 2011), the effect of TNFα was tested on membrane properties to determine if a change in this measure could be responsible for this increased rate. However, TNFα (60 ng/ml) did not change resting membrane potential [(control = −59.6 ± 2.9 mV; TNFα = −57.6 ± 3.0 mV; n = 7; t (6) = 1.63; p > 0.05]. Further, input resistance in controls (63.7 ± 5.6 MΩ) was not significantly altered during application of 60 ng/ml TNFα (58.8 ± 2.4 MΩ) [n = 6; t(5) = 0.90; p > 0.05]. Results indicate that these membrane properties were not changed by TNFα suggesting that these changes in neural resting membrane properties were not responsible for the TNFα increased rate of firing (Knapp et al., 2011). Because TNFα did not affect the resting membrane properties, the effect of TNFα (60-ng/ml) was tested on the threshold for initiating an action potential with current injection. Pre-test membrane potential of −60 mV was maintained by injecting 8.8 ± 5.4 pA prior to TNFα application (n=7). This current was not significantly changed for pre-test following TNFα (8.0 ± 5.0 pA [t(6)=0.55; p>0.05), consistent with the lack of an effect of TNFα on both resting membrane potential and input resistance, as shown above. When a current-step reached a threshold or beyond, single or multiple (2–8) action potentials were evoked. The mean increment for each current step was 0.12 ± 0.02 nA (n=7).
Under control conditions and in the presence of TNFα, Figs. 2A and 2B illustrate representative current traces in which TNFα reduced the current required to induce spiking. Fig. 2C demonstrates statistically that TNFα (60-ng/ml) significantly lowerd the current threshold required to initiate an action potential [t(6)=3.02; p<0.05; n=7]. Fig. 2D establishes that a combination of glutamate receptor antagonists [CNQX (10 μM)] and NMDA [AP-5 (50-μM)] completely blocked the TNFα (60-ng/ml)-induced decrease in threshold required to elicit an action potential [t(5)=0.93; p>0.05; n=6], a finding indicative that glutamate is involved in this neural change by TNFα. The solvent for TNFα (BSA) did not significantly alter the current threshold to trigger action potentials [control=0.21 ± 0.06 nA; BSA=0.23 ± 0.0.04 nA (t (5)=0.542; p>0.05, n=6)].
3.3. Testing of Selected Second-Messenger Antagonists on the TNFα-Induced Increase in the Amplitude of mEPSCs from CeA Neurons
Stellwagen et al. (2005) demonstrated that TNFα increased the amplitude of mEPSCs from hippocampal neurons through a phosphatidyl-inositol-3-kinase (PI-3-K)- dependent process, without finding involvement of several other kinases. Based upon this latter observation (Stellwagen et al., 2005), the PI-3-kinase inhibitor (wortmannin) and the p38-MAP-kinase inhibitor (SB 202190) were tested against the TNFα-induced increase in mEPSC-amplitude from CeA neurons.
Wortmannin (1-μM) incubation for 30 min to block PI-3-kinase activity abolished the increased mEPSC-amplitude induced by TNFα (Fig. 3; n=6). Percent change in amplitude of mEPSCs by TNFα alone (24.3 ± 7.2%; n=7) was significantly greater than that induced by TNFα when incubated with the wortmannin (1.9 ± 4.3 %; n=6) [post hoc LSD test, p<0.05; F(2,16)=5.07; p<0.05 for all treatments in Fig. 3]. The wortmannin solvent (0.1% DMSO) had no effect on the percent increase in the mEPSC amplitude by TNFα [DMSO + TNFα=22.7 ± 7.6% (n=6); TNFα alone=24.3 ± 7.2%; (n=7); p>0.05 post hoc LSD test)]. The SB 202190 (500-nM) incubation for 2 hours did not change the increase in amplitude of the mEPSCs by TNFα (60-ng/ml) (13.3 ± 1.5 pA for control; 17.0 ± 1.1 pA for TNFα) [t(4)=3.01; p<0.05]. Thus, in accord with the TNFα findings by Stellwagen et al. (2005) from hippocampal neurons, the PI-3-K-antagonist, wortmannin, prevented the TNFα-induced increase in amplitude of mEPSCs from CeA neurons, without the P38-MAP-kinase-inhibitor, SB 201190, having an effect.
Figure 3. Blockade of the TNFα-induced increase in mEPSC-amplitude by wortmannin (a PI-3-K inhibitor).
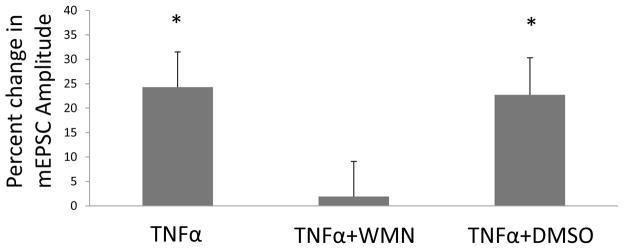
Shown are percent changes in amplitude of mEPSCs after TNFα (60-ng/ml) (TNFα), TNFα (60 ng/ml) with 1 μM wortmannin (WMN) incubation for 30-minutes (TNFα + WMN), and TNFα (60 ng/ml) with 0.1% DMSO incubation for 30 minutes (TNFα + DMSO; a control). For all treatments, F(2,16) = 5.07, p < 0.05; n = 6–7. Post hoc LSD test shows that the percent change of the mEPSC-amplitude by TNFα. TNFα + WMN-exposure is significantly less than the change in amplitude after TNFα alone (p<0.05) and the TNFα with DMSO (p<0.05).
3.4. Effect of TNFα on mIPSCs from CeA Neurons
In confirmation that TNFα increases GABA-release from CeA neurons (Knapp et al., 2011), Fig. 4A illustrates that the mIPSCs were completely blocked by bicuculline, a finding indicative that this TNFα-induced increase in mIPSC-frequency is caused by elevated GABA release. Fig. 4B further demonstrates that this increase in mIPSC frequency by TNFα [F (4,33)=3.06; p<0.05; n=5–10] is dose-related with a significant linear trend [r (38)=0.51; p<0.05]. A post- hoc LSD test showed that the percent increase in mIPSC frequency by the 60-ng/ml concentration of TNFα was greater than that induced by 0 and 6-ng/ml of TNFα (Fig. 3B). Fig. 4C illustrates that the amplitude of mIPSCs was not affected by TNFα [F(4,30)=0.44; p>0.05; n=5–10]. Because the effect of TNFα on mIPSCs did not recover during the 5-min wash out, sham experiments were conducted with ACSF, which is shown as the “0” TNFα concentration in Figs. 4B & 4C. Percent changes in frequency and amplitude of mIPSCs after BSA addition to the ACSF was −3.0 ± 2.6%; t=0.86, p>0.05 and 0.8 ± 6.1%; t=0.078, p>0.05 respectively (n=4), indicating that BSA alone had no effect on either frequency or amplitude of mIPSCs. These results are consistent with TNFα increasing the release probability of GABA without affecting synaptic sensitivity to GABA.
Figure 4. TNFα-Induced an Increase in the Frequency of mIPSCs from CeA Neurons.

(A) Representative mIPSC recording from a neuron in CeA before (control) and during (TNFα) application of TNFα (60 ng/ml) in the presence of 1 μM TTX, 10 μM CNQX and 50 μM AP-5. The bottom current trace shows that 20 μM bicuculline completely blocked these miniature currents, indicating that they were GABAergic. (B) TNFα concentration dependently [r (38) = 0.51; p < 0.05] increased the frequency of mIPSCs [n = 5–10 for individual concentrations, one way ANOVA, F (4, 33) = 3.06, p < 0.05]. (C) TNFα did not to change the amplitude of the mIPSCs [F (4,30) = 0.44; p > 0.05; n = 5–10].
3.5. Effect of CRF-Receptor-(CRFR) Antagonists on TNFα Changes in mIPSCs, mEPSCs and Sensitivity to Current Application
Knapp et al. (2011) reported that a CRF-receptor-antagonist prevents the repeated cytokine facilitation of ethanol withdrawal-induced anxiety, providing a potential association of CRF with cytokine and neural function. Previous work has demonstrated that like TNFα (Knapp et al., 2011), CRF can increase the frequency of mIPSCs (see Roberto et al., 2010). Based upon the comparable increases in neural firing (Inagaki et al., 2012) and frequency of mIPSC induced by both CRF and TNFα, studies were undertaken to determine if CRFR-antagonists would influence the increased frequency of mIPSCs induced by TNFα.
In Fig. 5A, the increased frequency of mIPSCs induced by 60-ng/ml of TNFα (p<0.01 compared to control sham) was blocked by 1 μM of α-helical-CRF (αHCRF), a general CRFR-antagonist (p<0.001) compared to TNFα alone [F(2,20)=9.89, p<0.01; for all groups, n=6–8]. In Fig. 5B, CRFR-antagonists that selectively blocked either CRF1Rs or CRF2Rs were tested against this increased frequency of mIPSCs by TNFα. While the CRF2R-antagonist (AST=astressin-2B; 100-nM) was without effect (p>0.05; n=7), the CRF1R-antagonist (NBI=NBI27914; 500-nM) blocked the increased frequency of mIPSCs by TNFα (Fig. 5B; p<0.05; n=7), just as did the general CRFR-antagonist (αHCRF; Fig. 5A). NBI 27914 alone had no significant effect on the percent change in frequency of mIPSCs from CeA neurons, which was −4.3 ± 7.2% (n=5). Representative mIPSCs tracings for control, TNFα and TNFα with NBI-27914 are shown in Figs. 5C & 5D.
Figure 5. CRF Receptor (CRFR) Antagonists prevented the TNFα-induced increase in frequency of mIPSCs.

(A) Histograms depicting the percent changes in mIPSCs during applications of 0 (control), 60-ng/ml TNFα and TNFα (60-ng/ml) with the non- specific CRF receptor blocker, α-helical CRF 9–41 (αHCRF, 1 μM). Over all the percent changes by the three treatments are significantly different [n=6–8 for individual treatments; F (2, 20)=9.89, p<0.01]. Compared with control, TNFα increased the frequency of mIPSCs (post hoc, LSD test, p < 0.01) while α-helical CRF blocked the effects of TNFα (post hoc, LSD test, p<0.001). (B) Shown are Histograms depicting the TNFα (60 ng/ml) induced increase in frequency of mIPSCs and either a CRF-2- receptor- (TNFα+Ast) or a CRF-1-receptor- (TNFα+NBI) antagonist on this TNFα (60-ng/mL)-induced increase in frequency of mIPSCs. Treatments altered modulation of mIPSCs [n = 6–7 for individual treatments; F (2,17)=5.85, p < 0.05]. While astressin 2B (100 nM) did not have effects on the function of TNFα (post hoc, LSD test no difference between TNFα and TNFα with astressin 2B, p>0.05), NBI 27914 (500 nM) blocked the effects of TNFα (post hoc, TNFα with NBI 27914 significantly less than either TNFα or TNFα with astressin 2B, p<0.01 and p<0.05, respectively). (C) Miniature IPSCs recording before (control) and during application of TNFα (60-ng/ml). Note increase in mIPSC frequency by TNFα. (D) Miniature IPSCs recording before (control) and during application of 60-ng/ml TNFα with 500 nM NBI 27914. In the presence of the CRF1RA, TNFα did not increase frequency of mIPSCs.
In Fig. 6A, TNFα (30 ng/ml) was confirmed to decrease the threshold for injected current to elicit an action-potential [t(6) = 3.34; p < 0.05; n = 7]—a change which was not blocked by 1-μM of the general CRFR-antagonist [αHCRF; (t (9)=2.57; p<0.05; n=10) Fig. 6B]. Furthermore, the increased amplitude of mEPSCs induced by TNFα was also not affected by the general CRFR-antagonist, αHCRF (Figs. 6C & 6D).
Figure 6. CRF-receptor (CRFR)-antagonists did not modulate the effect of TNFα on the current-injection threshold for eliciting an action potential or the amplitude of mEPSCs From CeA Neurons.

(A) TNFα 30-ng/ml lowered the threshold for current- induced action-potentials [n=7, t (6)=3.34, p<0.05]. (B) In the presence of α-helical CRF, TNFα 30-ng/ml continued to lower the threshold current for initiating action-potentials [t (9)=2.57, n=10, p<0.05]. (C) TNFα 30-ng/ml increased in the amplitude of mEPSCs [t (7)=4.67, n=8, p<0.01]. (D) In the presence of α-helical CRF, TNFα 30-ng/ml continued to increase the amplitude of mEPSCs [t (7)=3.07, n=8, p<0.05].
3.6. Effect of Minocycline on CRF Involvement in the TNFα-Induced Increase in the Frequency of mIPSCs from CeA Neurons
The involvement of CRF in the increased frequency of mIPSCs by TNFα was unexpected given that recording was in the presence of tetrodotoxin—a compound which prevents neural transmission. However, astrocytes are reported to contain CRF (Tian and Bishop, 2003) and CRF-binding protein (Maciejewski et al., 1996; McClennen and Seasholtz 1999). Furthermore, Ubink et al. (2003) reported that many other peptides and peptide mRNAs are present in astrocytes. Therefore, an experiment was initiated to determine if glial elements, which contain TNFα-receptors (Yan et al., 2008), could be releasing CRF.
Based upon minocycline being able to attenuate microglial (Giuliani et al. 2005; Nikodemova et al. 2007; Nutile-McMenemy et al., 2007) and astrocytic activity (McAllister and Miller, 2010), minocycline was tested to determine if the TNFα-induced increase in frequency of mIPSCs would be prevented. Fig. 7 shows that the TNFα-induced increase in mIPSC frequency after minocycline (60-μM) incubation for 1-hr was significantly decreased [(F(2,24)=4.17); n=7–13; p<0.05 with post hoc LSD test]. Minocycline (60 μM) incubation alone for 1-hour had no effect on the frequency of mIPSCs (i.e., percent change in frequency of mIPSCs was 9.80 ± 8.67%) (Fig. 7). Thus, TNFα-induced release of CRF from glial elements might underlie the apparent involvement of CRF suggested by the CRF-receptor antagonists blocking the TNFα-induced increase in frequency of mIPSCs.
Fig. 7. Effect of Minocycline on the TNFα-induced Increase in mIPSCs from CeA Neurons.
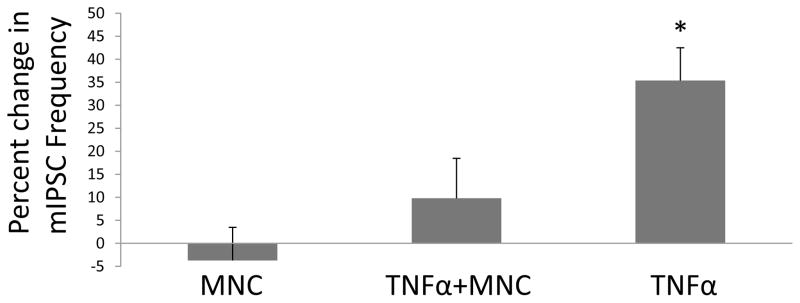
The change in frequency of mIPSCs are shown after 60-ng/ml TNFα (TNFα), −60-ng/ml TNFα with one hour incubation of 60-μM minocycline (TNFα + MNC) and 60-μM moncycline incubation in the absence of TNFα (MNC). The frequency of mIPSCs was affected by the minocycline treatment [n =7–13; F(2,24)=4.17, p<0.05]. The percent change in frequency of mIPSCs induced by TNFα alone is significantly greater than the change following MNC incubation with TNFα and MNC incubation alone (without TNFα) (p<0.01 and p<0.05, respectively).
Discussion
Because restraint stress (Porterfield et al., 2011; Whitman et al., 2013) and social defeat stress (Wohleb et al., 2011) increase cytokine-mRNAs in brain, this investigation of neural actions of TNFα on CeA neurons was undertaken because the CeA has a known involvement in stress-induced negative affect (Breese et al., 2004, 2011; Knapp et al., 2011; Koob, 2008). Initially, TNFα was found to increase the amplitude of mEPSCs, without affecting mEPSC frequency of CeA neurons—comparable to the earlier reports that TNFα increased the amplitude of mEPSCs from hippocampal neurons (De et al., 2003; Stellwagen et al., 2005). Even though TNFα was without an effect on membrane properties, TNFα was found to reduce the injected current required to induce spiking. This capacity of TNFα to reduce the current injection necessary to initiate action-potentials in the absence of an influence on membrane properties was unexpected. Importantly, the glutamate antagonist cocktail (CNQX + AP-5) not only prevented the effectiveness of glutamate at the synapse, but also blocked the TNFα-induced reduction in current required to initiate action potentials. Thus, the decreased current threshold for eliciting a spike appears to be glutamate-dependent. Because TTX was absent during the spike-threshold determination, TNFα could have this glutamate-dependent effect by increasing the effectiveness of glutamate neurotransmission at either the local synapse or at a distant site. Thus, the mechanism by which TNFα alters the current threshold for production of an action-potential needs further study.
Because both AMPA and NMDA receptors were blocked by the antagonist cocktail, either receptor type could be involved. However, NMDA currents have extremely slow rise-times (6–10 ms; Naylor et al.,2013) compared to AMPA currents (See Fig. 1B) and would not likely contribute to the TNFα-induced increase in the amplitude of the mEPSC. Regardless, this latter finding associated with glutamate is consistent with the earlier demonstration that TNFα increased firing of CeA neurons (Knapp et al., 2011). Because the frequency of mEPSCs was not increased, the most likely explanation for the increased amplitude of the mEPSCs by TNFα was a direct TNFα action on the neuron to increase the number of glutamate-receptors on the cellular membrane at the synapse (Collingridge et al., 2004; Heine et al., 2008; Rainey-Smith et al., 2010; Santello and Volterra, 2012; Stellwagen et al., 2005). In support of this increased amplitude of mEPSCs by TNFα being due to receptor trafficking, Beattie et al. (2002) demonstrated that glial TNFα induced surface expression of AMPA receptors on neurons—a finding supported by Stellwagen et al. (2005) who demonstrated directly that TNFα increased AMPA-receptor number on hippocampal neurons. One explanation for the TNFα-induced increase in amplitude of mEPSCs from CeA neurons would be an intracellular activation of a second messenger that would allow AMPA receptors to pass to the cell surface. In support of this supposition, Stellwagen et al. (2005) found that the TNFα-induced increase in amplitude of mEPSCs was dependent upon AMPA receptor transfer to the cell surface by a phosphatidylinositol-3-kinase (PI-3-K)-dependent process. To assess if the TNFα-induced increased amplitude of mEPSCs from CeA neurons was also related to this second messenger system, a PI-3-K antagonist and a P38-MAP-kinase inhibitor were tested against the TNFα-induced increase in the mEPSC amplitude from CeA neurons. In agreement with TNFα results from hippocampal neurons (Stellwagen et al., 2005), the PI-3-K-antagonist blocked the increase in mEPSC-amplitude of CeA-neurons without the P38-MAP-kinase inhibitor altering this response. Thus, this finding is consistent with the TNFα-induced increase in amplitude of mEPSCs from CeA neurons being dependent upon a PI-3-kinase process.
Accompanying the TNFα influence on glutamate function was confirmation that TNFα-increases the frequency of bicuculline-sensitive mIPSCs (Knapp et al., 2011). This observation was extended by demonstrating that this increase in mIPSC frequency by TNFα was dose-dependent and was not accompanied by a change in mIPSC amplitude. Thus, while TNFα elevates GABA-release, it does not influence post-synaptic sensitivity to GABA. An issue to be explored in the future is whether facilitation of GABA function by TNFα has any relation to the degree to which TNFα increases neural activity of CeA neurons (Knapp et al., 2011). Regardless, this increased frequency of mIPSCs from CeA neurons induced by TNFα is contrary to the reported reduction in mIPSC-frequency induced by TNFα from hippocampal neurons (Stellwagen et al., 2005) and the TNFα-induced increase in synaptic GABAA receptors on spinal-cord neurons (Stück et al. (2012). These reported differences between brain regions suggest that TNFα may have distinct effects on GABA function depending upon the brain region investigated—an aspect that should be explored further.
Based upon previous findings that systemic administration of a CRFR-antagonist prevented the in vivo action of stress and TNFα to facilitate ethanol-withdrawal anxiety (Breese et al., 2004; Knapp et al., 2011), the present study examined whether a CRFR-antagonist would influence any of the neural actions induced by TNFα. Of particular note is that the action of a general CRFR-antagonist did not alter the TNFα-induced increase in amplitude of mEPSCs or the reduced current injection required to induce spiking—both measures being related to glutamate function. In contrast to this lack of effect of the CRFR-antagonist on these measures of glutamate function, the general CRFR-antagonist and a CRF1R-antagonist prevented the TNFα-induced increase in mIPSC-frequency, whereas the CRF2R-antagonist was ineffective in preventing this TNFα-induced change. Hence, this experimental series provides evidence that CRF acts on a CRF1R to elevate the frequency of the mIPSCs induced by TNFα—a finding consistent with the demonstration that CRF applied to CeA neurons increases the frequency of mIPSCs (Roberto et al., 2010). Consequently, the CRF1R-antagonist blockade of the increased frequency of mIPSCs induced by TNFα is compatible with TNFα increasing a CRF synaptic presence that influences CRF-receptors to increase GABA release (see Roberto et al., 2010). Interestingly, while the CRFR-antagonists blocked the ability of TNFα to increase mIPSCs, the CRFR-antagonist had no effect on mIPSCs in the absence of TNFα. This latter finding indicates that under the present conditions ambient levels of CRF were not sufficient to influence GABA release (mIPSCs). The involvement of CRF in the increased mIPSC frequency in the CeA may explain why TNFα does not increase GABA release in all brain regions (see Stellwagen et al., 2005). An assessment for the future is whether the CRF1R-antagonist blockade of the TNFα-induced increase in mIPSCs has any relationship to CRF1Rs supporting stress and cytokine facilitation of ethanol-withdrawal anxiety (Breese et al., 2004, 2008; Knapp et al., 2011).
While the data collected with the CRFR-antagonists provide convincing evidence that CRF is involved in the TNF-induced increase in frequency of mIPSCs from CeA neurons, the mechanism for this CRF involvement in the presence of tetrodotoxin (TTX) was open to clarification. Because the presence of TTX logically eliminates TNFα from stimulating action-potentials to release CRF from neurons to increase mIPSC-frequency, one possible explanation considered for this present outcome was that the CRFR-antagonists prevent the TNFα–induced increase in frequency of mIPSCs by blocking the action of CRF released from cells other than neurons. Consistent with this latter possibility is the demonstration that astrocytes are reported to contain CRF (Tian and Bishop, 2003) as well as CRF-binding protein (Maciejewski et al., 1996; McClennen and Seasholtz, 1999). Further, a number of other peptides and/or their mRNAs have been localized in a variety of glial cell types including astrocytes (Ubink et al., 2003). Based upon the concept induced by Santello et al. (2011; 2012) that TNFα can enhance the abilty of astrocytes to release transmitter, it was reasoned that TNFα acting on TNFα-receptors on astrocytes or other glial elements (Yan et al., 2008), reported to contain CRF (Tian and Bishop, 2003), might be involved in making CRF available to account for the CRFR-antagonists preventing the TNFα-induced frequency increase in mIPSCs. Whereas this potential influence of TNFα on glia to influence CRF availability could be either direct or indirect (Santello et al., 2011), in either case glia would be involved in this action of TNFα.
Because minocycline can disrupt activation of microglia and astrocytes (McAllister and Miller, 2010), brain slices were pre-incubated with minocycline to test if the TNFα-induced increase in mIPSC-frequency would be affected. Following minocycline exposure, TNFα did not cause a significant increase in the mIPSC-frequency. Therefore, TNFα activation of astrocytes to initiate release CRF could be a part of the mechanism by which CRF is involved in the TNFα-induced increase in frequency of mIPSCs—a finding consistent with the many other peptides reported to be present in astrocytes (see Ubink et al., 2003). This potential involvement of CRF release from glia supporting the TNFα-induced increase in mIPSCs is illustrated in Fig. 8.
Fig. 8. Possible mechanism of TNFα-induced-CRF-dependent GABA release.

As noted, TNFα could act on a glial cell to induce glial release of CRF. This is consistent with the TNFα-induced release of GABA being blocked either by blocking glial activation with minocycline or by blocking the effect of CRF at its receptor on the presynaptic terminal with a CRF-1-antagonist [X]. Noted is that TNFα also acts on the post-synaptic neuron to activate the PI-3-K to allow amplitude of mEPSCs to be increased (Fig. 3)
In conclusion, the present electrophysiological studies provide convincing evidence that TNFα is capable of having an effect on neural function in the CeA. Because CCL2 has also been demonstrated to increase firing of CeA neurons (unpublished data), it would appear that cytokines in general may have the capability of influencing neural function in this brain site. Furthermore, the means by which TNFα increases the amplitude of mEPSCs was addressed. The present study did not find an effect of a CRFR-antagonist on induction of action-potentials by current injection, even though it blocked the effect of TNFα on GABA release in the CeA. A mechanism by which the CRFR-antagonist blocked the TNFα-induced increase in frequency of mIPSCs led to evidence with minocycline that CRF may be released from glia. Nonetheless, as intriguing as the present minocycline results are for implicating glial involvement of CRF in TNFα action on mIPSCs, one must consider that minocycline has other actions that might account for this drug preventing the increase in GABA release induced by TNFα (see Soczynska et al., 2012).
Even though cytokines released by stress in brain have the capability of influencing neural function, it has yet to be determined how stress in a “sterile” environment (i.e., in the absence of infection) can increase these cytokines in brain. This latter aspect too must be addressed before the full impact of stress in relation to cytokine influences on neural function is understood. The means by which cytokines are involved in stress is important because cytokines and stress are implicated in pain (Fu and Neugebauer, 2008; Vachon-Presseau et al., 2013; Watkins et al., 2001), sleep (Krueger et al., 2007; Churchill et al 2008), depression (Caspi et al., 2003; Hammen, 2005), anxiety (Breese et al., 2008; Shin and Liberzon, 2010) and drug addiction (Breese et al., 2011; Koob, 2008). A further understanding of the unknowns by which cytokines contribute to stress to alter neural function may allow a better appreciation of the complexity by which stress induces changes in neural transmission that contribute to the functional changes associated with stress-linked central disorders.
Acknowledgments
Funding: This work was supported by the National Institutes of Health, National Institute on Alcoholism and Alcohol Abuse [AA11605, AA14949 & AA017462] and Bowles Center for Alcohol Studies.
The authors acknowledge assistance of Dr. Darin Knapp in preparation of illustrations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler M, Geller E, Chen X, Rogers T. Viewing chemokines as a third major system of communication in the brain. AAPS J. 2006;7:E865–870. doi: 10.1208/aapsj070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFα. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Behan DP, Maciejewski D, Chalmers D, De Souza EB. Corticotropin releasing factor binding protein (CRF-BP) is expressed in neuronal and astrocytic cells. Br Res. 1995;698:259–264. doi: 10.1016/0006-8993(95)01014-m. [DOI] [PubMed] [Google Scholar]
- Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci Lett. 1994;172:151–154. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- Belkouch M, Dansereau MA, Réaux-Le Goazigo A, Van Steenwinckel J, Beaudet N, Chraibi A, Melik-Parsadaniantz S, Sarret P. The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gβγ-dependent mechanism. J Neurosci. 2011;31:18381–18390. doi: 10.1523/JNEUROSCI.3386-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Knapp DJ, Overstreet DH, Navarro M, Wills TA, Angel RA. Repeated lipopolysaccharide (LPS) or cytokine treatments sensitize ethanol withdrawal-induced anxiety-like behavior. Neuropsychopharmacology. 2008;33:867–876. doi: 10.1038/sj.npp.1301468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Knapp DJ, Overstreet DH. Stress sensitization of ethanol withdrawal-induced reduction in social interaction: inhibition by CRF-1 and benzodiazepine receptor antagonists and a 5-HT1A receptor agonist. Neuropsychopharmacology. 2004;29:470–482. doi: 10.1038/sj.npp.1300282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Overstreet DH, Knapp DJ. Conceptual framework for the etiology of alcoholism: a “kindling”/stress hypothesis. Psychopharmacology. 2005a;178:367–380. doi: 10.1007/s00213-004-2016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Overstreet DH, Knapp DJ, Navarro M. Prior multiple ethanol withdrawals enhance stress-induced anxiety-like behavior: Inhibition by CRF-1 and benzodiazepine-receptor antagonists and a 5-HT1A receptor antagonist. Neuropsychopharmacology. 2005b;30:1662–1669. doi: 10.1038/sj.npp.1300706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Sinha R, Heilig M. Chronic alcohol neuroadaptation and stress contribute to susceptibility for alcohol craving and relapse. Pharmacol Ther. 2011;129:149–171. doi: 10.1016/j.pharmthera.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaere C, Banisadr G, Desarmènien MG, Mechighel P, Kitabgi P, Rostène WH, Mèlik Parsadaniantz S. The chemokine SDF-1/CXCL12 modulates the firing pattern of vasopressin neurons and counteracts induced vasopressin release through CXCR4. Proc Natl Acad Sci. 2006;103:8221–8226. doi: 10.1073/pnas.0602620103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JTR, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:957–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington HL, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: Moderation by a polymorphism in the 5-HT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Churchill L, Rector DM, Yasuda K, Fix C, Rojas MJ, Yasuda T, Krueger JM. Tumor necrosis factor alpha: activity dependent expression and promotion of cortical column sleep in rats. Neuroscience. 2008;156:71–80. doi: 10.1016/j.neuroscience.2008.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De A, Krueger JM, Simasko SM. Tumor necrosis factor alpha increases cytosolic calcium responses to AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res. 2003;981:133–42. doi: 10.1016/s0006-8993(03)02997-4. [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Wang J, Ando T. Effects of cytokines on cerebral neurotransmission: comparison with the effects of stress. Adv Exp Med Biol. 1999;461:117–127. doi: 10.1007/978-0-585-37970-8_8. [DOI] [PubMed] [Google Scholar]
- Emch GS, Hermann GE, Rogers RC. TNFα activates solitary nucleus neurons responsive to a gastric distension. Am J Physiol GS Liver Physiol. 2000;279:G582–586. doi: 10.1152/ajpgi.2000.279.3.G582. [DOI] [PubMed] [Google Scholar]
- Emch GS, Hermann GE, Rogers RC. TNFα induced c-Fos generation in the nucleus of the solitary tract is blocked by NBQX and MK-801. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1394–1400. doi: 10.1152/ajpregu.2001.281.5.R1394. [DOI] [PubMed] [Google Scholar]
- Fu Y, Neugebauer V. Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci. 2008;28:3861–3876. doi: 10.1523/JNEUROSCI.0227-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani F, Hader W, Yong VW. Minocycline attenuates T cell and microglia activity to impair cytokine production in t cell-microglia interaction. J Leukocyte Biol. 2005;78:135–143. doi: 10.1189/jlb.0804477. [DOI] [PubMed] [Google Scholar]
- Gruol DL, Nelson TE. Purkinje neuron physiology is altered by the inflammatory factor interleukin-6. The Cerebellum. 2005;4:198–205. doi: 10.1080/14734220500199987. [DOI] [PubMed] [Google Scholar]
- Hammen C. Stress and Depression. Annu Rev Clin Psychol. 2005;1:293–319. doi: 10.1146/annurev.clinpsy.1.102803.143938. [DOI] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Béïque JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of post-synaptic AMPAR tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Hebert SL, Van Meter MJ, Holmes GM, Rogers RC. TNFα-p55 receptors: medullary brainstem imuunocytochemical localization in normal and vagus nerve-transected rats. Brain Res. 2004;1004:156–166. doi: 10.1016/j.brainres.2003.11.078. [DOI] [PubMed] [Google Scholar]
- Holmes GM, Hebert SL, Rogers RC, Hermann GE. Immunocytochemical localization of TNF type 1 and 2 receptors in the rat spinal cord. Brain Res. 2004;1025:210–219. doi: 10.1016/j.brainres.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Huang MM, Overstreet DH, Knapp DJ, Angel R, Wills TA, Navarro M, Rivier J, Vale W, Breese GR. Corticotropin-Releasing Factor CRF sensitization of ethanol withdrawal-induced anxiety-like behavior is brain site specific and mediated by CRF-1 Receptors: Relation to stress-induced sensitization. J Pharmacol Exp Ther. 2010;332:298–307. doi: 10.1124/jpet.109.159186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki TK, Muscatell KA, Irwin MR, Cole SW, Eisenberger NI. Inflammation selectively enhances amygdala activity to socially threatening images. Neuroimage. 2012;59:3222–3226. doi: 10.1016/j.neuroimage.2011.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: Distinct and overlapping role of IL1-β. IL-6 and TNFα in regulating synaptic and neuronal activity in superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp DJ, Whitman BA, Wills TA, Angel RA, Overstreet DH, Criswell HE, Ming Z, Breese GR. Cytokine involvement in stress may depend on corticotrophin releasing factor to sensitize ethanol withdrawal anxiety. Brain Behav Immun. 2011;25:S146–54. doi: 10.1016/j.bbi.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Rector DM, Churchill L. Sleep and cytokines. In: Vgontzas An., editor. Sleep medicine clinics of North America: sleep, sleep disorders and hormones. 2007. pp. 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukáts B, Egyed R, Karádi Z. Single neuron activity changes to interleukin-1β in the orbitalfrontal cortex of the rat. Brain Res. 2005;1038:243–246. doi: 10.1016/j.brainres.2005.01.038. [DOI] [PubMed] [Google Scholar]
- Lindqvist D, Janelidze S, Hagell P, Erhardt S, Samuelsson M, Minthon L, Hansson O, Björkqvist M, Träskman-Bendz L, Brundin L. Interleukin-6 is elevated in CSF of suicide attempters and relate to symptom severity. Biol Psychiatry. 2009;66:287–292. doi: 10.1016/j.biopsych.2009.01.030. [DOI] [PubMed] [Google Scholar]
- Maciejewski D, Crowe PD, De Souza EB, Behan DP. Regulation of coricotropin-releasing factor-binding protein expression in cultured rat astrocytes. J Pharmac Exp Ther. 1996;278:455–461. [PubMed] [Google Scholar]
- McAllister JP, Miller JM. Minocycline inhibits glial proliferation in the H-Tx rat model of congenital hydrocephalus. Cerebrospinal Fluid Res. 2010;7:7–8. doi: 10.1186/1743-8454-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClennen SJ, Seasholtz AF. Transcription regulation of corticotropin-releasing hormone-binding protein gene expression in astrocyte cultures. Endocrinology. 1999;140:4095–4103. doi: 10.1210/endo.140.9.6978. [DOI] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Niquet J, Wasterlain CG. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis. 2013;54:225–238. doi: 10.1016/j.nbd.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikodemova M, Watters JJ, Jackson SJ, Yang SK, Duncan ID. Minocycline down regulates MHCII expression in microglia and macrophages through inhibition of IRF-1 and protein kinase C (PKC)α/βII. J Biol Chem. 2007;282:15208–15216. doi: 10.1074/jbc.M611907200. [DOI] [PubMed] [Google Scholar]
- Nutile-McMenemy N, Elfenbein A, DeLeo JA. Minocycline decreases in vitro microglial motility, beta1-integrin, and Kv1.3 channel expression. J Neurochem. 2007;103:2035–2046. doi: 10.1111/j.1471-4159.2007.04889.x. [DOI] [PubMed] [Google Scholar]
- Oh SB, Cho C, Miller RJ. Electrophysiological analysis of neural chemokine receptors. Methods. 2003;29:335–344. doi: 10.1016/s1046-2023(02)00357-2. [DOI] [PubMed] [Google Scholar]
- Porterfield VM, Zimomra ZR, Caldwell EA, Camp RM, Gabella KM, Johnson JD. Rat strain differences in restraint stress-induced brain cytokines. Neuroscience. 2011;188:48–54. doi: 10.1016/j.neuroscience.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager G, Hadamitzky M, Engler A, Doenlen R, Wirth T, Pacheco-López G, Krügel U, Schedlowski M, Engler H. Amygdaloid signature of peripheral immune activation by bacterial lipopolysaccharide or Staphylococcal enterotoxin B. J Neuroimmune Pharmacol. 2012 doi: 10.1007/s11481-012-9373-0. In press. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuro-inflammation and progressive. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey-Smith SR, Andersson DA Williams RJ, Rattray M. Tumor necrosis factor-α induces rapid reduction in AMPA receptor-mediated calcium entry in motor neurons by increasing cell surface expression of the GluR2 subunit: relevance to neurodegeneration. J Neurochem. 2010;113:692–703. doi: 10.1111/j.1471-4159.2010.06634.x. [DOI] [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, Cottone P, Madamba SG, Stouffer DG, Zorrilla EP, Koob GF, Siggins GR, Parsons LH. Corticotopin-releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry. 2010;67:831–839. doi: 10.1016/j.biopsych.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostène W, Kitabgi P, Parsadaniantz SM. Chemokines: a new class of neuromodulator? Nat Rev Neurosci. 2007;8:895–903. doi: 10.1038/nrn2255. [DOI] [PubMed] [Google Scholar]
- Santello M, Bezzi P, Volterra A. TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron. 2011;69:988–1001. doi: 10.1016/j.neuron.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Santello M, Volterra A. TNFα in synaptic function: switching gears. TINS Trends Neurosci. 2012;35:638–547. doi: 10.1016/j.tins.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Shin LM, Liberzon I. The neurocircuitry of fear, stress and anxiety disorders. Neuropsychopharmacology. 2010;35:169–191. doi: 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soczynska JK, Mansur RB, Brietzke E, Swardfager W, Kennedy SH, Woldeyohannes HO, Powell AM, Manierka MS, McIntyre RS. Novel therapeutic targets in depression: minocycline as a candidate treatment. Behav Brain Res. 2012;235:302–317. doi: 10.1016/j.bbr.2012.07.026. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by TNFα. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stück ED, Christensen RN, Huie JR, Tovar CA, Miller BA, Knout YS, Bresnahan JC, Beattie MS, Ferguson AR. Tumor necrosis factor alpha mediates GABAA receptor trafficking to the plasma membrane of spinal cord neurons In Vivo. Neural Plats. 2012 doi: 10.1155/2012/261345. Article number-261345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabarean IV, Koran H, Bartram T. Interleukin-1β induces hyper-polarization and modulates synaptic inhibition in preoptic and anterior hypothalamic neurons. Neuroscience. 2006;141:1685–1695. doi: 10.1016/j.neuroscience.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Tian JB, Bishop GA. Frequency-dependent expression of corticotopin releasing factor in rat’s cerebellum. Neuroscience. 2003;121:363–377. doi: 10.1016/s0306-4522(03)00493-7. [DOI] [PubMed] [Google Scholar]
- Ubink R, Calza L, Hökfelt T. Neuro’-peptides in glia: focus on NPY and galanin. Trends Neurosci. 2003;26:604–609. doi: 10.1016/j.tins.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Vachon-Presseau E, Martel M-O, Roy M, Caron E, Albouy G, Marin M-F, Plante I, Sullivan MJ, Lupien SJ, Rainville P. Acute stress contributes to individual differences in pain and pain-related brain activity in healthy and chronic pain patients. J Neurosci. 2013;33:6826–6833. doi: 10.1523/JNEUROSCI.4584-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkovic L, Bockaert J, Jacque C. “Inflammatory” Cytokines: Neuromodulators in Normal Brain? J Neurochem. 2000;74:457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force of pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Whitman B, Knapp DJ, Werner DF, Crews FT, Breese GR. The Cytokine-mRNA Increase Induced by Withdrawal from Chronic Ethanol in the Sterile Environment of Brain is mediated by CRF and HMGB1 Release. Ethanol Clin Exp Res. 2013 doi: 10.1111/acer.12189. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman B, Knapp DJ, Zimomra Z, Breese GR. HMGB1-signaling supports stress-induction of cytokine-mRNAs after chronic-alcohol withdrawal, but not induction of cytokine-mRNAs in controls. Brain Behavior and Immunity. 2013 To be submitted. [Google Scholar]
- Wilkinson MF, Mathieson WB, Pittman QJ. Interleukin-1β has excitatory effects on neurons of the BNST. Brain Res. 1993;625:342–346. doi: 10.1016/0006-8993(93)91079-8. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, Nelson RJ, Godbout JP, Sheridan JF. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. 2011;31:6277–6288. doi: 10.1523/JNEUROSCI.0450-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Xia C, Niu S, Cheng C, Shao X, Shen A. The role of TNFα and its receptors in production of β-1,4-galactosyltransferase I mRNA by rat primary type-2 astrocytes. Cell Mol Neurobiol. 2008;28:223–236. doi: 10.1007/s10571-007-9182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]