

Histones are the main protein components of chromatin. They undergo numerous posttranslational modifications, including phosphorylation, acetylation, and methylation, which often affect each other[1, 2]. Such cross-regulation of histone modification is known to play a central role in many physiological processes. Phosphorylation of histone H3 at serine 10 (H3S10) and acetylation of N-terminal lysine residues in histone H3 are representative markers of transcriptional activation in eukaryotic cells. Many in vivo studies have suggested a possible link between these modifications[3-5], but this association is not yet established. Early in vitro studies using small synthetic H3 peptides provided mixed and conflicting results regarding the association between phosphorylation and acetylation[3, 4, 6], and recent peptide experiments have failed to find any correlation[7]. However, since synthetic peptides differ from the physiological substrates that undergo modifications, they can hardly represent the real physiological interactions between chromatin and modifying enzymes. Such peptides often show more than a thousand-fold lower binding abilities for modifying proteins compared to nucleosomal substrates[6]. Inconsistent results and a lack of direct evidence have led to the development of two opposing models (synergistic and independent) for these modifications[6, 8]. Thus, despite extensive studies, the correlation between phosphorylation and acetylation in histone H3 remains unclear.
Recombinant proteins with specific modifications can be generated by chemical ligation[9], but this method is restricted only to N- or C-terminal modification of a narrow range of proteins that lack cysteine residues and tolerate chemical treatment. In contrast, the co-translational approach is free of such restrictions, and applicable to a wide range of proteins[10-12]. We recently reported the genetic expansion of Escherichia coli with phosphoserine (Sep)[13]. Since serine phosphorylation is one of the most abundant post-translational modifications and affects many key cellular processes, including signaling and metabolism, an efficient method enabling site-specific Sep incorporation will be highly useful in answering many fundamental biological questions. The existing system, however, is inefficient; it generates only minute amounts of Sep proteins (e.g., 1 μg of fully active MEK1 per L culture)[13], which limits the general use of this method. Recently, a strain with enhanced Sep incorporation was made; however, cell viability was critically impaired[14].
Here, we present a much improved system of site-specific Sep incorporation by molecular evolution of phosphoseryl-tRNA synthetase (SepRS) and redesign of elongation factor (EF-Tu) (Scheme 1). By co-expressing these evolved orthogonal enzymes/proteins in the well known E. coli strain BL21(DE3), we produced mg quantities of recombinant Sep-containing histones. This then allowed us to investigate whether H3S10 phosphorylation affects the acetylation of H3 N-terminal lysine residues in order to explore the crosstalk mechanism underlying phosphorylation and acetylation.
Scheme 1.
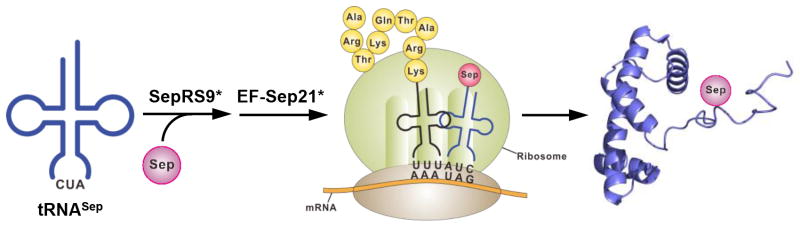
A facile strategy for producing recombinant histones with selective serine phosphorylation. The SepRS and EF-Tu mutants that were redesigned in this work are indicated with asterisks.
For efficient co-translational Sep incorporation we first attempted to improve tRNASep binding of Methanococcus maripaludis (Mmp) SepRS (Scheme 1), since mutations (G34C and C35U) introduced in the anticodon region of tRNASep caused a dramatic drop in the aminoacylation activity of Mmp SepRS (<5% in terms of initial velocity)[13, 15]. Based on the Archeaglobus fulgidus SepRS:tRNACys complex structure[16] (Figure 1a), we selected four equivalent residues (E412, E414, P495, and I496) in the anticodon-binding region of Mmp SepRS for complete randomization. Since the E. coli strain carrying Mmp SepRS, tRNASep and EF-Sep had a moderate background level of chloramphenicol (Cm) resistance[13], we generated a derivative of EF-Sep to facilitate the efficient selection of Mmp SepRS variants. We found that EF-Sep67S carrying Ser at 67 showed decreased Cm resistance (Figure 1a). Next, a library of MmpSepRS variants (1.6 × 106) was introduced into E coli TOP10ΔSerB carrying tRNASep and EF-Sep67S, and subjected to CAT-based selection, as previously described[13, 17]. Around 4,000 clones were collected from the selection agar plates. Molecular evolution (by error-prone PCR and DNA shuffling)[18] of these primary clones yielded 300 positive clones. The best clone (designated SepRS6) showed an IC50 of 25 ug/mL (Figure 1a). Additional DNA shuffling was conducted using these clones, and around 100 positive clones were selected and checked for Cm resistance. The best clone from the second shuffling (SepRS9) was found to exhibit an IC50 of 40 ug/mL (Figure 1a). SepRS9 has four mutations in the anticodon-binding region (E412S, E414I, P495R, and I496R) and three additional mutations (K356E, N361D, and L590I). The amino acid sequences of representative clones from each evolutionary step are listed in the Supplementary Information (Figure S1).
Figure 1.

Engineering of SepRS and EF-Sep for Sep incorporation. (a) (Left) Model of the anticodon-binding region of Archeaglobus fulgidus SepRS bound to tRNACys based on PDB file 2DU6. The mutations in the final mutant, SepRS9, are indicated with arrows. The numbering of amino acid sequence is based on A. fulgidus SepRS. (Right) In vivo aminoacylation activities of wild-type SepRS and the best mutants obtained from the first and second evolutionary steps. In vivo synthesis of chloramphenicol acetyltransferase (CAT) carrying an amber mutation by EF-Sep67S, tRNASep, and various types of SepRS was measured by their IC50 values. (b) (Left) The amino acid-binding pocket of E. coli EF-Tu complexed with Phe-tRNAPhe (adopted from the PDB file 1OB2). The mutations in EF-Sep are shown and an additional mutation generated in the final EF-Sep21 is indicated with an arrow. (Right) In vivo aminoacylation activity of various EF-Tu variants. (c) In vivo synthesis of CAT results from combining the best SepRS9 mutant and the engineered EF-Sep21.
As a second aim, we wanted to enhance the Sep-binding ability of EF-Sep (Scheme 1). First we tested the role of mutations that make up EF-Sep (H67R, E216N, D217G, F219Y, T229S, and N274W) by changing them to Ala (Figure 1b). Whereas the S229A (EF-Sep4) and W274A (EF-Sep5) mutations had modest effects, the Y219A mutation (EF-Sep3) dramatically reduced the activity of EF-Sep (Figure 1b). The H67 residue of EF-Tu is highly conserved across prokaryotic species, highlighting its role as a general binder for 20 natural amino acids. However, EF-Sep has Arg at this position, and the R67A mutation (EF-Sep1) seriously impaired Sep binding (Figure 1b). When R67 of EF-Sep was mutated back to His, Sep incorporation decreased substantially, indicating that R67 plays an essential role as a specific binder of Sep. Interestingly, when N216 was mutated to Ala, the resulting EF-Sep2 mutant cells displayed a large increase in Cm resistance, with the IC50 increasing from 30 to 80 μg/mL (Figure 1b). We thus performed random mutation at this position in an attempt to generate further improved variants. EF-Sep21, which carried mutation N216V, exhibited the highest in vivo activity (IC50 of 120 μg/mL) (Figure S2) and was selected for further study. Finally, we were able to obtain significantly enhanced in vivo Sep incorporation activity (IC50 value of 180 μg/mL) by combining the evolved SepRS9 with the mutant EF-Sep21 (Figure 1c). When EF-Sep21 was removed, the Cm resistance decreased to the basal level (IC50 of 3 μg/mL), illustrating the Sep selectivity of the engineered SepRS9 and EF-Sep21.
We then tested whether our advanced Sep incorporation system could site-specifically place Sep in a histone. An amber codon was introduced into Ser10 of the Xenopus laevis histone H3. Using E. coli BL21(DE3) carrying our system (engineered SepRS9/tRNASep and EF-Sep21), we were able to obtain significant amounts (3 mg per L culture) of recombinant full-length histone H3 carrying N-terminal His6 tag (Figure 2a). Unlike the previous report[14], we did not observe any noticeable cell growth damage since we used general E. coli strain BL21(DE3). In experiments run without EF-Sep21, only trivial amounts of full-length H3 were obtained. The production of H3 was also insignificant in experiments run using wild-type SepRS and the original EF-Sep, illustrating the importance of the engineered SepRS9 and EF-Sep21 proteins (Figure 2a). Western blot analysis using anti-H3S10 phosphorylation (H3S10ph) antibodies confirmed that the produced protein had Sep at position 10 (Figure 2b and 2c). Homogenous serine phosphorylation of the modified histone H3S10ph was verified by matrix-assisted deionization/ionization time-of-flight (MALDI-TOF) mass spectrometry (Figure S3). These results collectively demonstrate that our system using the engineered pair SepRS9/tRNASep and EF-Sep21 enables high-yield production of recombinant proteins with selective and homogenous serine phosphorylation. When His6 tag-removed H3S10ph (Figure S4) was assembled with H2A, H2B, and H4 to form histone octamers, the modified H3 displayed an efficiency comparable to that of unmodified H3 (Figure 2b). When nucleosomal arrays were reconstituted with the assembled histone octamers onto DNA template using the salt-dilution or glycerol-gradient method, there was no notable difference in yield or efficiency (Figure 2c).
Figure 2.

Production of serine-phosphorylated histone H3S10ph and assembly of histone octamers and nucleosomes. (a) SDS-PAGE analysis of full-length recombinant H3 produced by the EF-Sep and SepRS variants. (b) Assembly of histone octamers using wild-type histone H3 and modified H3S10ph. The presence of Sep in the modified histone is verified by Western blot analysis with anti-H3S10ph antibodies. (c) Reconstitution of nucleosomal arrays having unmodified H3 or H3S10ph-containing octamers into DNA template. Free and bound DNA were analyzed by 0.8% agarose gel electrophoresis.
Next, we investigated the effect of H3S10 phosphorylation on the histone acetyltransferase (HAT) activity of recombinant Gcn5p or the SAGA complex, using serine-phosphorylated histone H3, histone octamers, and nucleosomal arrays (Figure 3a). Both Gcn5p and the SAGA complex exhibited reduced activity on phosphorylated histone H3 compared to unmodified H3 (Figure 3b). When histone octamers containing Sep were tested, however, the SAGA complex displayed comparable activities for the phosphorylated and unmodified substrates (Figure 3b). Since epigenetic regulation takes place at the chromatin level, we next examined the acetylation activity of SAGA for the more physiologically relevant chromatin substrate: nucleosomal arrays. Intriguingly, SAGA showed a noticeably higher activity on nucleosomal arrays with H3S10 phosphorylation compared to unmodified nucleosomal arrays (Figure 3b). Previous experiments with chemically ligated histone failed to find a link between phosphorylation and acetylation in H3[19]. Here, the use of modified nucleosomal arrays with genetically incorporated Sep clearly demonstrated that H3S10 phosphorylation stimulates SAGA-mediated acetylation.
Figure 3.

The effect of H3S10 phosphorylation on the acetylation of N-terminal lysine residues in H3. (a) Schematic showing the interactions between different levels of chromatin substrates and Gcn5p or SAGA. (b) The histone acetyltransferase (HAT) activity of Gcn5p and SAGA for histone, octamers, and nucleosomal arrays. The HAT activity of SAGA for nucleosomal arrays is stimulated by H3S10 phosphorylation. (c) The acetylation of individual lysine residues (H3K9, K14, K18, and K23) was detected by position-specific anti-acetyl lysine antibodies.
This differential effect of phosphorylation on acetylation at different levels of chromatin substrates became more obvious when we performed Western blotting with position-specific antibodies to observe the acetylation of individual modification sites (K9, K14, K18, and K23 in H3). Western blotting for acetylation in K9 or K14 was carried out after alkaline phosphatase treatment, to prevent occlusion by neighboring S10 phosphorylation[20] (Figure S5). Previous experiments with small peptides of H3 showed that Gcn5p mainly acetylates the K14 position, whereas SAGA enables extended lysine acetylation[21]. Our experiments with unmodified histone H3 showed that both Gcn5p and SAGA acetylated all of the tested lysine residues (Figure 3c). Consistent with our HAT results (Figure 3b), the lysine acetylation of histone H3 by Gcn5p or SAGA was greatly hindered by the phosphorylation of H3S10 (Figure 3c). However, when the modified histone H3S10ph was assembled with other histones to form octamers, the phosphorylation-induced repression of acetylation was almost released, with only K9 acetylation remaining slightly reduced (Figure 3c). Furthermore, when the phosphorylated histone lay within the context of a nucleosomal array, H3S10 phosphorylation significantly (up to three-fold) enhanced SAGA-mediated acetylation of the lysine residues, K14, K18, and K23 (Figure 3c). Thus, our results establish that H3S10 phosphorylation directly stimulates acetylation in H3 under physiologically relevant conditions, and that cooperative interactions between the SAGA complex and nucleosomal arrays are crucial for the synergistic communication between phosphorylation and acetylation in H3, which is not possible with small peptides or free histone (Figure 3a).
Here, we developed a facile general method for site-specific Sep insertion into recombinant histones through inspired redesign of the orthogonal translation system involving SepRS and EF-Tu. Using this approach, we demonstrate that H3S10 phosphorylation directly stimulates H3 acetylation by SAGA in the context of nucleosomal arrays. We anticipate that our strategy will open new avenues for studying the specific roles of histone phosphorylation and the mechanisms of crosstalk with other modifications.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea [2011-0031363, 2011-0020322, and 2012R1A2A2A02047051 to H.-S.P], the Stem Cell Research Program [2012M 3A9B 4027953 to D.L], and DARPA [Contract N660-12-C-4211 to D.S.] and NIH [GM22854 to D.S.]..
Contributor Information
Sangsik Lee, Department of Chemistry, Korea Advanced Institute of Science and Technology, 335 Gwahak-ro, Yuseong-gu, Daejeon 305-701 (Republic of Korea), Fax: (+82) 42-350-2810.
Seunghee Oh, Department of Biological Sciences, Korea Advanced Institute of Science and Technology, 335 Gwahak-ro, Yuseong-gu, Daejeon 305-701 (Republic of Korea), Fax: (+82) 42-350-2610.
Aerin Yang, Department of Chemistry, Korea Advanced Institute of Science and Technology, 335 Gwahak-ro, Yuseong-gu, Daejeon 305-701 (Republic of Korea), Fax: (+82) 42-350-2810.
Jihyo Kim, Department of Molecular Biophysics and Biochemistry, Yale University, 266 Whitney Ave. New Haven, CT, 06511 (USA).
Prof. Dieter Söll, Department of Molecular Biophysics and Biochemistry, Yale University, 266 Whitney Ave. New Haven, CT, 06511 (USA)
Prof. Daeyoup Lee, Email: daeyoup@kaist.ac.kr, Department of Biological Sciences, Korea Advanced Institute of Science and Technology, 335 Gwahak-ro, Yuseong-gu, Daejeon 305-701 (Republic of Korea), Fax: (+82) 42-350-2610
Prof. Hee-Sung Park, Email: hspark@kaist.ac.kr, Department of Chemistry, Korea Advanced Institute of Science and Technology 335, Gwahak-ro, Yuseong-gu, Daejeon 305-701 (Republic of Korea), Fax: (+82) 42-350-2810
References
- 1.Fischle W, Wang Y, Allis CD. Curr Opin Cell Biol. 2003;15:172. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 2.Latham JA, Dent SY. Nat Struct Mol Biol. 2007;14:1017. doi: 10.1038/nsmb1307. [DOI] [PubMed] [Google Scholar]
- 3.Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD. Mol Cell. 2000;5:905. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- 4.Lo WS, Trievel RC, Rojas JR, Duggan L, Hsu JY, Allis CD, Marmorstein R, Berger SL. Mol Cell. 2000;5:917. doi: 10.1016/s1097-2765(00)80257-9. [DOI] [PubMed] [Google Scholar]
- 5.Thomson S, Clayton AL, Mahadevan LC. Mol Cell. 2001;8:1231. doi: 10.1016/s1097-2765(01)00404-x. [DOI] [PubMed] [Google Scholar]
- 6.Fry CJ, Shogren-Knaak MA, Peterson CL. Cold Spring Harb Symp Quant Biol. 2004;69:219. doi: 10.1101/sqb.2004.69.219. [DOI] [PubMed] [Google Scholar]
- 7.Liokatis S, Stutzer A, Elsasser SJ, Theillet FX, Klingberg R, van Rossum B, Schwarzer D, Allis CD, Fischle W, Selenko P. Nat Struct Mol Biol. 2012;19:819. doi: 10.1038/nsmb.2310. [DOI] [PubMed] [Google Scholar]
- 8.Nowak SJ, Corces VG. Trends Genet. 2004;20:214. doi: 10.1016/j.tig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 9.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 10.Liu CC, Schultz PG. Annu Rev Biochem. 2010;79:413. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 11.Chin JW. EMBO J. 2011;30:2312. doi: 10.1038/emboj.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu WR, Wang YS, Wan W. Mol Biosyst. 2011;7:38. doi: 10.1039/c0mb00216j. [DOI] [PubMed] [Google Scholar]
- 13.Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Soll D. Science. 2011;333:1151. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinemann IU, Rovner AJ, Aerni HR, Rogulina S, Cheng L, Olds W, Fischer JT, Soll D, Isaacs FJ, Rinehart J. FEBS Lett. 2012;586:3716. doi: 10.1016/j.febslet.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hohn MJ, Park HS, O’Donoghue P, Schnitzbauer M, Soll D. Proc Natl Acad Sci U S A. 2006;103:18095. doi: 10.1073/pnas.0608762103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukunaga R, Yokoyama S. Nat Struct Mol Biol. 2007;14:272. doi: 10.1038/nsmb1219. [DOI] [PubMed] [Google Scholar]
- 17.Umehara T, Kim J, Lee S, Guo LT, Soll D, Park HS. FEBS Lett. 2012;586:729. doi: 10.1016/j.febslet.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 18.Park HS, Nam SH, Lee JK, Yoon CN, Mannervik B, Benkovic SJ, Kim HS. Science. 2006;311:535. doi: 10.1126/science.1118953. [DOI] [PubMed] [Google Scholar]
- 19.Shogren-Knaak MA, Fry CJ, Peterson CL. J Biol Chem. 2003;278:15744. doi: 10.1074/jbc.M301445200. [DOI] [PubMed] [Google Scholar]
- 20.Lau PN, Cheung P. Proc Natl Acad Sci U S A. 2011;108:2801. doi: 10.1073/pnas.1012798108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant PA, Eberharter A, John S, Cook RG, Turner BM, Workman JL. J Biol Chem. 1999;274:5895. doi: 10.1074/jbc.274.9.5895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.