

Abstract
Background
Chronic alcohol consumption causes persistent oxidative stress in the lung, leading to impaired alveolar macrophage (AM) function and impaired immune responses. AMs play a critical role in protecting the lung from particulate matter (PM) inhalation by removing particulates from the airway and secreting factors which mediate airway repair. We hypothesized AM dysfunction caused by chronic alcohol consumption increases the severity of injury caused by particulate matter inhalation.
Methods
Age- and sex-matched C57BL6 mice were fed the Lieber-DeCarli liquid diet containing either alcohol or an iso-caloric substitution (control diet) for 8 weeks. Mice from both diet groups were exposed to combustion derived PM (CDPM) for the final 2 weeks. AM number, maturation, and polarization status were assessed by flow cytometry. Noninvasive and invasive strategies were used to assess pulmonary function and correlated with histomorphological assessments of airway structure and matrix deposition.
Results
Co-exposure to alcohol and CDPM decreased AM number and maturation status (CD11c expression) while increasing markers of M2 activation (IL-4Rα, Ym1, Fizz1 expression and IL-10 and TGF-β production). Changes in AM function were accompanied by decreased airway compliance and increased elastance. Altered lung function was attributable to elevated collagen content localized to the small airways and loss of alveolar integrity. Intranasal administration of neutralizing antibody to TGF-β during the CDPM exposure period improved changes in airway compliance and elastance while reducing collagen content caused by co-exposure.
Conclusion
CDPM inhalation causes enhanced disease severity in the alcoholic lung by stimulating the release of latent TGF-β stores in AMs. The combinatorial effect of elevated TGF-β, M2 polarization of AMs, and increased oxidative stress impairs pulmonary function by increasing airway collagen content and compromising alveolar integrity.
Introduction
The most common causes of alcohol-related morbidity and mortality are organ damage and susceptibility to infection (Nelson and Kolls, 2002). In the lungs, chronic alcohol consumption has a profound and negative impact on immune cell function and the development of immune defenses (Happel and Nelson, 2005). In particular, alveolar macrophage (AM) dysfunction has been observed in chronic alcoholics (Mehta and Guidot, 2012). Specifically, chronic alcohol intake impairs AM terminal differentiation through the production of oxidative stress, leading subsequently to deficits in surfactant clearance, phagocytosis, and cytokine production (Brown et al., 2009; Joshi and Guidot, 2007).
AMs, in addition to controlling immune function, also participate in tissue remodeling and repair dependent upon their polarization status (Gibbons et al., 2011). In response to stimuli such as IL-1β/LPS, IL-4/IL-13, and TGF-β/IL-10, AMs attain distinct activation/polarization states (reviewed in (Gordon and Martinez, 2010). Polarizing stimuli subsequently alter expression of cytokine and scavenger receptors, co-stimulatory molecules and major histocompatibility complex, cytokine/chemokine production, and production of reactive oxygen/nitrogen species and enzymes. In the local tissue compartment, polarization thus influences recruitment of cytotoxic verses helper T-cells, oxidative stress, as well as fibroblast proliferation and matrix deposition. Thus AM polarization status dictates repair and remodeling processes in airways and alveoli and influences pulmonary function. Macrophage polarization has been well studied with focus on specific disease states such as bacterial infection, cancer, and asthma; however, research into how alcohol intake modulates polarization states in vitro and in vivo is in its infancy. Recent literature shows that M2 polarization occurs in AMs cultured in ethanol containing media (Brown and Brown, 2012). M2 (or alternatively) activated macrophages have been shown to play major roles in airway remodeling and pulmonary fibrosis, due to polarization by IL-4/IL-13 and the production of TGF-β/IL-10 (Gibbons et al., 2011; Homer et al., 2011; Sun et al., 2011). Though in vivo evidence suggests AM polarization may be impaired as a result of alcohol intake (Brown et al., 2009), how alcohol consumption may influence polarization in the immature AM pools has yet to be defined.
Limited epidemiological studies suggest an association between chronic alcohol consumption and increased airflow obstruction independent of smoking history (Emirgil and Sobol, 1977; Sisson et al., 2005), a disease state characterized by small airway remodeling and fibrosis accompanied by destruction of the alveoli. Though little is known regarding the relationships between chronic alcohol intake and pulmonary function (Sisson, 2007), chronic M2 macrophage polarization appears to play a prominent role in alcohol morbidity and mortality by stimulating remodeling/fibrosis (altering pulmonary function). However, links between the chronic M2 macrophage polarization in alcoholic lungs and damage to the airways/alveolar space are not well understood.
Exposure to airborne particulate matter (PM) derived from cigarette smoke, industrial/urban, or automotive sources has been consistently linked with altered pulmonary function (Balakrishna et al., 2011; Gauderman et al., 2007; Moshammer et al., 2006) and predisposition or exacerbation of pulmonary diseases such as COPD and pulmonary fibrosis (Kelly and Fussell, 2011). Though the risks of PM exposure to pulmonary health are clear, limited epidemiological studies have examined chronic alcoholism as a co-morbidity to PM exposure (Sisson, 2007). The relationships between alcoholism and PM exposure are particularly concerning given an estimated 18 million people in the US with alcohol use disorders (Grant et al., 2004) and the high percentage of individuals who heavily consume alcohol and smoke (Batel et al., 1995). Though comorbid studies of alcohol and smoking are limited with conflicting observations, combinatorial effects have been observed in two clinical studies showing increased glutathione oxidation (Yeh et al., 2007) and exacerbation of COPD (Saric et al., 1977) in smoking alcoholics.
Our group has shown that combustion-derived PM (CDPM), such as that found in cigarette smoke and airborne PM derived from industrial treatment and refinery processes and consumption of biomass fuels, produce oxidative stress in biological environments (Balakrishna et al., 2011; Thevenot et al., 2013). Inhalation of CDPM leads to pulmonary oxidative stress, airway remodeling, increased airways resistance, and decreased compliance. Since chronic alcohol ingestion increases expression of TGFβ and IL-13 (Brown and Brown, 2012), which are markers for M2 polarization; we hypothesized that M2 polarization and TGF-β production along with injury induced by CDPM inhalation would lead to fibrotic remodeling of the airways. We tested this hypothesis in C57BL/6 mice fed a 5.3% liquid alcohol diet or iso-caloric control diet for eight weeks, an alcohol feeding period sufficient to induce clinical indicators of chronic alcoholism including liver microsteatosis, altered AST/ALT levels, and an increased pulmonary ratio of oxidized to reduced glutathione and elevated 8-IPs. During the last two weeks of the protocol, mice were exposed to a laboratory generated, endotoxin-free CDPM (referred to as DCB-230; Balakrishna et al., 2011; Thevenot et al., 2013) or filtered air. DCB-230 exposure at the concentration (200 μg/m3), exposure period and duration employed in this study induces airway hyperresponsiveness analogous to epidemiological reports (Banauch et al. 2003).
Materials and Methods
Mouse Model of Chronic Alcohol Ingestion
Female C57BL/6 mice (8 weeks of age, Harlan) were fed Lieber-DeCarli liquid diet (32% calories from ethanol, 5.3% by volume) or non-alcohol iso-caloric control liquid diet (#710261 and #710028, respectively, Dyets Inc., Bethlehem, PA) for a total of eight weeks. Morning blood alcohol content measured after six consecutive weeks of diet protocol ranged from 0.13 – 0.17% (130 – 170 mg/dL). Model efficacy was verified by histological examination of liver microsteatosis and increased serum ALT (4-fold with respect to control diet).
Combustion-derived PM0.2 (DCB-230) – Synthesis and Dispersion
The environmentally-persistent free radical containing PM0.2, DCB-230, was used as a model combustion-derived PM (Balakrishna et al., 2011; Thevenot et al., 2013). DCB-230 synthesis and chemical characterization were performed in the laboratories of Drs. Barry Dellinger and Slawo Lomnicki (Louisiana State University, Baton Rouge, LA) as previously described (Lomnicki et al., 2008). Briefly, copper oxide was reacted with amorphous cab-o-sil (<200 nm in diameter). These particles were then exposed to 1,2 dichlorobenzene (DCB) vapor at 230°C chemisorbing DCB onto the CuO-silica matrix. Particles were then cooled, dried and stored under vacuum until use. DCB-230 had an aerodynamic diameter of 0.2 μm and was free of endotoxin.
DCB-230 Exposure Model
During weeks 6–8, while remaining on alcohol or control diets, mice were exposed for 14 consecutive days to DCB-230 (200 μg/m3, 30 min/day) or filtered room air. DCB-230 was suspended by sonication in 0.9% saline solution containing 0.02% Tween-80. Mice were exposed to nebulized DCB-230 by nose-only inhalation using the Scireq InExpose system (Montreal, Canada).
Invasive Measurements of Pulmonary Function
Twenty-four hours after the last exposure, lung function was assessed using the forced oscillation technique (FlexiVent, Scireq, Montreal, Canada). Mice were anesthetized by intraperitoneal injection of ketamine/xylazine, tracheostomized, and cannulated. Mice were mechanically ventilated using a computer controlled ventilator that delivers a tidal volume of 10 ml/kg at frequency of 120 breath/min. A positive end expiratory pressure (PEEP) of (3 cm H2O) was established and Primewave-8 perturbations were collected to obtain values for central airway resistance (Rn), tissue damping (peripheral distortion, G), and tissue elastance (H).
Histology and Immunohistochemistry
Blood was removed from the lungs by retrograde saline perfusion. A cannula was inserted into the trachea and lungs were inflated to a pressure of 25 cm H2O with Zn-formalin, fixed, and paraffin embedded. Sections (4 μm) were taken of the lungs and stained with hemotoxylin and eosin (H&E; to visualize morphology and structure) or Masson’s Trichrome (to visualize collagen and fibrosis). Histomorphometry of H&E stained frontal sections was used to assess mean linear intercept and mean alveolar surface as previously described (Lyerla et al., 2003; Martin et al., 2007) by an unbiased observer. Morphometric analysis of collagen content was performed as previously described (Podowski et al., 2012) and quantified using a hydroxyproline assay kit (Biovision, Mountain View, CA) according to manufacturer’s instructions.
Flow Cytometry
Perfused lungs were minced and enzymatically digested with collagenase and DNase to obtain a single cell suspension using the gentleMACs Octo Dissociator (Miltenyi). Red blood cells were removed with RBC lysis buffer (eBioscience). Live cells were determined by trypan blue staining. Cells were stained with surface antibodies to F4/80-Pacific Blue, CD11b-APC, CD11c-PE-Cy7, MHCII-APC-efluor780 (eBioscience). AMs were defined as viable, CD11c+, F4/80+ (gating strategy in supplemental material). M2 polarization in gated AMs was assessed using biotinylated IL-4Rα (BD) labeled with PE-streptavidin (eBioscience). TGF-β production in AMs was determined in permeabilized cells using LAP-PerCP-efluor710 (Clone TW7-16B4, specific for LAP+TGF, not LAP or pro-TGF).
Bronchoalveolar Lavage, AM Isolation, and Ex Vivo Polarization and DCB-230 Exposure
Bronchoalveolar lavage (BAL) was performed by inflating the lungs via the trachea with 1 ml of PBS 3% BSA. After centrifuging to remove cells, the BAL fluid was then flash frozen in liquid nitrogen and stored at −80°C until analysis. Cells recovered from the BAL (~ 0.2 × 106 cells from naïve control and alcohol diet) were suspended in RPMI with 10% FBS and antibiotics and plated for 1 hr at 37°C to select for alveolar macrophages as previously described (Trujillo et al., 2008). Prior to plating, differential staining showed ≥ 98% of BAL cells were macrophage/monocytes in all groups. Following plating, flow cytometry analyses showed 99% of recovered cells were alveolar macrophages.
For ex vivo exposure experiments, non-adherent cells were removed and media containing vehicle alone (0.9% saline with 0.02% Tween-80) or DCB-230 was added for 24 hrs. M1 polarization was induced by culture in media containing LPS (100 ng/mL) for 6 hrs. M2 polarization was induced by culture in media containing IL-4 (10 ng/mL) for 48 hrs. After exposure or polarization, adherent cells were processed for RNA.
Quantitative Real-Time PCR
Total RNA was isolated from ex vivo cultured AMs and lung preparations using TriZol (Invitrogen). Obtained RNA was converted to cDNA using Bio-Rad iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed on the Bio-Rad IQ5 using Bio-Rad SYBR green supermix with gene specific intron-spanning primers (Ym1, Arg1, Fizz1, Nos2, Tgf-b1). Single species amplicons for each primer pair were verified by dissociation curves. Relative expression was calculated using the delta-delta Ct method and normalized to the reference gene Hprt1.
TGF-β ELISA
BAL and whole lung homogenates were analyzed for TGF-β by ELISA (eBioscience).
TGF-β Neutralization
Local neutralization of TGF-β was achieved by intranasal administration of anti-TGF-β1,2,3 IgG1 (clone 1D11, R&D Systems) as previously described (Alcorn et al., 2007). Briefly, 50 μg of antibody was suspended in 30 μL of PBS 0.1% BSA and delivered intranasally (i.n.) to isofluorane anesthetized mice. Neutralizing antibody was administered on exposure day 0, 2, 6, 10, and 14.
Oxidative Stress
Total oxidized and reduced glutathione (GSSG/GSH) were measured in the BAL fluid using HPLC electrochemical detection as previously described (Thevenot et al., 2013).
Statistical Analysis
All results are expressed as the mean ± SEM and data are shown as means ± SEM from five mice and three independent experiments. A two-way ANOVA and Bonferroni select comparisons test were used to detect statistical differences between the control + air, control + DCB-230, alcohol + air, and alcohol + DCB-230. Values of p ≤ 0.05 was considered statistically significant and graphically depicted with abbreviated letters of respective control group used in the comparison: (ca), p ≤ 0.05 relative to control + air; (aa), p ≤ 0.05 relative to alcohol + air; (cd), p ≤ 0.05 relative to control + DCB-230.
Results
Chronic Alcohol Ingestion Impairs AM Maturation and Exposure to CDPM Increases Oxidative Stress and Suppresses Pulmonary AM Numbers
Since chronic alcohol ingestion alters AM polarization, maturation, and function, we hypothesized that chronic alcohol ingestion would alter AM numbers in the lungs after exposure to CDPM. DCB-230 exposure in control fed mice led to a significant increase in the total number of AMs in the lungs, however the total number of AMs in the lungs of alcohol fed, DCB-230 exposed mice were significantly lower with respect to mice fed a control diet and exposed to DCB-230 (Figure 1A). Depressed AM levels in the alcohol fed, DCB-230 exposed mice were accompanied by a dramatic decrease in glutathione availability (GSH/GSSG) (Figure 1B). AM maturation status was assessed by flow cytometry via mean fluorescent intensity of CD11c and CD11b on gated AMs. CD11c expression was significantly decreased on AMs from alcohol fed, DCB-230 exposed mice compared to alcohol or DCB-230 exposure alone (Figure 1C), indicating impaired terminal maturation. Conversely, CD11b expression was significantly higher on AMs from alcohol fed, air exposed mice compared with mice fed a control diet and exposed to air (Figure 1D). This increase was absent in alcohol fed, DCB-230 exposed mice compared with mice fed a control diet and exposed to DCB-230, suggesting a comparatively more immature phenotype due to chronic alcohol ingestion that is decreased with DCB-230 exposure. Interestingly, an increase in the percentage of CD11bhi AMs (F4/80+ CD11c+) was observed in only the alcohol fed mice exposed to DCB-230. This indicates that, while overall expression of CD11b in AMs is not changed in this group, a larger population of very highly expressing immature cells are present (Figure 1E).
Figure 1.

Chronic alcohol ingestion impairs AM maturation and exposure to CDPM increases oxidative stress and suppresses pulmonary AM numbers. Animals were administered control/alcohol diet and exposed to air/DCB-230 as described in text. (A) Total isolated AMs (CD11c+ F4/80+) per lung as determined from the percentage of AMs multiplied by total live isolated cells. (B) Reduced:Oxidized glutathione ratio in the BAL fluid. (C) Mean fluorescent intensity (MFI) values for CD11c and (D) median fluorescent intensity for CD11b on gated AM populations. (E) Percentage of CD11bhi AMs (F4/80+ CD11c+). Significance (p<0.05, Two-way ANOVA, Bonferroni post-tests) indicated above sample column by the letters of the reference control group of the comparison. Control+Air (ca), Alcohol+Air (aa), Control+DCB-230 (cd), Alcohol+DCB-230 (ad).
Chronic Alcohol Ingestion Induces M2 Polarization Which is Exacerbated Following Exposure to CDPM
We hypothesized that chronic alcohol ingestion would prime AMs for M2 polarization and that exposure to CDPM would further enhance M2 polarization. Expression of IL-10 and IL-4Rα were used to assess M2 polarization by flow cytometry, following antibody optimization using IL-4 stimulated MH-S macrophages in vitro. Polarization status of gated AMs from whole lungs was analyzed for each treatment group (Figure 2). IL-10 and IL-4Rα levels were unchanged on AMs from the lungs of mice fed control diet alone or exposed to DCB-230 (Figure 2A). However, both IL-10 and IL-4Rα were significantly upregulated in mice fed the alcohol diet compared to the control diet, with a further significant increase in alcohol fed mice exposed to DCB-230.
Figure 2.

Chronic alcohol ingestion induces M2 polarization which is exacerbated following exposure to CDPM. (A) Mean fluorescent intensity and representative histogram of IL-10 and IL-4Rα in AMs. (B) M2 gene expression in BAL-isolated AMs. (C) M1 (Nos2) and M2 (Arg1) gene expression in whole lung RNA preps. Significance (p<0.05, Two-way ANOVA, Bonferroni post-tests) indicated above sample column by the letters of the reference control group of the comparison. Control+Air (ca), Alcohol+Air (aa), Control+DCB-230 (cd), Alcohol+DCB-230 (ad).
M2 polarization was further confirmed by expression of Ym1 and Fizz1 in AMs obtained from the BAL (Figure 2B). Both markers were significantly increased following alcohol ingestion and exposure to DCB-230 compared to both alcohol and DCB-230 exposure alone, suggesting that DCB-230 exposure increased M2 polarization following alcohol ingestion. Since no change in M2 markers was observed following DCB-230 exposure in control fed mice, we examined expression of the M1 marker inducible nitric oxide synthase 2 (Nos2) and the M2 marker Arginase 1 (Arg1) (Figure 2C). Arg1 levels were not statistically different among treatment groups, however, increased Nos2 expression and decreased Arg1:Nos2 ratios are consistent with an M2 phenotype. Nos2 expression was significantly elevated in control fed, DCB-230 exposed mice resulting in a significant decrease in the Arg1:Nos2 ratio compared to mice fed an alcohol diet and exposed to DCB-230, indicating that chronic alcohol ingestion alters macrophage polarization associated with exposure to CDPM.
Chronic Alcohol Ingestion during Exposure to CDPM Increase Active TGF-β Levels in the Lung
To assess TGF-β-driven pulmonary fibrosis, we examined levels of TGF-β in the lungs and expression and production of TGF-β in AMs (Figure 3) from the lungs of mice fed an alcohol diet and exposed to DCB-230. In the lung, total TGF-β was elevated in mice fed an alcohol diet and exposed to DCB-230, with respect to both alcohol and DCB-230 alone (Figure 3A). To determine whether AMs were contributing to the increased levels of TGF-β in the lung, Tgf-β1 mRNA expression was quantified in AMs obtained from BAL (Figure 3B). Tgf-β1 expression was significantly elevated in AMs from mice fed an alcohol diet and exposed to DCB-230 with respect to all other groups of mice. To determine whether chronic alcohol ingestion increased latent levels of TGF-β, we performed intracellular staining for latency-associated peptide (LAP) in AMs and analyzed by flow cytometry. Chronic alcohol ingestion resulted in a significant increase in the percentage of LAP+ cells (Figure 3C). This suggests chronic alcohol intake increases latent levels of TGF-β in AMs and subsequent exposure to CDPM results in elevated expression of TGF-β and increased levels of active TGF-β in the lung.
Figure 3.
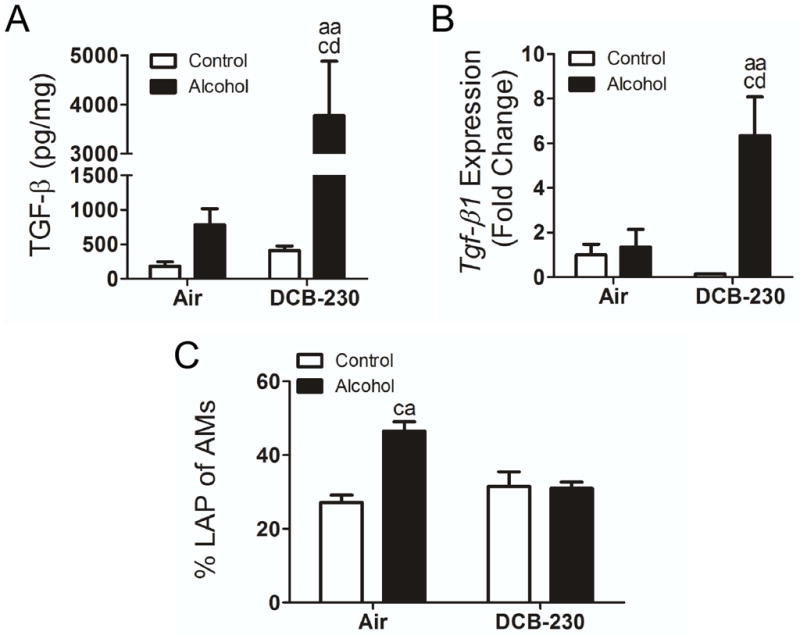
Chronic Alcohol ingestion during exposure to CDPM increases active TGF-β levels in the lung. (A) Total TGF-β in whole lung tissue homogenates. (B) Tgf-β gene expression in BAL-isolated AMs. (C) Percentage of LAP+ AMs in whole lung. Significance (p<0.05, Two-way ANOVA, Bonferroni post-tests) indicated above sample column by the letters of the reference control group of the comparison. Control+Air (ca), Alcohol+Air (aa), Control+DCB-230 (cd), Alcohol+DCB-230 (ad).
Exposure of Chronic Alcohol Fed Mice to CDPM Reduces Lung Function in a Manner Consistent with Fibrosis
Since DCB-230 exposure of mice fed a chronic alcohol diet led to increased TGF-β production, we hypothesized that co-exposure might result in pulmonary fibrosis worsening pulmonary function. Pressure-volume loops were collected to derive quasi-static compliance (Cst). Exposure to DCB-230 resulted in a significant decrease in Cst (associated with fibrosis), with no additional exacerbation due to chronic alcohol ingestion (Figure 4A). Newtonian Resistance (Rn; Figure 4B) was significantly reduced only in mice fed a chronic alcohol diet and exposed to DCB-230. Collectively, functional parameters in DCB-230 exposed mice trend toward fibrosis, due to loss of compliance and increased elastance (H; Figure 4C) regardless of diet. However, DCB-230 exposure in chronic alcohol fed mice appeared to elicit additional obstructive components as suggested by decreased Newtonian or central airway resistance.
Figure 4.

Exposure of chronic alcohol fed mice to CDPM reduces lung function in a manner consistent with fibrosis. Twenty-four hrs following the final exposure day, animals were assessed for baseline pulmonary function per methods section. (A) Quasi-static compliance (Cst) derived from pressure-volume loops. (B) Newtonian resistance (Rn) and (C) tissue elastance (H) derived from Primewave-8 perturbations. Significance (p<0.05, Two-way ANOVA, Bonferroni post-tests) indicated above sample column by the letters of the reference control group of the comparison. Control+Air (ca), Alcohol+Air (aa), Control+DCB-230 (cd), Alcohol+DCB-230 (ad).
Chronic Alcohol Ingestion and Exposure to CDPM Increases Airway Collagen
Pulmonary fibrosis was assessed by histological examination of collagen deposition in the lungs and quantification of lung collagen content using the hydroxyproline assay (Figure 5). Histological quantification resulted in an insignificant increase in airway collagen content only in the mice fed a chronic alcohol diet and exposed to DCB-230 (Figure 5A,B). However, hydroxyproline levels were significantly elevated only in the lungs of mice fed a chronic alcohol diet and exposed to DCB-230 (Figure 5C). These data suggest that the decreased compliance observed in mice fed an alcohol diet and exposed to DCB-230 is at least partially due to increased airway collagen deposition.
Figure 5.
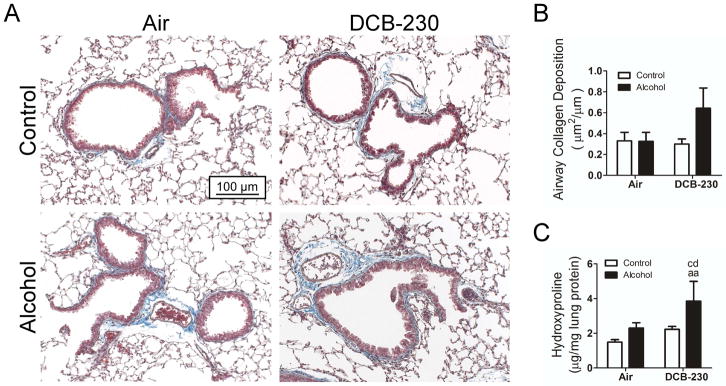
Chronic alcohol and exposure to CDPM increases airway collagen. (A) Representative histological sections of mouse lungs with total collagen visualized by Masson’s trichrome staining. (B) Histomorphological assessment of airway collagen content normalized to airway length. Sections analyzed in blindfolded manner. (C) Hydroxyproline assay for total collagen content in whole lung preps. Significance (p<0.05, Two-way ANOVA, Bonferroni post-tests) indicated above sample column by the letters of the reference control group of the comparison. Control+Air (ca), Alcohol+Air (aa), Control+DCB-230 (cd), Alcohol+DCB-230 (ad). Scale bar = 100 μm.
CDPM Exposure in Chronic Alcohol Fed Mice Causes Loss of Alveolar Integrity
We hypothesized that elevations in oxidative stress caused by chronic alcohol ingestion and exposure to DCB-230 may be accelerating parenchymal tissue destruction and emphysema compared to mice exposed to DCB-230 and fed a control diet. Therefore, alveolar integrity was examined by calculating space percentage and mean linear intercept (Figure 6). Mice on a chronic alcohol diet exposed to DCB-230 exhibited significant increases in both the percentage of alveolar space (Figure 6B) and mean linear intercept (Figure 6C), compared to mice exposed to DCB-230 and fed a control diet. This suggested that chronic alcohol ingestion and exposure to CDPM increases alveolar tissue damage and emphysema.
Figure 6.
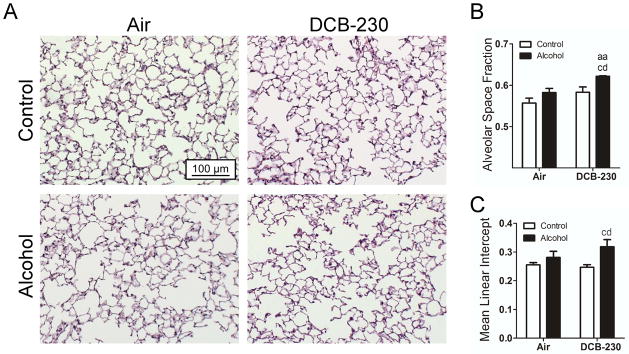
CDPM exposure in chronic alcohol fed mice causes loss of alveolar integrity. (A) Representative histological sections of mouse lungs stained by H&E. (B) Alveolar space fraction as determined from total image area minus interstitial fraction. (C) Mean linear intercept measured across the alveoli (3 measurements taken from 6 random images of each replicate). Significance (p<0.05, Two-way ANOVA, Bonferroni post-tests) indicated above sample column by the letters of the reference control group of the comparison. Control+Air (ca), Alcohol+Air (aa), Control+DCB-230 (cd), Alcohol+DCB-230 (ad). Scale bar = 100μm.
Neutralizing Antibody to TGF-β Improves Pulmonary Function and Reduces Collagen Deposition Due to Chronic Alcohol Ingestion and Exposure to CDPM
We hypothesized that increased TGF-β production by M2 polarized AMs was central to increased collagen deposition and reduced pulmonary function indicative of fibrosis in the mice fed a chronic alcohol diet and exposed to DCB-230. To test this, we administered TGF-β neutralizing antibody (i.n.) immediately following DCB-230 exposure on days 0, 2, 6, 10, and 14 of the exposure period. Pulmonary function and lung collagen levels were then analyzed (Figure 7). In mice fed an alcohol diet and exposed to DCB-230, TGF-β neutralization did not statistically effect Cst or Rn (Figure 7A,B). However, TGF-β neutralization significantly reduced tissue elastance in alcohol fed, DCB-230 exposed mice (Figure 7C). Additionally, neutralization of TGF-β significantly decreased collagen content in the lungs of mice fed an alcohol diet and exposed to DCB-230 (Figure 7D). In total, these data suggest that aberrant TGF-β production in mice is responsible for the pulmonary dysfunction observed following DCB-230 exposure in mice chronically fed an alcohol diet.
Figure 7.

Neutralizing antibody to TGF-β improves pulmonary function and reduces collagen deposition sue to chronic alcohol ingestion and exposure to CDPM. (A) Static compliance (Cst) derived from pressure-volume loops. (B) Newtonian resistance (Rn) and (C) tissue elastance (H) derived from Primewave-8 perturbations. (D) Hydroxyproline assay for collagen content in whole lung preps. Significance (p<0.05, Two-way ANOVA, Bonferroni post-tests) indicated above sample column.
Discussion
Chronic alcohol intake leads to tissue destruction and organ damage (Guidot and Hart, 2005), which in the lungs manifests as deficits in pulmonary function consistent with airflow obstruction (Emirgil and Sobol, 1977; Sisson et al., 2005). Alcohol mediates these effects by altering immune cell function (Happel and Nelson, 2005; Joshi and Guidot, 2007), increasing TGF-β production (Bechara et al., 2004; Guidot and Hart, 2005), and increasing oxidative stress (Moss et al., 2000; Yeh et al., 2007). Pathologically severe obstructive and restrictive lung diseases are also associated with oxidative stress and elevated TGF-β production, which stimulates remodeling, matrix deposition, and loss of alveolar structure (Bergeron et al., 2003; Podowski et al., 2012; Takizawa et al., 2001). Macrophages obtained from the lungs of patients with mild to moderate COPD (Shaykhiev et al., 2009) and fibrotic diseases (Furuhashi et al., 2010; Gibbons et al., 2011; Mathai et al., 2010; Murray et al., 2010) are M2 polarized, and several studies suggest the development of pulmonary fibrosis is directly related to elevated IL-4/IL-13 and TGF-β leading to persistent and/or exaggerated M2 polarization (Gibbons et al., 2011; Martinez et al., 2009; Pulichino et al., 2008; Yogo et al., 2009). In addition to stimulating remodeling and fibrosis, M2s increase TGF-β and IL-10 production (Mosser and Edwards, 2008). AM depletion or reversing M2 polarization greatly diminishes collagen deposition in many models of fibrosis (Gibbons et al., 2011; Homer et al., 2011).
Since chronic alcohol intake induces oxidative stress and TGF-β production in lungs, we hypothesized that exposure of the alcoholic lung to CDPM (at concentrations found in urban locales) would further increase M2 polarization and activation exacerbating deficits in pulmonary function and tissue destruction. We investigated this hypothesis using C57Bl6 mice using nose-only inhalation of a laboratory generated CDPM (DCB-230) in mice chronically fed a liquid alcohol diet.
We previously showed that nose-only exposure to PM in rodents induces inflammatory responses including increased AMs into the lung (Balakrishna et al., 2011; Thevenot et al., 2013). However chronic alcohol appears to interrupt AM influx by suppressing myeloid development (Melvan et al., 2012) and reducing the maturation status of AMs (Brown et al., 2009; Joshi et al., 2005). Therefore it was not surprising that AM levels in lungs of DCB-230-exposed mice chronically fed alcohol were reduced compared mice fed a control liquid diet and exposed to DCB-230. The AM maturation markers, CD11c and CD11b, were also decreased in DCB-230 exposed, alcohol fed mice compared to controls, demonstrating decreased AM maturation. These decreases could be the result of impaired differentiation from interstitial macrophages in the lung or from circulating monocyte pools, as AMs may derive from both.
Since AMs of chronic alcohol fed mice were less mature, we initially hypothesized M2 polarization in this setting would be impaired. However, steady state AM turnover in the lung is slow, with data suggesting that a mere 15% of AMs turnover at three months and at most 40% at one year (Janssen et al., 2011). Our data demonstrate that resident BAL-derived AMs, whether obtained from mice fed a control or alcohol diet, are capable of polarizing to the M2s during extended culture in media containing alcohol. We therefore formulated the refined hypothesis that chronic alcohol intake increases M2 polarization of resident AMs.
IL-10 expression in AMs is used as a marker of M2 polarization, and IL-4Rα expression has also been a factor strongly correlated with M2. Chronic alcohol ingestion caused increased expression and production of these M2 associated factors in AMs consistent with an M2 phenotype. Exposure to DCB-230 in alcohol fed mice led to further elevations in IL-10 and IL-4Rα with respect to alcohol intake alone as well as increased expression of the M2 genes Ym1 and Fizz1, demonstrating increased capacity for AMs to polarize into M2s. Conversely, DCB-230 exposure in mice fed a control liquid diet led to an increase in Nos2 expression, indicative of M1 polarization. Several conflicting observations with regard to the effects of CDPM on AM polarization have been reported, in which AM phenotype is likely attributable to route of exposure, chemical/endotoxin contamination, and perhaps most influenced by the degree and duration of exposure (Brown et al., 2011; Miyata and van Eeden, 2011). Our results suggest that alcohol predisposes AMs to the M2 phenotype and that is exacerbated upon CDPM exposure.
Mounting evidence suggests AMs, specifically M2 polarized AMs, are the predominant source of TGF-β and depleting M2 polarized macrophages during the progressive phase of fibrosis reduces collagen deposition by limiting TGF-β production (Gibbons et al., 2011). TGF-β alone has been shown to predispose to M2 polarization by increasing expression of IL-4Rα expression (Homer et al., 2011). Increased levels of TGF-β, contribute to the immature phenotype observed in AMs in this and previous chronic alcohol ingestion models (Joshi and Guidot, 2007). We observed elevated levels of total TGF-β and IL-4Rα in the lungs of alcohol fed mice and this was enhanced upon exposure to DCB-230.
With chronic alcohol we observed increases in latent TGF-β in AMs with no increase in active TGF-β (similar to others; Bechara et al., 2004). CDPM exposure in chronic alcohol fed mice led to elevated TGF-β levels in lung digests, elevated TGF-β1 transcripts in AMs, and decreased GSH/GSSG ratios and increased collagen content in the lungs. Oxidative stress alone has been shown to play an important role in the development of fibrosis through activation of latent TGF-β (reviewed in Liu and Gaston Pravia, 2010). In agreement with clinical data from smoking chronic alcoholics (Yeh et al., 2007), we found that co-exposure to CDPM and chronic alcohol induced oxidative stress. Therefore, it is plausible that increased oxidative stress activates latent TGF-β in addition to increasing TGF-β production through increased M2 activation.
Using pulmonary functional assessments and morphometric analyses of lung sections, we demonstrate that CDPM exposure decreased lung function in alcohol fed mice by increasing fibrosis and disrupting alveolar integrity. Concordantly, recent studies on alcohol and lung function demonstrate that alcohol consumption alone decreases airway smooth muscle contraction (Oldenburg et al., 2009; 2012). We have previously shown that DCB-230 exposure reduced compliance and increased tissue elastance regardless of diet (Balakrishna et al., 2011), indicating an increase in stiffness. As anticipated, increased collagen deposition in the perivascular regions of the airways was observed in mice fed an alcohol diet and exposed to DCB-230 and correlated with a significant increase in lung hydroxyproline content. Histologcial examination of the alveolar spaces showed significant loss of alveolar tissue in alcohol fed, DCB-230 exposed mice. Collectively, function and histological data illustrate how increased M2 polarization coupled with elevations in TGF-β increased disease severity in alcohol fed mice exposed to CDPM.
Coupling our data with the work of others, we propose the following sequence of events which ultimately lead to enhanced fibrosis and lung dysfunction in CDPM-exposed chronic alcohol consuming mice. AMs in chronic alcohol fed mice are primed to develop into M2s capable of producing TGF-β. Subsequent pulmonary disturbances demonstrated here with CDPM further increase pulmonary oxidative stress and enhance M2 polarization/activation, leading to significant elevations in total active TGF-β. Similar to the phenotypes observed in pulmonary bleomycin and TGF-β over-expression models, M2 polarization and increased TGF-β lead to fibrosis, which here appears localized to the airways, in addition to destruction of the alveolar space. Collectively, these tissue architectural changes lead to decrements in pulmonary function. We hypothesized that excess TGF-β was critical in manifesting deficits in pulmonary function in alcohol fed and DCB-230 exposed mice, and show that decreasing TGF-β by administration of neutralizing antibody effectively reduced collagen content in the lungs and improved pulmonary function. This suggests targeting M2 polarization may be of therapeutic value to inhibiting tissue damage in chronic alcoholics exposed to CDPM. In conclusion, our data demonstrate alcohol consumption increases M2 predisposition in the lungs, and leads to increased tissue damage and lung dysfunction.
Supplementary Material
Acknowledgments
A portion of this work was presented as a talk and poster at the Alcohol and Immunology Research Interest Group (AIRIG) Meeting, November 18th, Chicago, IL, USA and an abstract is published from this presentation in Alcohol, 46(2):178, 2012.
This work was supported by an NIAAA grant to PT (AA007577), a Louisiana State Board of Regents grant to JS (LEQSF(2009-14)-GF-08), NIEHS grants to TD (P42ES013648) and to SAC (R01ES015050 and P42ES013648), NIAAA grant to SAC and KH (P60 AA009803) and NIAID grant to SAC (R01AI090059). The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of NIH or the state of Louisiana. Special thanks to Terry Ahlert for assistance with exposures and training.
References
- Alcorn JF, Rinaldi LM, Jaffe EF, van Loon M, Bates JH, Janssen-Heininger YM, Irvin CG. Transforming growth factor-beta1 suppresses airway hyperresponsiveness in allergic airway disease. Am J Respir Crit Care Med. 2007;176:974–982. doi: 10.1164/rccm.200702-334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishna S, Saravia J, Thevenot P, Ahlert T, Lominiki S, Dellinger B, Cormier SA. Environmentally persistent free radicals induce airway hyperresponsiveness in neonatal rat lungs. Part Fibre Toxicol. 2011;8:11. doi: 10.1186/1743-8977-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batel P, Pessione F, Maitre C, Rueff B. Relationship between alcohol and tobacco dependencies among alcoholics who smoke. Addiction. 1995;90:977–980. doi: 10.1046/j.1360-0443.1995.90797711.x. [DOI] [PubMed] [Google Scholar]
- Bechara RI, Brown LA, Roman J, Joshi PC, Guidot DM. Transforming growth factor beta1 expression and activation is increased in the alcoholic rat lung. Am J Respir Crit Care Med. 2004;170:188–194. doi: 10.1164/rccm.200304-478OC. [DOI] [PubMed] [Google Scholar]
- Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, Hance AJ, Tazi A. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J. 2003;22:69–76. doi: 10.1183/09031936.03.00014703. [DOI] [PubMed] [Google Scholar]
- Brown BN, Price IM, Toapanta FR, DeAlmeida DR, Wiley CA, Ross TM, Oury TD, Vodovotz Y. An agent-based model of inflammation and fibrosis following particulate exposure in the lung. Math Biosci. 2011;231:186–196. doi: 10.1016/j.mbs.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SD, Brown LA. Ethanol (EtOH)-Induced TGF-beta(1) and Reactive Oxygen Species Production Are Necessary for EtOH-Induced Alveolar Macrophage Dysfunction and Induction of Alternative Activation. Alcohol Clin Exp Res. 2012;36:1952–1962. doi: 10.1111/j.1530-0277.2012.01825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SD, Gauthier TW, Brown LA. Impaired terminal differentiation of pulmonary macrophages in a Guinea pig model of chronic ethanol ingestion. Alcohol Clin Exp Res. 2009;33:1782–1793. doi: 10.1111/j.1530-0277.2009.01017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emirgil C, Sobol BJ. Pulmonary function in former alcoholics. Chest. 1977;72:45–51. doi: 10.1378/chest.72.1.45. [DOI] [PubMed] [Google Scholar]
- Furuhashi K, Suda T, Nakamura Y, Inui N, Hashimoto D, Miwa S, Hayakawa H, Kusagaya H, Nakano Y, Nakamura H, Chida K. Increased expression of YKL-40, a chitinase-like protein, in serum and lung of patients with idiopathic pulmonary fibrosis. Respir Med. 2010;104:1204–1210. doi: 10.1016/j.rmed.2010.02.026. [DOI] [PubMed] [Google Scholar]
- Gauderman WJ, Vora H, McConnell R, Berhane K, Gilliland F, Thomas D, Lurmann F, Avol E, Kunzli N, Jerrett M, Peters J. Effect of exposure to traffic on lung development from 10 to 18 years of age: a cohort study. Lancet. 2007;369:571–577. doi: 10.1016/S0140-6736(07)60037-3. [DOI] [PubMed] [Google Scholar]
- Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van Rooijen N, Haslett C, Howie SE, Simpson AJ, Hirani N, Gauldie J, Iredale JP, Sethi T, Forbes SJ. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184:569–581. doi: 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Grant BF, Dawson DA, Stinson FS, Chou SP, Dufour MC, Pickering RP. The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug Alcohol Depend. 2004;74:223–234. doi: 10.1016/j.drugalcdep.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Guidot DM, Hart CM. Alcohol abuse and acute lung injury: epidemiology and pathophysiology of a recently recognized association. J Investig Med. 2005;53:235–245. doi: 10.2310/6650.2005.53506. [DOI] [PubMed] [Google Scholar]
- Happel KI, Nelson S. Alcohol, immunosuppression, and the lung. Proc Am Thorac Soc. 2005;2:428–432. doi: 10.1513/pats.200507-065JS. [DOI] [PubMed] [Google Scholar]
- Homer RJ, Elias JA, Lee CG, Herzog E. Modern concepts on the role of inflammation in pulmonary fibrosis. Arch Pathol Lab Med. 2011;135:780–788. doi: 10.5858/2010-0296-RA.1. [DOI] [PubMed] [Google Scholar]
- Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, Henson PM. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med. 2011;184:547–560. doi: 10.1164/rccm.201011-1891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi PC, Applewhite L, Ritzenthaler JD, Roman J, Fernandez AL, Eaton DC, Brown LA, Guidot DM. Chronic ethanol ingestion in rats decreases granulocyte-macrophage colony-stimulating factor receptor expression and downstream signaling in the alveolar macrophage. J Immunol. 2005;175:6837–6845. doi: 10.4049/jimmunol.175.10.6837. [DOI] [PubMed] [Google Scholar]
- Joshi PC, Guidot DM. The alcoholic lung: epidemiology, pathophysiology, and potential therapies. Am J Physiol Lung Cell Mol Physiol. 2007;292:L813–823. doi: 10.1152/ajplung.00348.2006. [DOI] [PubMed] [Google Scholar]
- Kelly FJ, Fussell JC. Air pollution and airway disease. Clin Exp Allergy. 2011;41:1059–1071. doi: 10.1111/j.1365-2222.2011.03776.x. [DOI] [PubMed] [Google Scholar]
- Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med. 2010;48:1–15. doi: 10.1016/j.freeradbiomed.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomnicki S, Truong H, Vejerano E, Dellinger B. Copper oxide-based model of persistent free radical formation on combustion-derived particulate matter. Environ Sci Technol. 2008;42:4982–4988. doi: 10.1021/es071708h. [DOI] [PubMed] [Google Scholar]
- Lyerla TA, Rusiniak ME, Borchers M, Jahreis G, Tan J, Ohtake P, Novak EK, Swank RT. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol. 2003;285:L643–653. doi: 10.1152/ajplung.00024.2003. [DOI] [PubMed] [Google Scholar]
- Martin S, Dawidowski L, Mandalunis P, Cereceda-Balic F, Tasat DR. Characterization and biological effect of Buenos Aires urban air particles on mice lungs. Environ Res. 2007;105:340–349. doi: 10.1016/j.envres.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, Murray LA, Siner JM, Antin-Ozerkis DE, Montgomery RR, Reilkoff RA, Bucala RJ, Herzog EL. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest. 2010;90:812–823. doi: 10.1038/labinvest.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AJ, Guidot DM. Alcohol abuse, the alveolar macrophage and pneumonia. Am J Med Sci. 2012;343:244–247. doi: 10.1097/MAJ.0b013e31823ede77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvan JN, Siggins RW, Stanford WL, Porretta C, Nelson S, Bagby GJ, Zhang P. Alcohol impairs the myeloid proliferative response to bacteremia in mice by inhibiting the stem cell antigen-1/ERK pathway. J Immunol. 2012;188:1961–1969. doi: 10.4049/jimmunol.1102395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata R, van Eeden SF. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol Appl Pharmacol. 2011;257:209–226. doi: 10.1016/j.taap.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Moshammer H, Hutter HP, Hauck H, Neuberger M. Low levels of air pollution induce changes of lung function in a panel of schoolchildren. Eur Respir J. 2006;27:1138–1143. doi: 10.1183/09031936.06.00089605. [DOI] [PubMed] [Google Scholar]
- Moss M, Guidot DM, Wong-Lambertina M, Ten Hoor T, Perez RL, Brown LA. The effects of chronic alcohol abuse on pulmonary glutathione homeostasis. Am J Respir Crit Care Med. 2000;161:414–419. doi: 10.1164/ajrccm.161.2.9905002. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray LA, Rosada R, Moreira AP, Joshi A, Kramer MS, Hesson DP, Argentieri RL, Mathai S, Gulati M, Herzog EL, Hogaboam CM. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS One. 2010;5:e9683. doi: 10.1371/journal.pone.0009683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S, Kolls JK. Alcohol, host defence and society. Nat Rev Immunol. 2002;2:205–209. doi: 10.1038/nri744. [DOI] [PubMed] [Google Scholar]
- Oldenburg PJ, Poole JA, Sisson JH. Alcohol reduces airway hyperresponsiveness (AHR) and allergic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2012;302:L308–315. doi: 10.1152/ajplung.00077.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg PJ, Wyatt TA, Factor PH, Sisson JH. Alcohol feeding blocks methacholine-induced airway responsiveness in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296:L109–114. doi: 10.1152/ajplung.00487.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podowski M, Calvi C, Metzger S, Misono K, Poonyagariyagorn H, Lopez-Mercado A, Ku T, Lauer T, McGrath-Morrow S, Berger A, Cheadle C, Tuder R, Dietz HC, Mitzner W, Wise R, Neptune E. Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J Clin Invest. 2012;122:229–240. doi: 10.1172/JCI46215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulichino AM, Wang IM, Caron A, Mortimer J, Auger A, Boie Y, Elias JA, Kartono A, Xu L, Menetski J, Sayegh CE. Identification of transforming growth factor beta1-driven genetic programs of acute lung fibrosis. Am J Respir Cell Mol Biol. 2008;39:324–336. doi: 10.1165/rcmb.2007-0186OC. [DOI] [PubMed] [Google Scholar]
- Saric M, Lucic-Palaic S, Horton RJ. Chronic nonspecific lung disease and alcohol consumption. Environ Res. 1977;14:14–21. doi: 10.1016/0013-9351(77)90061-5. [DOI] [PubMed] [Google Scholar]
- Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, O’Connor TP, Crystal RG. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183:2867–2883. doi: 10.4049/jimmunol.0900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson JH. Alcohol and airways function in health and disease. Alcohol. 2007;41:293–307. doi: 10.1016/j.alcohol.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson JH, Stoner JA, Romberger DJ, Spurzem JR, Wyatt TA, Owens-Ream J, Mannino DM. Alcohol intake is associated with altered pulmonary function. Alcohol. 2005;36:19–30. doi: 10.1016/j.alcohol.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, Shanley TP. New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L341–353. doi: 10.1152/ajplung.00122.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, Okada Y, Yamasawa F, Nakahara K, Umeda A. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD) Am J Respir Crit Care Med. 2001;163:1476–1483. doi: 10.1164/ajrccm.163.6.9908135. [DOI] [PubMed] [Google Scholar]
- Thevenot PT, Saravia J, Jin N, Giaimo JD, Chustz RE, Mahne S, Kelley MA, Hebert VY, Dellinger B, Dugas TR, Demayo FJ, Cormier SA. Radical-Containing PM0.2 Initiates Epithelial-to-Mesenchymal Transitions in Airway Epithelial Cells. Am J Respir Cell Mol Biol. 2013;48:188–197. doi: 10.1165/rcmb.2012-0052OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo G, O’Connor EC, Kunkel SL, Hogaboam CM. A novel mechanism for CCR4 in the regulation of macrophage activation in bleomycin-induced pulmonary fibrosis. Am J Pathol. 2008;172:1209–1221. doi: 10.2353/ajpath.2008.070832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh MY, Burnham EL, Moss M, Brown LA. Chronic alcoholism alters systemic and pulmonary glutathione redox status. Am J Respir Crit Care Med. 2007;176:270–276. doi: 10.1164/rccm.200611-1722OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogo Y, Fujishima S, Inoue T, Saito F, Shiomi T, Yamaguchi K, Ishizaka A. Macrophage derived chemokine (CCL22), thymus and activation-regulated chemokine (CCL17), and CCR4 in idiopathic pulmonary fibrosis. Respir Res. 2009;10:80. doi: 10.1186/1465-9921-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.