

Abstract
Identification of Endocrine Disrupting Chemicals is one of the important goals of environmental chemical hazard screening. We report on the development of validated in silico predictors of chemicals likely to cause Estrogen Receptor (ER)-mediated endocrine disruption to facilitate their prioritization for future screening. A database of relative binding affinity of a large number of ERα and/or ERβ ligands was assembled (546 for ERα and 137 for ERβ). Both single-task learning (STL) and multi-task learning (MTL) continuous Quantitative Structure-Activity Relationships (QSAR) models were developed for predicting ligand binding affinity to ERα or ERβ. High predictive accuracy was achieved for ERα binding affinity (MTL R2=0.71, STL R2=0.73). For ERβ binding affinity, MTL models were significantly more predictive (R2=0.53, p<0.05) than STL models. In addition, docking studies were performed on a set of ER agonists/antagonists (67 agonists and 39 antagonists for ERα, 48 agonists and 32 antagonists for ERβ, supplemented by putative decoys/non-binders) using the following ER structures (in complexes with respective ligands) retrieved from the Protein Data Bank: ERα agonist (PDB ID: 1L2I), ERα antagonist (PDB ID: 3DT3), ERβ agonist (PDB ID: 2NV7), ERβ antagonist (PDB ID: 1L2J). We found that all four ER conformations discriminated their corresponding ligands from presumed non-binders. Finally, both QSAR models and ER structures were employed in parallel to virtually screen several large libraries of environmental chemicals to derive a ligand- and structure-based prioritized list of putative estrogenic compounds to be used for in vitro and in vivo experimental validation.
Keywords: Endocrine Disrupting Chemicals, Estrogen Receptor, Quantitative Structure-Activity Relationships modeling, Multi-Task Learning, Docking, Virtual Screening
Introduction
Endocrine disrupting chemicals (EDCs) interfere with the synthesis, secretion, transport, metabolism, binding, or elimination of hormones (Diamanti-Kandarakis et al., 2009). Adverse health effects of EDCs in humans have been demonstrated to include developmental, reproductive, neurological, cardiovascular, metabolic and immune systems (Schug et al., 2011). A wide range of natural and man-made chemical substances may be causing endocrine disruption and are considered as both human health and environmental hazards (Diamanti-Kandarakis et al., 2009). Costly testing of chemicals for their endocrine disruption potential is required in most industrialized countries (Adler et al., 2011). Because the mechanisms of endocrine disruption are diverse and complex (e.g., interactions with hormone and non-steroid receptors, activation of enzymatic and signaling pathways, etc.), a wide array of in vitro and in vivo tests is used to identify EDCs (Jacobs et al., 2008; Shanle and Xu, 2011; Sung et al., 2012; Rotroff et al., 2013).
Estrogen-like activity is one of the most common adverse effects of EDCs. Estrogen receptors (ER) have been extensively studied (Mueller and Korach, 2001; Shanle and Xu, 2011) and two subtypes of ER have been identified, ERα and ERβ. These subtypes have overlapping yet unique physiological roles depending on the tissue and cell type, presence of cofactors, and ligands (Minutolo et al., 2011). With regards to the sequences of the ER subtypes, they are most similar in the DNA-binding domain (97%), while there is less conservation in the C-terminal ligand binding domain (56%) and N-terminal transactivation domain (18%) (Hall et al., 2001; Koehler et al., 2005). The amino acid differences, both inside the binding cavity and in other regions of ligand binding domain, may be responsible for the subtype selectivity of some ER ligands.
Structure-activity modeling plays an important role in government programs in support of protecting human populations from exposure to environmental contaminants (Demchuk et al., 2011). Specifically, computational methods to identify chemicals that may pose endocrine disruption hazard for additional in vitro or in vivo testing are important prioritization approaches (Lo Piparo and Worth, 2010; Tsakovska et al., 2011). Because of the diversity and complexity of endocrine disruption mechanisms, as well as the limited data available for in silico modeling, most studies have focused on EDCs that act via estrogen or androgen receptors. These modeling approaches include Quantitative Structure-Activity Relationship (QSAR) (Tong et al., 2004; Salum et al., 2007), molecular dynamics simulations (van Lipzig et al., 2004), docking (Celik et al., 2007; Celik et al., 2008) and pharmacophores (Taha et al., 2010). Consequently, many of the models have been implemented as computational tools that are available either publicly or commercially (Table 1). Most of these tools only provide hazard classification of chemicals, e.g., as either receptor binders or non-binders of ER subclasses or other receptors, while only few provide quantitative estimation of the relative binding affinity [e.g., the Endocrine Disruptor Knowledge Base (EDKB) (Ding et al., 2010), ADMET Predictor (http://www.simulations-plus.com/), and MolCode (http://molcode.com/)].
Table 1.
Software/toolbox/web portals capable of predicting endocrine disrupting potential of chemicals.
| In silico System | Availability | Description |
|---|---|---|
|
ChemBench (Walker et al., 2010) http://chembench.mml.unc.edu/ |
Publicly available | Quantitative prediction of binding affinity to ERα and ERβ (this work) |
|
Endocrine Disruptor Knowledge Base (EDKB) (Tong et al., 2004) http://www.fda.gov/scienceresearch/bioinformaticstools/endocrinedisruptorknowledgebase/default.htm |
Publicly available | Quantitative prediction of binding affinity to ER and Androgen Receptor (AR) |
|
OECD (Q)SAR Toolbox http://www.oecd.org/document/54/0,3746,en_2649_34379_42923638_1_1_1_1,00.html |
Publicly available | Binary prediction of ER binders/non-binders |
|
ACD/Tox Suite (ToxBoxes) http://www.acdlabs.com/products/pc_admet/tox/tox/modules.php |
Subscription-based | Binary prediction of ERα binders/non-binders |
|
ADMET Predictor http://www.simulations-plus.com/ |
Subscription-based | Qualitative and quantitative prediction of binding affinity to ER |
|
Derek Nexus http://www.lhasalimited.org/ |
Subscription-based | Classification models (different levels of likelihood) based on 23 alerts for developmental toxicity; 4 alerts for estrogenicity |
|
MolCode Toolbox http://molcode.com/ |
Subscription-based | Quantitative prediction of binding affinity to ER and aryl hydrocarbon receptor (AhR) |
|
TIssue MEtabolism Simulator (TIMES) (Serafimova et al., 2007) http://oasis-lmc.org/ |
Subscription-based | Binary prediction of ER, AR and AhR binders/non-binders |
|
VirtualToxLab (Vedani and Smiesko, 2009; Vedani et al., 2009) http://www.biograf.ch |
Subscription-based | Prediction of endocrine disruption potential based on simulations of compound interactions with AR, AhR, ER, thyroid, glucocorticoid, liver X, mineralo-corticoid, peroxisome proliferator-activated receptors, as well as CYP450 3A4 and 2A13 enzymes |
This table is sorted by availability and alphabetical order. More extensive description of the available tools may be found in (Lo Piparo and Worth, 2010).
There are several limitations of existing computational tools for prediction of ER-mediated endocrine disruption. First, most available computational tools focus on ERα since they were developed using a dataset of 232 compounds tested against ERα, which is available from the Endocrine Disruptor Screening Program (EDSP) (Branham et al., 2002). Second, relatively few chemicals have been tested for ERβ-specific activity and therefore there are no current tools that can predict ERβ binding, or distinguish between ER subtypes. Finally, tools that can predict ER binding affinity are not capable of distinguishing the type of functional activity (i.e., agonism vs. antagonism). To address these limitations, we have assembled a large dataset including data on ER binding affinity (546 compounds for ERα and 137 for ERβ) and functional activity (67 agonists/39 antagonists for ERα, 48 agonists/32 antagonists for ERβ) and developed QSAR models for each ER subtype. Next, QSAR and ER subtype-specific docking were used in parallel to virtually screen a library of environmental chemicals to identify putative ER binders and predict their selectivity and functional activity.
Materials and Methods
Datasets
ERα and ERβ binding affinity
For QSAR modeling (see below), we used binding affinity (ERα and/or ERβ) data from publicly available sources (Table 2). For ERα, 414 unique organic compounds were identified in EDKB (Ding et al., 2010) and ChEMBL (Gaulton et al., 2012), and additional 132 compounds with ERα binding affinity data were extracted from the published literature (Kuiper et al., 1997; Taha et al., 2010). For ERβ, binding affinity data were available for 137 chemicals (Kuiper et al., 1997; Taha et al., 2010). For both ERα and ERβ datasets, the relative binding affinity (RBA), as compared to 17β-estradiol (E2) from ER competitive-binding assays, was calculated using Equation (1).
Table 2.
Datasets of ER binding affinity and functional activity used in this study.*
| Modeling Method | Dataset | ER Subtype | Number of Compounds |
|---|---|---|---|
| QSAR |
Binding Affinity (logRBA) (Kuiper et al., 1997; Ding et al., 2010; Taha et al., 2010; Gaulton et al., 2012) |
ERα | 546 |
| ERβ | 137 | ||
| Docking |
Known Agonists, Known Antagonists, and Presumed Decoys (Friesner et al., 2004; Huang et al., 2006; Minutolo et al., 2011) |
ERα | 67 agonists 39 antagonists |
| 2570 agonist decoys 1448 antagonist decoys | |||
| ERβ | 48 agonists 32 antagonists |
||
| 1000 agonist decoys 1000 antagonist decoys |
See Supplemental Table 1 for a complete list.
| (1) |
ERα and ERβ functional activity (agonism/antagonism)
For docking studies (see below) we collected information on whether a compound is a known agonist/antagonist, or is a presumed decoy, with regards to a particular receptor (Table 2). Presumed decoys are defined as chemicals that have similar physical properties but are topologically dissimilar from the known ligand structures and are expected not to bind the respective receptor. For ERα, 67 known agonists and 39 known antagonists were obtained from the Directory of Useful Decoys (Huang et al., 2006). In addition, 2570 presumed agonist decoys and 1448 presumed antagonist decoys were from the same source (Huang et al., 2006). For ERβ, 48 known agonists and 32 known antagonists were obtained from (Minutolo et al., 2011). Because there were no publicly available decoy datasets for ERβ agonists or antagonists at the time of this study, the dataset of 1000 drug-like compounds (Friesner et al., 2004) was used as presumed ERβ non-binders. Partial ER agonists or antagonists were not used. The chemical structures of agonists, antagonists, and their corresponding presumed decoys/non-binders for both ER subtypes are included in Supplemental Table 1.
The three-dimensional conformations of the ER ligands and presumed decoys/non-binders were prepared using LigPrep (LigPrep 2011). The ionization state of each molecule was calculated assuming the pH value of 7.0±2.0. All molecules were subjected to energy minimization using the MMFF force field before docking.
ER agonist and antagonist conformations for docking
Four ER crystal structures were used for docking in this study: ERα agonist conformation with bound ERα agonist R,R-tetrahydro-chrysene [PDB ID: 1L2I (Shiau et al., 2002)], ERα antagonist conformation with bound ERα antagonist GW2368 [PDB ID: 3DT3 (Fang et al., 2008)], ERβ agonist conformation with bound ERβ agonist naphthalene [PDB ID: 2NV7 (Mewshaw et al., 2007)], and ERβ antagonist conformation with bound ERβ antagonist (5R,11R)-5,11-diethyl-5,6,11,12-tetrahydrochrysene-2,8-diol [PDB ID: 1L2J (Shiau et al., 2002)]. These ER crystal structures were selected because they had the highest resolution according to PDB. The protein-ligand crystal structures are shown in Supplemental Figure 1. Protein structures were prepared using Protein Preparation wizard (Protein Preparation Wizard, 2011) by following the protocols in (Mouchlis et al., 2010). A grid for each protein was calculated using Grid Generation in Maestro (Maestro 2011), with the binding site defined by the location of co-crystallized ligands.
Data curation
The structures for all compounds employed in this study were manually examined and curated according to the guidelines described in (Fourches et al., 2010). All curated structures were stored in SDF format for further analysis.
Molecular Descriptors
Molecular descriptors (represented with explicit hydrogen atoms) were computed for each compound using Dragon software (version 5.4; Talete s.r.l., Milan, Italy). Descriptors with low variance (standard deviation lower than 0.0001) or missing values were removed. Furthermore, if the squared correlation coefficient (R2) between values of two descriptors over the entire data set exceeded 0.95, one of the descriptors was randomly removed. The final descriptor set used in this study contained 432 descriptors range-scaled to the [0, 1] interval.
QSAR Modeling Approaches
Training, test, and external evaluation sets selection
To avoid well-documented limitations of QSAR models developed with training sets (Golbraikh and Tropsha, 2002), each binding affinity dataset (consisting of 546 unique organic compounds for ERα and of 137 for ERβ) was subjected to 5-fold external cross-validation procedure as detailed in (Sedykh et al., 2011). Specifically, each dataset was randomly partitioned into 5 subsets of similar size. Models were then independently developed such that compounds in 4 of the 5 subsets were used as modeling set and the compounds in the remaining subset were used as an external evaluation set. The modeling set was further subdivided into 36–50 diverse internal training and test sets of different sizes, using a sphere-exclusion method (Golbraikh et al., 2003). Individual models were developed based on each internal training set and internally validated by predicting the corresponding test set. All individual models showing acceptable performance on internal training/test sets were retained in an ensemble for the application to the external evaluation set; thus, the latter set was not used in any way in model development or internal validation. This procedure was repeated 5 times such that each of the five subsets was employed as external evaluation set once and the remaining subsets were used as a modeling set.
Variable selection k-nearest neighbors (kNN) QSAR modeling
Candidate models were built using variable selection kNN method (Zheng and Tropsha, 2000). Specifically, a set of nvar descriptors are randomly selected (nvar is set to multiple values during successive modeling attempts in order to find the best fitted model). The activity of any compound can then be predicted by averaging the activities of the k compounds most similar to it, as measured by Euclidean distance calculated in the multidimensional space defined by nvar selected descriptors (k ranges from 1 to 5 and is also subject to independent optimization). This candidate model is then evaluated by leave-one-out cross-validation, where each compound is eliminated from the training set and its ER binding affinity is predicted from that of its k nearest neighbors. The correlation between the predicted affinities and the actual values is then calculated using leave-one-out cross-validated R2 (q2) as metric. The descriptor selection is optimized to achieve the highest q2 using simulated annealing approach with Metropolis-like model acceptance criteria Further details of this approach can be found elsewhere (Zheng and Tropsha, 2000).
Single-task learning and multi-task learning kNN QSAR modeling
In traditional QSAR modeling, a correlation between values of multiple chemical descriptors (or independent variables) and a single type of biological activity, e.g., estrogenic activity, is sought: this type of modeling can be termed single task learning (STL). However, it is often feasible that two partially overlapping groups of compounds may interact with related biological targets causing similar biological response (e.g., two receptor subtypes); in this case it may be advantageous to build a model correlating descriptor values with both biological responses simultaneously, i.e., employ multi-task learning (MTL). Herein, in addition to traditional STL-kNN methodology (Zheng and Tropsha, 2000), we used MTL approaches (Figure 1). MTL trains a model on multiple tasks in parallel and benefits from the inductive transfer of knowledge between related tasks (Varnek et al., 2009). In our implementation of MTL-kNN method, compounds from all tasks are mapped onto the same chemical descriptor space, and the variable-selection procedure is driven by optimizing the cumulative fitness-value calculated across all tasks. The algorithm considers neighbors of each compound only within the same task; thus, the predictions for each task are based on its own data, but all tasks drive the variable-selection jointly. The MTL-kNN modeling approach was applied to the simultaneous modeling of ERα and ERβ binding affinities.
Figure 1. QSAR modeling workflow for STL- and MTL-kNN QSAR modeling.

Numbers of ligands included in each dataset used in model development are shown within the circles at the top of the figure.
Selection and validation of QSAR models
The kNN QSAR models were considered acceptable using the following criteria as detailed in (Zhang et al., 2008; Tropsha, 2010): (i) leave one out cross-validated q2; (ii) square of the correlation coefficient between the predicted and observed activities; (iii) coefficients of determination (predicted versus observed activities, and observed versus predicted activities); and (iv) slopes k and k′ of regression lines (predicted versus observed activities, and observed versus predicted activities) forced through the origin.
Applicability domain
Since kNN models interpolate activities from the nearest neighbor compounds in training sets, a special applicability domain (i.e., similarity threshold) was introduced to avoid classifying test set compounds that differ from the training set molecules. Applicability domains for all models were calculated according to (Golbraikh et al., 2003).
Robustness of QSAR models
Y-randomization (randomization of response) is widely used to establish robustness of QSAR models (Rucker et al., 2007). The process involves rebuilding models using randomized response values in the training set and subsequent assessment of the model prediction accuracy. It is expected that models obtained for the training set with randomized response values should have significantly lower prediction accuracy. Y-randomization was applied in duplicate to all training/test dataset divisions considered in this study as detailed in (Golbraikh and Tropsha, 2002).
Docking Studies
All ERα and ERβ agonists and antagonists, as well as their respective presumed decoys/non-binders, were docked into the corresponding protein structures using Glide XP (Friesner et al., 2004) with default flexible ligand docking settings and the ligands were ranked by docking scores, using Glide XP scoring function.
Virtual Screening
QSAR and docking models developed in this study were used to virtually screen (i.e., predict RBA values and agonist/antagonist docking scores with respect to ERα and ERβ) two additional libraries of chemicals. First, we used a list of compounds in EDKB (Ding et al., 2010) that have reported log relative potency (logRP) value (with E2 as the reference compound) in uterotrophic assay (Supplemental Table 5). There were 1707 compounds with logRP values in EDKB; of these, 970 were unique (see curation procedure described above) and were not overlapping with the list of compounds used to develop either QSAR or docking models. Performance of the QSAR and docking models developed in this study was evaluated only for these 970 compounds, 34 had logRP≥0 (considered as “active”), and 936 had logRP<0 (considered as “inactive”). Second list of compounds was obtained from the Endocrine Disruptor Screening Program at the US Environmental Protection Agency (EPA) (http://epa.gov/endo/pubs/edsp_chemical_universe_list_11_12.pdf). After chemical structure curation, 3557 unique compounds were used for virtual screening and prioritization.
QSAR-based virtual screening
QSAR models developed in this study were used to virtually screen both uterotrophic and EDSP datasets in order to identify potential ERα and/or ERβ ligands. Specifically, MTL-kNN models were used to predict the ERα and ERβ binding affinity for every compound identified within the respective models’ applicability domain. Consensus prediction of ERα or ERβ binding affinity was calculated by averaging the individual predictions across all models that passed internal validation criteria in five-fold external cross-validation procedure.
Docking-based virtual screening
Procedures described above were followed and the results of docking runs were organized into the following four ranked lists: 1) ERα agonists; 2) ERα antagonists; 3) ERβ agonists; 4) ERβ antagonists.
Virtual screening performance metrics
To compare the relative efficiency of QSAR and docking methods, enrichment factors and receiver operating characteristic (ROC) curves were calculated. Both of these metrics assess the ability of a method to distinguish known ligands from a larger pool of tested compounds. The enrichment factor (Equation 2) reflects how many seed compounds (or known ligands) were found within a defined “early recognition” fraction of the ranked list relative to a random distribution where Hscr is the number of target-specific ligands recovered at a specific % level of the ligand/decoy datasets; Htot is the total number of ligands for the target; Dscr is the number of compounds screened at a specific % level of the database; Dtot is the total number of compounds in the database.
| (2) |
The ROC curves were generated by plotting sensitivity (Equation 3) against [1–specificity (Equation 4)] for a binary classifier system as its discrimination threshold is varied. In the case of virtual screening for recovering the ith known active from the inactive decoys (or presumed decoys), the sensitivity and specificity were defined as follows:
| (3) |
| (4) |
The area under the ROC curve is the metric widely accepted for assessing the likelihood that a screening method assigns a higher rank to known actives than to inactive compounds. The area under the curve values at a specific percentage of the ranked database were calculated from Equation (5) where n is the total number of known actives in the screening database.
| (5) |
In addition, in order to compare or combine the performances among different models, the predicted binding affinity by QSAR models or the calculated docking score by docking program were converted into Z-scores respectively. The consensus prediction for each chemical by different types of models was then calculated by averaging all individual Z-scores.
Results and Discussion
QSAR Modeling
The ERα and ERβ binding affinity (logRBA) datasets assembled for this study include 546 ERα ligands and 137 ERβ ligands, respectively. These datasets are among the largest reported thus far [Table 1 and (Lo Piparo and Worth 2010)]. For the ERα dataset, logRBA ranged from −4.50 to 2.81; the logRBA range for ERβ dataset was −2.00 to 2.91. Because ERα binding affinity data was derived from several public sources, for some compounds multiple measurements were reported. A concordance analysis of these duplicate measurements from different sources revealed high correlation (R2=0.86, N=18) of binding affinity suggesting a reasonably high reliability and consistency of the information.
There were 131 overlapping compounds between ERα and ERβ binding affinity datasets. Although the correlation between ERα and ERβ binding affinity for these 131 compounds is significant (R2=0.46, p<0.001), a number of these ER ligands still have largely different binding affinities to ERα and ERβ (i.e., many are ER subtype-selective ligands). Therefore separate computational predictors for ERα and ERβ binding affinity were developed.
QSAR models for ERα and ERβ were built separately using conventional STL QSAR modeling approach. STL QSAR models of ERα binding affinity showed high external predictive accuracy (R2=0.73 for 5-fold external cross validation) (Figure 2). This result appears similar to or better than those reported previously (Lo Piparo and Worth, 2010). However, direct comparison among models is difficult because our dataset was larger than any of the previous modeling sets with continuous binding affinity data. Generally, the increased size of a modeling set should enlarge the AD of resulting models. This is particularly important for QSAR models that aim to predict environmental chemical hazards, because those compounds tend to be structurally diverse. For instance, the AD of our ERα models (based on 546 compounds) was compared with the AD of the models reported in the literature for the well-known EDKB binding affinity dataset (232 chemicals). ADs were calculated based on the same set of 432 Dragon descriptors for both datasets. The comparison showed that all compounds found in the EDKB binding affinity dataset were within the AD of our ERα models, while only 73% of the 546 compounds in our ERα dataset were within the AD of models developed based on the EDKB dataset. Relatively high concordance between the ADs of these datasets is probably due to the fact that the majority of ER binders belong to a small number of chemical classes (phenols, steroids, etc.); thus, once a dataset has representatives from these classes, inclusion of additional compounds may have minor impact. Still, it can be concluded that the large size of our newly compiled dataset resulted in an extended AD for the new models.
Figure 2. External prediction accuracy of MTL (black bars) and STL (white bars) QSAR models estimated from 5-fold external cross validation.

AD - applicability domain; STL - conventional single-task learning QSAR modeling approach; MTL - multi-task learning QSAR modeling approach. Vertical lines above bars indicate the mean absolute prediction error. *, Significantly different (p<0.05) between models as indicated by brackets.
Predictive accuracy of STL ERβ models (R2=0.32 for 5-fold external cross validation results) was considerably less than that of ERα. One possible explanation of this result is the smaller size and higher diversity of the ERβ dataset (the average Tanimoto coefficient between each chemical and its 10 nearest neighbor compounds (i.e., local similarity) ERβ was 0.81, while it was 0.85 for ERα).
Next, we explored whether MTL method would improve the accuracy of the ERβ models. MTL method was substituted for STL kNN in the standard QSAR modeling workflow, which allowed for the simultaneous modeling of ERα and ERβ binding affinity. Previous MTL QSAR studies suggested that this method, along with other inductive knowledge transfer approaches, improves prediction accuracy when the tasks are related (Varnek et al., 2009). ERα and ERβ can be considered as related since they belong to the same protein family, have moderately conserved ligand binding domains, and binding affinity for common ligands is moderately correlated (R2=0.46, N=131, p<0.001). We found that MTL method significantly improved predictive accuracy of ERβ binding affinity models (Figure 2, R2 increased from 0.32 to 0.53, p<0.05 by two-tailed Student’s t-test between prediction errors by STL vs. MTL models). At the same time, predictive accuracy of MTL ERα models (R2=0.71) was not different from that of STL ERα models (R2 =0.73).
Several factors that may be responsible for the improvements in predictive accuracy achieved by MTL model have been suggested (Caruana, 1997). Our results show that overlapping compounds [present in both ERα and ERβ binding affinity datasets, so-called “representation bias” (Caruana, 1997)] were not essential for the improved predictive accuracy by MTL models (Supplemental Figure 2). Alternative explanation of the improvement is data amplification: the size of ERβ dataset may be insufficient to afford predictive conventional (STL) QSAR models. However, by training ERα and ERβ models simultaneously using MTL method, the ERβ dataset was effectively enlarged by additional information from the ERα dataset (joint fitness function and common variable selection). Thus, predictive accuracy of ERβ, but not ERα, models was significantly improved. Similar findings were reported by (Varnek et al., 2009) who found that MTL improves model predictivity, especially for relatively smaller datasets.
Filtering by applicability domain yields only a modest increase in predictive accuracy of both STL and MTL models (Figure 2). Y-randomization test demonstrated the robustness of all QSAR models. For example, there were 276 MTL models satisfying the acceptance criteria (see Methods) for both training and test sets; however, no models satisfying these criteria were obtained in the Y-randomization test.
Model Interpretation by the Means of Descriptor Analysis and Implications for the Design of Chemicals to Minimize Endocrine Disruption Potential
Some descriptors occurred repeatedly in both STL and MTL models, suggesting that they represent important chemical features for predicting ER binding affinity. We focused on a set of 87 most frequently utilized descriptors that were shared by STL ERα and both MTL models (STL ERβ models were excluded due to inferior predictivity). For these descriptors (see Supplemental Tables 2 and 3), we compared mean values for the strong and the weak ER binders (defined by activity thresholds of logRBA=1 and −1, respectively). Figure 3 (left panel) shows that for ERα, many descriptors exhibited substantially different mean values between the two groups when all chemicals were considered. Such variation implies that these descriptors could potentially serve as determinants of ERα binding affinity. Similar observation was made for ERβ (figure not shown, data is included in Supplemental Table 3). Indeed, Figure 4 shows patterns of chemical descriptor profiles for several examples of strong and weak ERα or ERβ binders. These plots clearly illustrate that not only average, but also individual chemical’s descriptor profiles show appreciable divergence between certain descriptor values for strong vs. weak binders.
Figure 3. Frequency profiles of chemical descriptors for ER binders.

Activity threshold to define strong and weak binders is logRBA=1 and logRBA=−1, respectively. Shown are the 87 descriptors used most frequently in STL ERα and MTL (both receptors) models. Descriptor values were normalized to fall within the range of [0, 1]. Left panel, descriptor profile comparison for strong (solid line) vs. weak (dashed line) binders of ERα. Right panel, impact of the individual descriptors on the relative binding affinity to ERα vs. ERβ. Each bar shows the difference between mean descriptor values for strong vs. weak binders for ERα (black bars) and ERβ (white bars).
Figure 4.

Examples of the descriptor profiles for strong and weak ERα (left panel) and ERβ (right panel) binders.
We posit that such analysis affords the interpretation of QSAR models in terms of inherent chemical properties that may contribute to the chemical’s endocrine disruption potential. Indeed, the nature of Dragon descriptors employed in this study (e.g., topological descriptors or connectivity indices, or atom and bond counts) does not allow for straightforward model interpretation in terms of common ER scaffolds, such as steroids or phenols. However, those descriptors that map onto relatively small chemical features and are discriminatory can be useful in assessing what chemical modification may affect ER binding potential. For illustration, several examples of chemical modifications (suggested by the descriptor analysis as described above) that reduce the binding affinity of a known ERβ binder are shown in Table 3. For example, ARR (aromatic ratio) and B01[NO] (presence of N-O motif) have a negative impact on binding affinity to ERβ, while B09[CO] (presence of C…O motif at topological distance of 9) and nArOH (number of OH groups attached to aromatic ring) have a positive impact. In order to reduce the binding affinity of this ER binder, the values of descriptors with negative impacts should be increased, or the values of descriptors with positive impacts should be decreased. For instance, the aromatic ratio (descriptor ARR) could be increased by removing aliphatic atoms (Table 3). Indeed, by doing so, both predicted and experimentally-derived binding affinity of the resulting compound are −0.44 and −0.68, which is about 1 log unit less than the affinity of the original chemical.
Table 3.
Examples of structural modifications that are expected to reduce ER binding affinity (based on the analysis of significant descriptors in QSAR models).
| ERβ Binder | Structural features | Modified Chemicals | Predicted ERβ binding affinity by MTL models | Experimental ERβ binding affinity | |
|---|---|---|---|---|---|
| Descriptor | Suggested modification | ||||
 logRBAERβ=0.64 ARR=0.79 B01[NO]=0 B09[OC]=1 nArOH=0.2 |
ARR (negatively correlated with binding affinity) | Increase aromatic ratio by removing alkyl chain |
 ARR=0.89; % Dscr profile changed=92 |
−0.44 | −0.68 |
| B01[NO] (negatively correlated with binding affinity) | Add N-O motif |
 B01[NO]=1; %Dscr profile changed=85 |
−0.42 | N/A | |
| B09[OC] (positively correlated with binding affinity) | Remove O-C at topological distance 9 |
 B09[OC]=0; %Dscr profile changed=60 |
−0.15 | N/A | |
| Modifications Not Suggested |
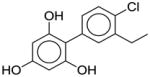 nArOH=0.6; %Dscr profile changed=16 |
1.17 | N/A | ||
| nArOH (positively correlated with binding affinity) | Add OH groups attached to aromatic ring | ||||
% Dscr profile changed indicates the percentage of descriptors changed in the favorable direction due to the structural modifications. In order to decrease a compound binding affinity, the values should be decreased (increased) for descriptors that positively (negatively) correlate with ER binding affinity. The values of “%Dscr profile changed” were calculated as (number of positively correlated descriptors whose values decreased + number of negatively correlated descriptors whose values increased)/(total number of descriptors).
N/A, not available.
While the potency-defining descriptors may guide structural modifications and lead to the enhancement or reduction of ER binding potency, it is usually difficult to determine whether and by how much the binding affinity of a compound can be changed merely by considering few individual descriptors. Instead, these descriptors (and underlying chemical features) should be viewed as having high priority for chemists to consider when structural modifications modulating the estrogenic potential of chemicals are desired. It must be emphasized that resulting changes to the entire descriptor profiles need to be considered in order to determine whether a structural modification is desirable or not. For example, if structural modifications leading from strong to weak binding are taken as favorable, and aromatic ratio is to be increased as in the first example of Table, 92% of the 87 frequent descriptors will also change in the favorable direction (i.e., the values of descriptors with positive (negative) impacts are reduced (increased)). This understandably leads to a large decrease in binding affinity for the compound of interest (logRBA from 0.64 to −0.68).
Figure 3 (right panel) also illustrates a comparison between the impact of selected descriptors on ERα vs. ERβ binding affinity, when assessed as the differences of mean descriptor values of strong and weak binders. Interestingly, most of the selected descriptors have a similar impact on binding affinity for both ER subtypes (black and white bars are in the same direction). However, the impacts of B09[CO] (topological index) and ARR are subtype-independent, while JG16 (topological charge index) and nPyrazoles (number of pyrazoles) are subtype-selective. These comparisons could also provide more specific suggestions for structural modifications to chemists who aim to convert potential EDCs into safer compounds in an ER-subtype specific manner. For the purpose of identifying potential environmental hazards, which could be ERα and/or ERβ binders, subtype selectivity may not be as critical as in drug design selective ER modulation is desired.
Docking Studies
It is important to predict functional behavior of chemicals, i.e., whether ER binders will act as receptor agonists or antagonists; thus, structure-based docking studies using agonist- or antagonist-bound receptor conformations were conducted. ER agonists/antagonists (67/39 for ERα and 48/32 for ERβ) were compared to presumed decoys (2570/1448 for ERα, and 1000/1000 for ERβ). We confirmed that agonists/antagonists and their respective presumed decoys have similar physico-chemical properties (Supplemental Figure 3).
Compounds were docked to the ERα or ERβ agonist or antagonist protein conformations and their enrichment factors and AUCs were compared to establish the discriminatory power of structure-based functional annotation of ER ligands. We found that all protein conformations were able to discriminate their corresponding ligands from presumed decoys (Figure 5). Moreover, all protein conformations could successfully enrich their corresponding ligands (agonists or antagonists) with high selectivity (EFmax ranges from 15 to 229) (Figure 5). These results indicate that each receptor conformation is capable of accurately recognizing the type of molecules it is expected to bind.
Figure 5. ROC and enrichment factor curves obtained as a result of docking studies using ER agonist and antagonist protein conformations.
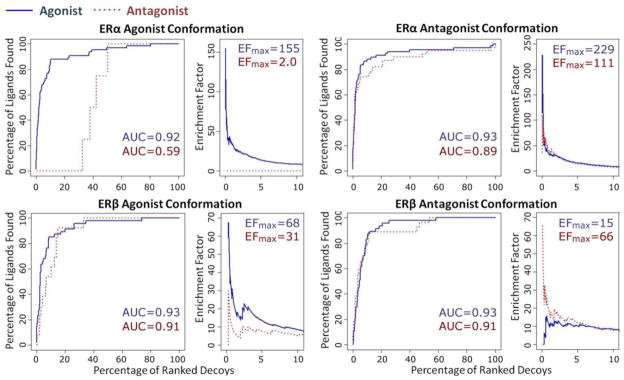
Blue solid lines and numbers indicate ER agonists, and red dotted lines and numbers indicate ER antagonists. Enrichment Factor (EF) is defined in the Methods. The numbers of known ER ligands and presumed decoys/non-binders are stated in Table 2.
ERα agonist conformation was superior for separating ERα agonists from antagonists (Figure 5 top left, AUCs for agonists/antagonists by ERα agonist confirmation are 0.92/0.59). Both ER antagonist confirmations were less capable than ER agonist confirmations of separating their respective ligands (Figure 5, AUCs for recognizing antagonists by ERα and ERβ antagonist conformations are 0.89 and 0.91, with no significant difference from their AUCs for agonists, 0.93 and 0.93, respectively). For comparison, AUCs for agonists by ERα and ERβ agonist confirmations are 0.92 and 0.93, higher than their AUCs for antagonists, 0.59 and 0.91, respectively. A possible explanation is that ER agonist conformations have relatively smaller ligand binding pocket than antagonist conformations due to the position of the helix 12 (see Supplemental Figure 1); as a result, it is quite difficult for antagonists to fit into agonist conformations as they are usually larger molecules than agonists (for instance, the average molecular weight is 286 and 427 for the ERα agonists and antagonists employed in this study, respectively).
QSAR- and Docking-based Virtual Screening
All four receptors were used, along with QSAR models, for parallel virtual screening of several external datasets. We reasoned that the consensus of all models represents a more reliable approach for discriminating ER binders from non-binders. After putative ER binders are selected by the consensus QSAR predictions of binding affinity, the docking models may be used to establish the functional activity of the binders. Based on our results described above, ERα agonist conformation demonstrated the best performance with regards to estimating putative binding affinity and characterizing agonism/antagonism of for ERα ligands.
The uterotrophic assay is an in vivo (rats or mice) endocrine disruption screening program screening test (EPA, 2012) for evaluating the ability of a chemical to elicit biological activities consistent with agonists or antagonists of natural estrogens (e.g., 17β-estradiol). A high correlation (R2=0.76) between logRP and logRBA was observed for 32 compounds in EDKB that were tested in both uterotrophic and ER binding affinity assays, which confirms the utility of this dataset in the validation of ER models. Both QSAR models and docking were used to evaluate their retrieving power for the 34 estrogenic chemicals from this virtual screening dataset. We found that QSAR models and ERα agonist conformation were capable of enriching active compounds (Figures 6A and C, AUC>0.7). Interestingly, a fairly high enrichment power by ERβ QSAR model (Figure 6A, AUC=0.89) indicates that this model is able to differentiate ERβ binders vs. non-binders. This observation further suggests that the relatively low external predictive accuracy of the ERβ model (R2=0.53) was probably due to the small size and structural diversity of the modeling and test datasets. In addition, 10 out of the 34 estrogenic compounds were ranked high by ERβ models but not ERα models (the predicted ERβ binding affinity was at least 1 log unit higher than the predicted ERα binding affinity), which suggests the ability of QSAR models to detect subtype-selective ER ligands. As expected, when comparing the performance of the four protein models in virtual screening, ERα agonist protein conformation outperformed others (Figures 6C and D). A possible explanation for this is that most of the active compounds in uterotrophic assay act via the activation of ERα (i.e., they are ERα agonists) which favors the ERα agonist conformation.
Figure 6. Results of virtual screening of the EDKB uterotrophic dataset using both QSAR models and ER docking.

ROC curves resulting from the use of QSAR models and docking models to identify 34 known estrogenic compounds in the uterotrophic dataset. (A) QSAR models; (B) enrichment factor curves of QSAR models; (C) Docking studies; (D) enrichment factor curves of docking studies. Consensus All: consensus results using all six models; Consensus QSAR: consensus prediction using both ERα and ERβ QSAR models; Consensus Docking: consensus results of docking using all four protein models.
It should be noted, however, that it is difficult to select the best model that would be most predictive for identifying compounds with a potential for ER activation-related hazard. Previous studies [e.g., aquatic toxicity (Zhu et al., 2008)] suggest that the consensus prediction based on the results obtained by all predictive models provide the most stable and reliable solutions. Therefore, the consensus predictions for the uterotrophic dataset compounds by QSAR models (CONSQSAR), docking models (CONSdock) and both QSAR and docking models (CONSall) were compared. Enrichment factors and ROC curves were plotted for each consensus predictor in order to compare their performance with each other, as well as with the individual models (Figures 6B and D). Indeed, we found that consensus prediction by both QSAR and docking models outperformed other models (Figure 6 and Supplemental Table 5).
Based on the encouraging results observed in the virtual screening of the uterotrophic dataset, both QSAR and docking models were then applied to the chemical library of ~3,500 compounds in the EDSP dataset. By averaging the predictions by MTL for ERα and ERβ QSAR models as well as by using four protein models, the consensus predictor initially ranked all compounds. Application of a conservative threshold (consensus Z-score=1) resulted in selection of 286 chemicals from this chemical library as potential ER-active EDCs with the highest confidence by the consensus between all models (Supplemental Table 4). These chemicals may be considered of the highest priority for further in vitro and in vivo endocrine disruption testing.
Conclusions
We have developed QSAR models for quantitative prediction of binding affinity to both subtypes of ER and showed the use of MTL was critical to improve the model prediction power for the smaller ERβ dataset. We have analyzed the models for significant chemical descriptors. As a result of this analysis, we posit that descriptors that were most frequently used in QSAR models may be interpreted as chemical features that influence ER binding affinity and thus may be used to suggest structural modifications to diminish potential ER binding hazard. Several examples (Table 3) were used to illustrate how such descriptor analysis as part of model interpretation may facilitate the design of safer chemicals. Another important methodological outcome of this study is the concurrent use of structure-based docking as a complement to QSAR models for binding affinity. All four protein conformations were able to discriminate corresponding ligands from presumed decoys/non-binders. In addition, ERα agonist conformation worked best in discriminating agonists from antagonists. Thus, structure-based methods (when possible due to the availability of target protein structures) may serve as a crucial complement to ligand-based approaches for understanding the mechanism of endocrine disruption and facilitating the identification of previously unknown ligands in chemical libraries. Indeed, virtual screening of the EDKB uterotrophic dataset demonstrated that the consensus predictions by QSAR and protein models outperformed individual models. A prioritized (i.e., potential ER-mediated endocrine disruptors) set of 286 compounds was generated from a large library of EDSP chemicals to show that models developed and employed in this study (publicly available from (http://chembench.mml.unc.edu/)) can be used as effective computational pre-screening tool to prioritize chemicals for further experimental testing.
Supplementary Material
HIGHLIGHTS.
This is the largest curated dataset inclusive of ERα and β (the latter is unique)
New methodology that for the first time affords acceptable ERβ models
A combination of QSAR and docking enables prediction of affinity and function
The results have potential applications to green chemistry
Models are publicly available for virtual screening via a web portal
Acknowledgments
This work was supported, in part, by grants from NIH (GM076059) and EPA (RD83499901, RD83382501). The content does not necessarily reflect the views and policies of the EPA and mention of trade names or commercial products does not constitute endorsement or recommendation for use.
List of abbreviations
- AD
Applicability Domain
- ADMET
Absorption, Distribution, Metabolism, Excretion, and Toxicity
- AhR
Aryl Hydrocarbon Receptor
- AR
Androgen Receptor
- AUC
Area Under the Curve
- E2
17β-estradiol
- ER
Estrogen Receptor
- EDCs
Endocrine Disrupting Chemicals
- EDKB
Endocrine Disruptor Knowledge Base
- EDSP
Endocrine Disruptor Screening Program
- EF
Enrichment Factor
- EPA
US Environmental Protection Agency
- kNN
k-Nearest Neighbors
- MTL
Multi-Task Learning
- PDB
Protein Data Bank
- QSAR
Quantitative Structure-Activity Relationships
- RBA
Relative Binding Affinity
- ROC
Receiver Operating Characteristic
- RP
Relative Potency
- SE
Sensitivity
- SP
Specificity
- STL
Single-Task Learning
Footnotes
Conflict of Interest:
Antreas Afantitis, Varnavas D. Mouchlis and Georgia Melagraki are employed by Novamechanics, Ltd., an in silico drug design company. Other authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler S, Basketter D, Creton S, Pelkonen O, van Benthem J, Zuang V, Andersen KE, Angers-Loustau A, Aptula A, Bal-Price A, Benfenati E, Bernauer U, Bessems J, Bois FY, Boobis A, Brandon E, Bremer S, Broschard T, Casati S, Coecke S, Corvi R, Cronin M, Daston G, Dekant W, Felter S, Grignard E, Gundert-Remy U, Heinonen T, Kimber I, Kleinjans J, Komulainen H, Kreiling R, Kreysa J, Leite SB, Loizou G, Maxwell G, Mazzatorta P, Munn S, Pfuhler S, Phrakonkham P, Piersma A, Poth A, Prieto P, Repetto G, Rogiers V, Schoeters G, Schwarz M, Serafimova R, Tahti H, Testai E, van Delft J, van Loveren H, Vinken M, Worth A, Zaldivar JM. Alternative (non-animal) methods for cosmetics testing: current status and future prospects-2010. Arch Toxicol. 2011;85:367–485. doi: 10.1007/s00204-011-0693-2. [DOI] [PubMed] [Google Scholar]
- Branham WS, Dial SL, Moland CL, Hass BS, Blair RM, Fang H, Shi L, Tong W, Perkins RG, Sheehan DM. Phytoestrogens and mycoestrogens bind to the rat uterine estrogen receptor. J Nutr. 2002;132:658–664. doi: 10.1093/jn/132.4.658. [DOI] [PubMed] [Google Scholar]
- Caruana R. Multitask learning. Machine Learning. 1997;28:41–75. [Google Scholar]
- Celik L, Davey J, Lund D, Schiott B. Exploring interactions of endocrine-disrupting compounds with different conformations of the human estrogen receptor alpha ligand binding domain: a molecular docking study. Chem Res Toxicol. 2008;21:2195–2206. doi: 10.1021/tx800278d. [DOI] [PubMed] [Google Scholar]
- Celik L, Lund JD, Schiott B. Conformational dynamics of the estrogen receptor alpha: molecular dynamics simulations of the influence of binding site structure on protein dynamics. Biochemistry. 2007;46:1743–1758. doi: 10.1021/bi061656t. [DOI] [PubMed] [Google Scholar]
- Demchuk E, Ruiz P, Chou S, Fowler BA. SAR/QSAR methods in public health practice. Toxicol Appl Pharmacol. 2011;254:192–197. doi: 10.1016/j.taap.2010.10.017. [DOI] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D, Xu L, Fang H, Hong H, Perkins R, Harris S, Bearden ED, Shi L, Tong W. The EDKB: an established knowledge base for endocrine disrupting chemicals. BMC Bioinf. 2010;11(Suppl 6):S5. doi: 10.1186/1471-2105-11-S6-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA. Endocrine Disruptor Screening Program Test Guidelines - OPPTS 890.1600: Uterotrophic Assay [EPA 740-C-09-0010] 2012 [Google Scholar]
- Fang J, Akwabi-Ameyaw A, Britton JE, Katamreddy SR, Navas F, 3rd, Miller AB, Williams SP, Gray DW, Orband-Miller LA, Shearin J, Heyer D. Synthesis of 3-alkyl naphthalenes as novel estrogen receptor ligands. Bioorg Med Chem Lett. 2008;18:5075–5077. doi: 10.1016/j.bmcl.2008.07.121. [DOI] [PubMed] [Google Scholar]
- Fourches D, Muratov E, Tropsha A. Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J Chem Inf Model. 2010;50:1189–1204. doi: 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1 Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models. J Comput Aided Mol Des. 2003;17:241–253. doi: 10.1023/a:1025386326946. [DOI] [PubMed] [Google Scholar]
- Golbraikh A, Tropsha A. Beware of q2! J Mol. Graph Model. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: A new approach for rapid, accurate docking and scoring. 2 Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs MN, Janssens W, Bernauer U, Brandon E, Coecke S, Combes R, Edwards P, Freidig A, Freyberger A, Kolanczyk R, Mc Ardle C, Mekenyan O, Schmieder P, Schrader T, Takeyoshi M, van der Burg B. The use of metabolising systems for in vitro testing of endocrine disruptors. Curr Drug Metab. 2008;9:796–826. doi: 10.2174/138920008786049294. [DOI] [PubMed] [Google Scholar]
- Koehler KF, Helguero LA, Haldosen LA, Warner M, Gustafsson JA. Reflections on the discovery and significance of estrogen receptor beta. Endocr Rev. 2005;26:465–478. doi: 10.1210/er.2004-0027. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- LigPrep, Suite 2011: LigPrep, version 2.5. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- Lo Piparo E, Worth A. Tools for predicting developmental and reproductive toxicity. Institute for Health and Consumer Protection, Joint Research Centre, European Commission; Luxembourg: 2010. http://ihcp.jrc.ec.europa.eu/our_labs/predictive_toxicology/doc/EUR_24522_EN.pdf. [Google Scholar]
- Maestro, Suite 2011: Maestro, version 9.2. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- Mewshaw RE, Bowen SM, Harris HA, Xu ZB, Manas ES, Cohn ST. ERbeta ligands. Part 5: synthesis and structure-activity relationships of a series of 4′-hydroxyphenyl-aryl-carbaldehyde oxime derivatives. Bioorg Med Chem Lett. 2007;17:902–906. doi: 10.1016/j.bmcl.2006.11.066. [DOI] [PubMed] [Google Scholar]
- Minutolo F, Macchia M, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor beta ligands: recent advances and biomedical applications. Med Res Rev. 2011;31:364–442. doi: 10.1002/med.20186. [DOI] [PubMed] [Google Scholar]
- Mouchlis VD, Mavromoustakos TM, Kokotos G. Molecular docking and 3D-QSAR CoMFA studies on indole inhibitors of GIIA secreted phospholipase A(2) J Chem Inf Model. 2010;50:1589–1601. doi: 10.1021/ci100217k. [DOI] [PubMed] [Google Scholar]
- Mueller SO, Korach KS. Estrogen receptors and endocrine diseases: lessons from estrogen receptor knockout mice. Curr Opin Pharmacol. 2001;1:613–619. doi: 10.1016/s1471-4892(01)00105-9. [DOI] [PubMed] [Google Scholar]
- Protein Preparation Wizard, Suite 2011: Schrödinger Suite 2011 Protein Preparation Wizard; Epik version 2.2. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]; Impact version 5.7. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]; Prime version 3.0. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- Rotroff DM, Dix DJ, Houck KA, Knudsen TB, Martin MT, McLaurin KW, Reif DM, Crofton KM, Singh AV, Xia M, Huang R, Judson RS. Using in vitro high throughput screening assays to identify potential endocrine-disrupting chemicals. Environ Health Perspect. 2013;121:7–14. doi: 10.1289/ehp.1205065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucker C, Rucker G, Meringer M. y-Randomization and its variants in QSPR/QSAR. J Chem Inf Model. 2007;47:2345–2357. doi: 10.1021/ci700157b. [DOI] [PubMed] [Google Scholar]
- Salum LB, Polikarpov I, Andricopulo AD. Structural and chemical basis for enhanced affinity and potency for a large series of estrogen receptor ligands: 2D and 3D QSAR studies. J Mol Graph Model. 2007;26:434–442. doi: 10.1016/j.jmgm.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Schug TT, Janesick A, Blumberg B, Heindel JJ. Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol. 2011;127:204–215. doi: 10.1016/j.jsbmb.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedykh A, Zhu H, Tang H, Zhang L, Richard A, Rusyn I, Tropsha A. Use of in vitro HTS-derived concentration-response data as biological descriptors improves the accuracy of QSAR models of in vivo toxicity. Environ Health Perspect. 2011;119:364–370. doi: 10.1289/ehp.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafimova R, Todorov M, Nedelcheva D, Pavlov T, Akahori Y, Nakai M, Mekenyan O. QSAR and mechanistic interpretation of estrogen receptor binding. SAR QSAR Environ Res. 2007;18:389–421. doi: 10.1080/10629360601053992. [DOI] [PubMed] [Google Scholar]
- Shanle EK, Xu W. Endocrine disrupting chemicals targeting estrogen receptor signaling: identification and mechanisms of action. Chem Res Toxicol. 2011;24:6–19. doi: 10.1021/tx100231n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat Struct Biol. 2002;9:359–364. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- Sung E, Turan N, Ho PW, Ho SL, Jarratt PD, Waring RH, Ramsden DB. Detection of endocrine disruptors - from simple assays to whole genome scanning. Int J Androl. 2012;35:407–414. doi: 10.1111/j.1365-2605.2012.01254.x. [DOI] [PubMed] [Google Scholar]
- Taha MO, Tarairah M, Zalloum H, Abu-Sheikha G. Pharmacophore and QSAR modeling of estrogen receptor beta ligands and subsequent validation and in silico search for new hits. J Mol Graph Model. 2010;28:383–400. doi: 10.1016/j.jmgm.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Tong W, Xie Q, Hong H, Shi L, Fang H, Perkins R. Assessment of prediction confidence and domain extrapolation of two structure-activity relationship models for predicting estrogen receptor binding activity. Environ Health Perspect. 2004;112:1249–1254. doi: 10.1289/txg.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropsha A. Best practices for QSAR model development, validation, and exploitation. Mol Inf. 2010;29:1868–1751. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- Tsakovska I, Pajeva I, Alov P, Worth A. Recent advances in the molecular modeling of estrogen receptor-mediated toxicity. Adv Protein Chem Struct Biol. 2011;85:217–251. doi: 10.1016/B978-0-12-386485-7.00006-5. [DOI] [PubMed] [Google Scholar]
- van Lipzig MM, ter Laak AM, Jongejan A, Vermeulen NP, Wamelink M, Geerke D, Meerman JH. Prediction of ligand binding affinity and orientation of xenoestrogens to the estrogen receptor by molecular dynamics simulations and the linear interaction energy method. J Med Chem. 2004;47:1018–1030. doi: 10.1021/jm0309607. [DOI] [PubMed] [Google Scholar]
- Varnek A, Gaudin C, Marcou G, Baskin I, Pandey AK, Tetko IV. Inductive transfer of knowledge: application of multi-task learning and feature net approaches to model tissue-air partition coefficients. J Chem Inf Model. 2009;49:133–144. doi: 10.1021/ci8002914. [DOI] [PubMed] [Google Scholar]
- Vedani A, Smiesko M. In silico toxicology in drug discovery - concepts based on three-dimensional models. Altern Lab Anim. 2009;37:477–496. doi: 10.1177/026119290903700506. [DOI] [PubMed] [Google Scholar]
- Vedani A, Smiesko M, Spreafico M, Peristera O, Dobler M. VirtualToxLab - in silico prediction of the toxic (endocrine-disrupting) potential of drugs, chemicals and natural products. Two years and 2,000 compounds of experience: a progress report. ALTEX. 2009;26:167–176. doi: 10.14573/altex.2009.3.167. [DOI] [PubMed] [Google Scholar]
- Walker T, Grulke CM, Pozefsky D, Tropsha A. Chembench: a cheminformatics workbench. Bioinformatics. 2010;26:3000–3001. doi: 10.1093/bioinformatics/btq556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhu H, Oprea TI, Golbraikh A, Tropsha A. QSAR modeling of the blood-brain barrier permeability for diverse organic compounds. Pharm Res. 2008;25:1902–1914. doi: 10.1007/s11095-008-9609-0. [DOI] [PubMed] [Google Scholar]
- Zheng W, Tropsha A. Novel variable selection quantitative structure--property relationship approach based on the k-nearest-neighbor principle. J Chem Inf Comput Sci. 2000;40:185–194. doi: 10.1021/ci980033m. [DOI] [PubMed] [Google Scholar]
- Zhu H, Tropsha A, Fourches D, Varnek A, Papa E, Gramatica P, Oberg T, Dao P, Cherkasov A, Tetko IV. Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis. J Chem Inf Model. 2008;48:766–784. doi: 10.1021/ci700443v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.