

Abstract
Therapeutically engineered stem cells (SC) are emerging as an effective tumor-targeted approach for different cancer types. However, the assessment of the long-term fate of therapeutic SC post-tumor treatment is critical if such promising therapies are to be translated into clinical practice. In this study, we have developed an efficient stem cell based therapeutic strategy that simultaneously allows killing of tumor cells and assessment and eradication of SC post highly malignant glioblastoma multiforme (GBM) treatment. Mesenchymal stem cells (MSC) engineered to co-express the prodrug converting enzyme, herpes simplex virus thymidine kinase (HSV-TK) and a potent and secretable variant of tumor necrosis factor apoptosis-inducing ligand (S-TRAIL), induced caspase-mediated GBM cell death and showed selective MSC sensitization to the prodrug ganciclovir (GCV). A significant decrease in tumor growth and a subsequent increase in survival were observed when mice bearing highly aggressive GBM were treated with MSC co-expressing S-TRAIL and HSV-TK. Furthermore, the systemic administration of GCV post-tumor treatment selectively eliminated therapeutic MSC expressing HSV-TK in vitro and in vivo, which was monitored in real time by positron emission-computed tomography (PET) imaging utilizing 18F-FHBG, a substrate for HSV-TK. These findings demonstrate the development and validation of a novel therapeutic strategy that has implications in translating stem cell based therapies in cancer patients.
Keywords: stem cells, HSV-TK, GBM, in vivo imaging, TRAIL
INTRODUCTION
Despite considerable advances in brain tumor therapy, glioblastoma multiforme (GBM) remains one of the most challenging diseases, particularly because of its highly invasive nature, which precludes surgical removal, and its resistance to a number of antitumor agents [1]. The ability of mesenchymal stem cells (MSCs) to preferentially migrate towards local and disseminated malignant disease and their non-immunogenic nature presents them as the most attractive candidates for cell based therapies in humans [2]. Recent evidence by our laboratory and others have shown that neural stem cells (NSC) and MSC migrate toward GBMs [3-5]. MSC mediated delivery of anti-tumor agents like a potent and secretable variant of tumor necrosis factor apoptosis-inducing ligand (S-TRAIL) [6, 7] is a very effective method of delivering this tumor specific anticancer agent to both established and resected tumors in our recently created mouse model of GBM resection [8]. However, in order to avoid continuous access of anti-tumor agents to normal tissue and to circumvent any malignant transformation of MSC, it is critical to develop and test MSCs that simultaneously allow killing of tumor cells, follow the fate of therapeutic MSCs with a clinically relevant non-invasive imaging method and ultimately selectively eradicate MSC post-tumor treatment.
Suicide gene therapy is based on transferring a gene encoding a suicide protein into cells for their selective elimination, and herpes simplex virus thymidine kinase (HSV-TK) is the most widely used in suicide therapy [9]. Expression of HSV-TK within a cell selectively sensitizes it to the prodrug ganciclovir (GCV) by preferential monophosphorylation of nontoxic GCV into a toxic compound by the viral TK enzyme [9]. This toxic metabolite can be transferred from a cell expressing the HSV-TK to adjacent cells that do not express HSV-TK by diffusion through gap junctions inducing neighboring cell death mediated by bystander effect. In addition, since HSV-TK has the capacity to phosphorylate a variety of substrates that cannot be phosphorylated by the mammalian TK, HSV-TK can be utilized as a marker for positron emission-computed tomography (PET) imaging [10] in combination with different radioactive substrates such as 18F-FHBG [11] 18F-FEAU [12] or 124I-FAIU [13], which will be trapped intracellularly due to HSV-TK-mediated phosphorylation.
Recently, MSC have been used to deliver suicide gene therapies such as HSV-TK/GCV or cytosine deaminase/5-fluorouracil (CD/5-FU) to different types of tumors [14-16] including GBMs [17-19] and have led to a reduction of tumor growth and an increase in survival in mice post-GCV treatment. However, this anti-tumor therapy approach involves immediate killing of therapeutic stem cells before the complete elimination of the tumor. In the current study, we have developed an efficient stem cell based therapeutic strategy that simultaneously allows killing of tumor cells as well as assessment and eradication of stem cells post-tumor treatment. To our knowledge, this is the first report that describes stem cell-based therapeutic approach that simultaneously allows tumor cell specific killing, clinically relevant imaging of the fate of stem cells and assessment of the safety of therapeutic MSCs by selectively sensitizing the stem cells to the prodrug GCV.
MATERIAL AND METHODS
Cell Culture and reagents
Human bone marrow-derived mesenchymal stem cells (hMSC) were obtained from A&M Health Science Center Institute for Regenerative Medicine (Temple, TX, USA) and grown in Alpha- modified Eagle’s medium (Invitrogen, Carlsbad, CA; www.invitrogen.com/gibco) with 20% fetal bovine serum (FBS), 2-4 mM L-glutamine and 1% penicillin/streptomycin 100 U/mL penicillin and 100μg/mL streptomycin (P/S). Human GBM cells, U87-MG and Gli36 expressing a constitutively active variant of Epidermal growth factor receptor (EGFR) (Gli36vIII) were grown as described [7]. 3T3 murine fibroblast cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA; www.atcc.org) and grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS and 1% of (P/S). Human and mouse MSC were kindly provided by Dr. Darwin Prockop, University of Texas. Human MSC were grown as previously described (20) and mouse (m) MSC were grown in DMEM containing 10% FBS, 10% horse serum and 1% of (P/S). GCV was obtained from the in-patient pharmacy at Massachusetts General Hospital, Boston, MA. A stock solution at 100mg/mL and dilutions were prepared in phosphate buffered saline (PBS) according to the manufacturer’s instructions.
Generation of viral vectors and transduction of cells
The following lentiviral (LV) and retroviral (RV) vectors were used in this study: LV-pico2-Fluc-mCherry expressing Firefly luciferase and mCherry (FmC), a kind gift from Dr. Andrew Kung (Dana Farber Cancer Institute; Boston, MA), LV-GFP-Fluc (GFl), LV-HSV-TK (TK), LV-S-TRAIL (TR), RV-GFP-Fluc, RV-HSV-TK and RV-S-TRAIL. Lentiviral vector, pLV-CS-CGW bearing an internal ribosomal entry site-green fluorescent protein (IRES-GFP) element [21], was used as a backbone. HSV-TK was PCR amplified using HSV-TK genome (kindly provided by Dr. Hiroaki Wakimoto, Massachusetts General Hospital; Boston, MA) as a template and primers with restriction sites NheI and XhoI at 5’ and 3’ ends respectively. The resulting HSV-TK fragment was digested with NheI and XhoI and ligated in-frame into NheI/XhoI digested pLV-CSC-IG vector. Retroviral vector, MGRi bearing an IRES-GFP (kindly provided by Dr. Manuel Caruso, Centre Hospitalier Universitaire de Québec, Canada) was used as a backbone. CMV-HSV-TK and CMV-S-TRAIL fragments were purified by digestion of LV-HSV-TK and LV-S-TRAIL. The resulting fragments were ligated in-frame into a MGRi vector. All lentiviral constructs were packaged as LV vectors in 293T cells using a helper virus-free packaging system as previously described [22]. All retroviral constructs were packaged as RV vectors by transient transfection of 293T cells. Briefly, cells (15×106) were seeded in 150mm2 tissue culture plates with DMEM with 10% FBS 24h before transfection and cells were refed with fresh medium 4h before transfection. Transfection was performed by calcium phosphate precipitation method using 50μg of transfer plasmid DNA and the RV helper plasmid pCL-Eco (25μg; Addgene). Cells were washed with fresh medium 16–18h after transfection, and vector supernatants were harvested 48h and 72h after transfection. The supernatants were centrifuged and filtered (using a 0.45 μm filter) and loaded in a Beckman Quick-Seal ultracentrifuge tube (Beckman Coulter, Fullerton, CA) and centrifuged at 28,000 × g for 90 min. Pellets were re-suspended in desired media and stored at -80°C. Titers of the GFP expressing vectors were determined by counting fluorescent transduced 3T3 cells. GBM cells (U87-MG and Gli36vIII) were engineered to express FmC (U87-FmC and Gli36vIII-FmC) by transduction of LV-FmC at a multiplicity of infection (MOI) of 2 in medium containing protamine sulfate (8μg/mL) and selected with puromycin (4μg/mL). hMSC and mMSC were transduced with LV or RV respectively at a MOI of 10 and after 48h were sorted by GFP expression with a fluorescence-activated cell sorting (FACSAria Cell-Sorting System, BD Biosciences).
Cell viability and caspase assays
To determine the effects of GCV in MSC-TK cells, tumor or MSC cells were seeded on 96-well plates (5×103/well) and treated with different doses of GCV (0-40 μg/mL) 24h after plating. Cell viability was assessed at 48h post GCV treatment by using an ATP-dependent luminescent reagent (CellTiterGlo, Promega, Madison, WI; www.promega.com). All experiments were performed in triplicates. To assess bystander experiments, different proportions MSC-TK cells were co-cultured with MSC in 96-well plates (5×103 total cells/well) and treated with GCV (10 μg/mL) 24h after plating. The effect of GCV on viability was assessed at 72h post GCV treatment. Caspase 3/7 activity of tumor cells in the co-culture experiment was determined 24h after plating the cells using DEVD tetrapeptide-aminoluciferin (CaspaseGlo 3/7, Promega, Madison, WI; www.promega.com) according to manufacturer’s instructions. All experiments were performed in triplicates and repeated twice.
Conditioned media (CM) and co-culture experiments
To determine the effect of mMSC-TR and mMSC-TR-TK conditioned media (CM), different proportions of CM (obtained 48h post-plating of MSC) were mixed with previously plated U87-FmC and Gli36vIII-FmC in 96-well plates (2.5×103/well). The co-culture experiments of MSC with GBM cells were performed by plating U87-FmC and Gli36vIII-FmC in 96-well plates (2.5×103/well) with different proportions of engineered MSC-TR or MSC-TR-TK (0-50%). The effect of TRAIL secreted from engineered MSC on GBM cells was assessed after 3 days by measuring the Fluc activity of GBM cells as described previously [23]. To determine the fate of therapeutic MSC after tumor treatment, MSC, MSC-TR and MSC-TR-TK expressing GFl (MSC-TR-GFl or MSC-TR-TK-GFl) were co-cultured with GBM cells (1:1) and 2 days later, GCV (10 μg/mL) was added to the cultures. Three days post treatment, Fluc signal was measured and fluorescent images were taken. All experiments were performed in triplicates and repeated twice.
Western Blotting Analysis
GBM cells exposed to conditioned media of mMSC, mMSC-TR and mMSC-TR-TK for 24h were lysed with NP40 buffer supplemented with protease (Roche) and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO; www.sigmaaldrich.com). Ten μg of harvested proteins from each lysate were denatured and resolved on 10% SDS-PAGE, immunoblotted with antibodies against poly (ADP-ribose) polymerase (PARP) (Cell Signaling; 1:1000 dilution) or α-tubulin (Sigma-Aldrichm, St. Louis, MO; www.sigmaaldrich.com; 1:5000 dilution) and detected by chemiluminescence after incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies.
Intracranial cell implantation and bioluminescence imaging (BLI) imaging in vivo
To follow the fate of MSC after GCV treatment, hMSC-TK-GFl (2×105 cells) or mMSC-TK-GFl (5×105 cells) were implanted stereotactically (from bregma, AP: -2mm, ML: 1.5mm V (from dura): 2mm) (n= 8 mice in each case) into right frontal lobe of adult severe combined immunodeficiency (SCID) or nude mice brains, respectively. Mice were injected intraperitoneally with 4.5mg/mouse of D-luciferin, distributed in two groups and treated with GCV (50mg/kg) (n= 4) or control PBS (n= 4). MSC fate was determined by BLI as described earlier [8]. To determine the efficacy and safety of MSC-TR-TK, mice bearing Gli36vIII-FmC tumors (1×105 cells) were implanted stereotactically (from bregma, AP: -2mm, ML: 1.5mm V (from dura): 2mm) into the right frontal lobe of adult nude mice brains and 3 days later mice were imaged to confirm the presence of GBMs by BLI. The mice were then distributed into three groups and implanted intratumorally (from bregma, AP: -2mm, ML: 1.5mm V (from dura): 2mm) on two consecutive days with 3×105 mMSC-TR-TK (n=7), mMSC-TR (n=7) or injected with control PBS (n=14). The next day post- mMSC treatment, PBS treated mice were distributed into 2 groups (n=7 each) and injected intraperitonally with either GCV (50mg/kg) or PBS daily for 10 days. GBM burden in mice was followed by Fluc imaging over time as described previously [8]. Fourteen days post-MSC implantation, one mouse from each group was sacrificed and H&E staining was performed on brain sections as described below. The rest of the mice (n=6 per group) were followed for survival and sacrificed when neurological symptoms became apparent. Fourteen days post therapeutic MSC implantation, other set of Gli36vIII tumor bearing mice treated with mMSC-TR-TK or mMSC-TR were administered daily with GCV (50mg/kg) (n=4) or control PBS (n=4) intraperitonally during 10 days. Eighteen days post-GCV administration, mice from each group were sacrificed and mMSC viability was determined by GFP expression in brain sections. To quantify GFP intensity (indicative of mMSC survival), four random fields in a viable tissue zones for each tumor were captured at original magnification x10 and GFP cells were counted. All in vivo procedures were approved by the Subcommittee of Research Animal Care at Massachusetts General Hospital.
PET imaging
Mice implanted intracranially with mMSC-TK-GFl were fasted for 4h prior to imaging, anesthetized with 2% isoflurane and 100% oxygen, and injected with 500-600μCi of 18F-FHBG via tail vein. Two hours later, a static dataset was acquired for 15 minutes in one bed position (FOV 4.2 cm) using an energy window of 250-700 keV for each mouse. Images were reconstructed using a 2D OSEM algorithm with 2 iterations and 16 subsets. For measuring the mean and maximum standardized uptake values (SUVmean and SUVmax) and metabolic tumor volume (MTV), three-dimensional (3D) regions of interest were drawn over the tumors and uptake values were measured using the eXplore Vista software (GE Healthcare, Boston, MA; www.gehealthcare.com). The maximum intensity projection images were generated using ImageJ (NIH, Besthesda, MD; www.nih.gov). After the baseline PET scan, mice were treated daily with GCV (50mg/kg) and imaged again by PET at day 11 post GCV treatment.
Tissue processing, H&E staining and immunohistochemistry
Mice bearing GBMs, MSC or both GBMs and MSC were perfused and brains were removed and sectioned for histological analysis. Brain sections on slides were washed in PBS and mounted for microscopy to be visualized for fluorescence on a confocal microscope (LSM Pascal, Zeiss, Oberkochen, Germany; www.zeiss.com). For H&E staining, sections were incubated with Hematoxylin and EosinY (1% alcohol), dehydrated with 95% and 100% EtOH and mounted in xylene-based media. For CD31 staining, sections were incubated for 1 hour in a blocking solution (0.3% bovine serum albumin (BSA), 8% goat serum and 0.3% Triton-X100) at room temperature, followed by incubation at 4°C overnight with CD31 antibody diluted in blocking solution (Abcam; 1:50 dilution). Sections were washed three times with PBS, incubated at 4°C in Alexa Fluor 649 goat anti-rabbit secondary antibody diluted in blocking solution (Invitrogen, Carlsbad, CA; www.invitrogen.com/gibco; 1:500 dilution) and visualized as described above.
Statistical Analysis
Data were analyzed by Student’s t test when comparing 2 groups and by ANOVA, followed by Dunnetts post-test when comparing greater than 2 groups. Data were expressed as mean +SD in vitro studies and +SEM in vivo studies, and differences were considered significant at P< 0.05. Survival times of the mouse groups treated with mMSC-TR-TK, mMSC-TR or control PBS were compared using the log-rank test.
RESULTS
GCV induces selective cell killing in MSC-TK
Human (h) MSC and mouse (m) MSC were efficiently transduced with engineered LV and RV constructs encoding HSV-TK (supplemental Fig. 1A). A dose dependent decrease in hMSC-TK (Fig. 1A) and mMSC-TK (Fig. 1B) viability was observed in cultures treated with GCV. MSC-TK co-cultured with MSC showed that even low proportions of MSC-TK (25% in hMSC-TK and 1.6% in mMSC-TK) induced a significant decrease in MSC viability (Fig. 1C-D). Next, we engineered MSC-TK to express a bimodal fluorescent and bioluminescent marker, GFP-Fluc (MSC-TK-GFl). A direct correlation between MSC cell numbers, Fluc signal intensity and GFP-positive cells was seen in vitro within the ranges tested (supplemental Fig. 1B). In vivo BLI of mice bearing hMSC-TK-GFl (Fig. 1E) or mMSC-TK-GFl (Fig. 1F) treated with GCV showed a significantly reduced MSC viability as early as 6 days post-GCV treatment in comparison to control PBS treatment. As expected, mMSCs survived significantly longer than hMSCs in vivo, which is in line with the previous studies [20, 24]. These results show that MSC expressing HSV-TK can be eradicated after GCV treatment both in vitro and in vivo.
Fig.1. GCV induces selective cell killing in MSC-TK.
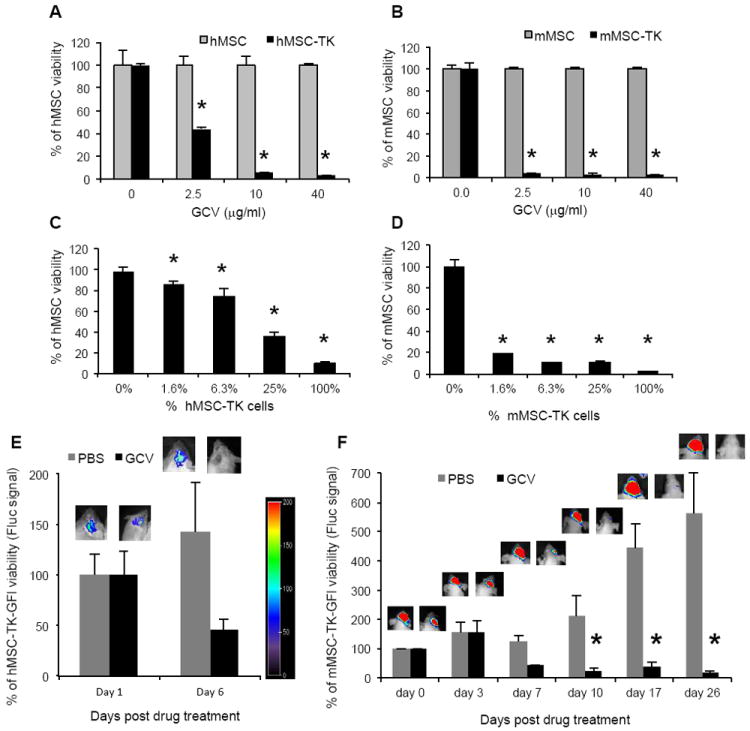
(A-B) hMSCs (A) or mMSC (B) were transduced with LV-TK or RV-TK respectively and 48hrs later were plated and incubated with serial dilutions of GCV (0-40 μg/mL). Plots show MSC viability at 72 hrs post GCV treatment. Bars, +SD. (C-D) Different proportions of hMSC-TK (C) or mMSC-TK (D) were plated with non-transduced hMSC or mMSC respectively and incubated with GCV (10μg/mL). Plots show MSC viability 72hrs post GCV treatment. Bars, + SD. All experiments were performed in triplicates and repeated twice. (E-F) hMSC-TK-GFl (E) or mMSC-TK-GFl (F) were implanted intracranially in adult SCID or nude mice respectively and treated with GCV (50mg/kg). Plots show percentage of MSC viability measured by Fluc intensities of control PBS and GCV group. Bars, +SE. One representative image of a mouse from each group and from each time point is shown. In all panels, *, p < 0.05 versus controls. Abbreviations: hMSC, human mesenchymal stem cells; mMSC, mouse mesenchymal stem cells; GCV, ganciclovir; GFl, green fluorescent protein and firefly luciferase fusion; TK, Thymidine Kinase; PBS, phosphate buffered saline.
PET imaging allows assessment of the fate of engineered MSC in vivo
To follow the MSC fate post GCV treatment, we performed PET imaging using 18F-FHBG, an optimal radiotracer to monitor HSV-TK activity that has high uptake rate and low background activity [4]. PET imaging allowed specific detection of intracranially implanted mMSC-TK in non-tumor bearing mice (Fig. 2A-B). As shown in representative images and summary graph, a significant decrease in 18-FHBG accumulation was seen in mMSC-TK bearing mice treated during 11 days with GCV (Fig. 2C-E). High levels of 18-FHBG were also observed in the gallbladder and kidneys, as renal clearance is the major route of elimination for the radiotracer (Fig. 2A-D). Fluorescence confocal microscopy pictures on brain sections from a representative mouse of each group confirmed the long-term significant decrease in number of GFP-positive cells in mice treated with GCV as compared to controls 50 days post GCV treatment (Fig. 2F-I). Together, these results show that MSC fate can be detected by clinically relevant PET imaging and confirm that MSC expressing HSV-TK can be eliminated selectively with GCV in vivo.
Fig.2. PET imaging allows assessment of the fate of engineered MSC in vivo.
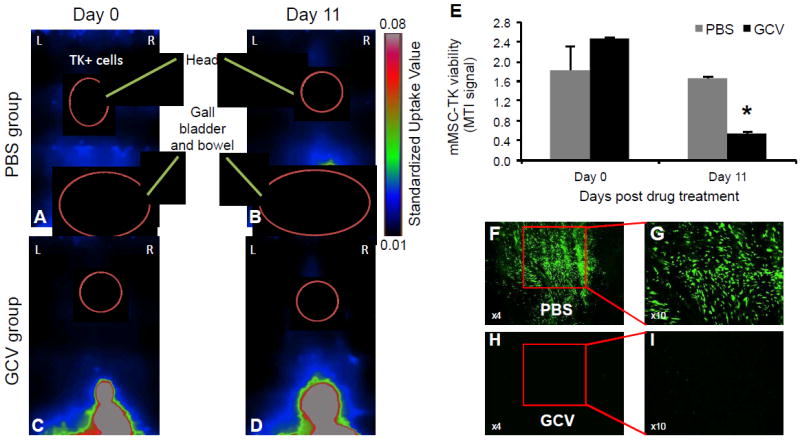
(A-D) Mice bearing mMSC-TK were imaged by PET using 18F-FHBG pre (A,B)- and post (C,D)- GCV treatment. Representative images of a mouse from each group are shown. (E) Plot shows MTI signal values pre and post GCV treatment from the control PBS and GCV group. MTI represents the amount of 18F-FHBG uptake in the entire tumor. Bars, +SD. (F-I) Low and high resolution photomicrographs (original magnification, x4 and x10 respectively) from brain sections show the long-term fate of mMSC-TK-GFP in control PBS and GCV injected mice at day 50 post GCV treatment. In all panels, *, p < 0.05 versus controls. Abbreviations: mMSC, primary mouse mesenchymal stem cells; GCV, ganciclovir; TK, Herpes Simplex Virus Thymidine Kinase; MTI, molecular tumor index; PBS, phosphate buffered saline.
Imaging MSC-TR-TK antitumor effects and fate in vitro
To develop therapeutic stem cells that allow simultaneous tumor specific killing and eradication of MSC post-tumor treatment, we engineered both human and murine MSC to co-express tumor specific, secretable (S)-TRAIL and HSV-TK. We also created human GBM lines U87-MG and Gli36vIII expressing FmC (U87-FmC and Gli36vIII-FmC) by transducing GBM cells with LV-FmC to monitor the effect of engineered MSCs on GBM growth in culture and in vivo. A direct correlation between GBM cell number, Fluc signal intensity and mCherry-positive cells was seen in vitro within the ranges tested (Supplemental Fig. 1C-D). A dose dependent decrease in GBM cell viability was seen in U87-FmC (Fig. 3A) and Gli36vIII-FmC (Fig. 3B) exposed to mMSC-TR and mMSC-TR-TK conditioned media (CM) in vitro. The expression of high levels of cleaved PARP, one of the effector proteins downstream of the caspase apoptosis pathway, in GBM cells exposed to mMSC-TR and mMSC-TR-TK CM confirmed the induction of apoptosis in the GBM cells (Fig. 3C). In addition, co-cultures of different percentages of mMSC-TR and mMSC-TR-TK with U87-FmC (Fig. 3D) and Gli36vIII-FmC (Fig. 3E) showed a decrease in cell viability measured by Fluc signal and confirmed by a decrease in mCherry-positive tumor cells. The increase in caspase-3/7 activity in Gli36vIII cells up to 4 fold of basal levels in co-cultures with mMSC-TR and mMSC-TR-TK confirmed the induction of the caspase activation pathway in the GBM cells (Fig. 3F). Similar results were obtained when these experiments were performed using human MSC engineered to express TRAIL (hMSC-TR) or TRAIL and HSV-TK (hMSC-TR-TK) (supplemental Fig. 2A-B).
Fig.3. MSC-TR and MSC-TR-TK have antitumor effect in vitro.
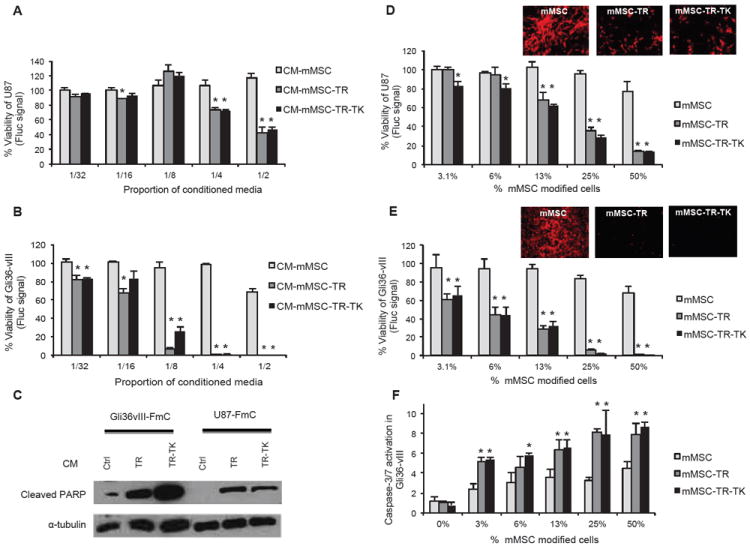
(A-E) U87-FmC and Gli36vIII-FmC were cultured with different proportions of conditioned media (CM) from engineered mMSC (A-B) or co-cultured with mMSC (white bars), mMSC-TR (grey bars) or mMSC-TR-TK (black columns) (D-E). Plots show percentage of tumor cell viability measured by Fluc signal 3 days later. Photomicrographs (original magnification, x4) showing mCherry positive cells were obtained from the co-cultures with 50% modified mMSC. All experiments were performed in triplicates and repeated twice. (C) Western Blotting analysis shows changes in PARP cleavage in Gli36vIII and U87 GBM cells exposed to CM from engineered mMSC. (F) Plot shows caspase-3/7 activation in the co-cultures with Gli36vIII and mMSC (white bars), mMSC-TR (grey columns) or mMSC-TR-TK (black columns) measured by CaspaseGlo assay at day one post plating. In all panels, Bars, +SD. Abbreviations: CM, conditioned media; mMSC, primary mouse mesenchymal stem cells; GCV, ganciclovir; Fluc, firefly luciferase; TK, Thymidine Kinase; TR, tumor necrosis factor apoptosis-inducing ligand; PARP, poly (ADP-ribose) polymeras; FmC, Fluc and mCherry.
To determine the feasibility of killing therapeutic MSCs post TRAIL therapy, MSC-TR-GFl and MSC-TR-TK-GFl were co-cultured with U87 or Gli36vIII tumor cells and treated with GCV 48hrs later. A significant decrease of Fluc signal and GFP-positive mMSCs was seen in mMSC-TR-TK treated with GCV in comparison to controls (Fig. 4A-B). Similar results were obtained when these studies were performed with hMSC-TR-TK (supplemental Fig. 2C-G). These results reveal that MSCs co-expressing S-TRAIL and HSV-TK simultaneously allow killing of tumor cells and specific elimination of therapeutic MSCs post tumor therapy.
Fig. 4. MSC fate post TRAIL therapy in vitro.
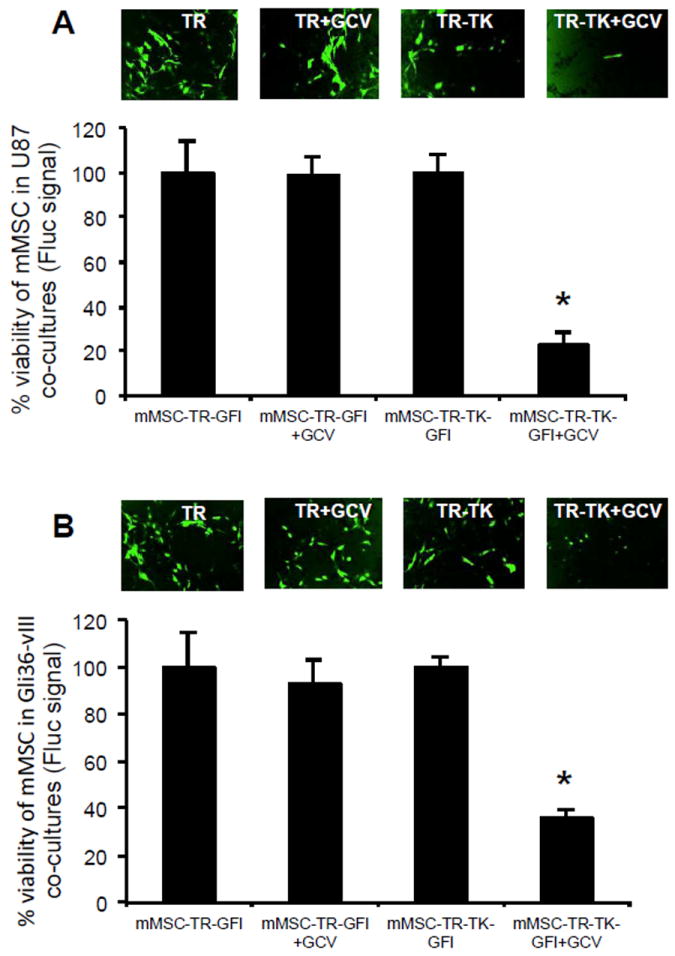
(A-B) Co-culture experiments of U87-FmC (A) or Gli36vIII-FmC (B) with engineered mMSC-TR-GFl or mMSC-TR-TK-GFl were performed as mentioned before in a 1:1 ratio and two days later GCV (10 μg/mL) was added to the cultures. Photomicrographs (original magnification, x4) show mMSC GFP-positive cells three days post GCV treatment. Plots show percentage of viability of modified mMSC-Fluc cells measured by Fluc signal at day three. In all panels, *, p < 0.05 versus controls. Bars, +SD. Abbreviations: mMSC, primary mouse mesenchymal stem cells; GCV, ganciclovir; Fluc, firefly luciferase; TK, Thymidine Kinase; TR, tumor necrosis factor apoptosis-inducing ligand; GFl, green fluorescent protein and Fluc fusion.
MSC-TR-TK has antitumor effect and its fate can be imaged in vivo
We tested the effect of engineered MSC-TR-TK in a highly aggressive malignant GBM model, Gli36vIII. Mice bearing Gli36vIII-FmC tumors treated intratumorally with mMSC-TR-TK or mMSC-TR showed a significant reduction in tumor growth as compared to controls (p < 0.05; degrees of freedom: 17; F values: 23,32; posthoc Dunnett’s test values: 5 (control vs mMSC-TR-TK); and 5.193 (control vs mMSC-TR) at day 14; Fig. 5A). Representative BLI images (Fig. 5B) and H&E staining on brain sections at day 14 post MSC treatment (Fig. 5C-F) showed significant tumor reduction in the mMSC-TR-TK and mMSC-TR treated mice as compared to the control mice. Kaplan-Meier survival analysis revealed that the decrease in tumor growth was correlated with a statistically significant prolongation of survival in mice treated with mMSC-TR-TK or mMSC-TR (median survival time: 31.5 days and 39 days respectively) compared to the control group (median survival time: 17 days; log-rank test, p < 0.002; Fig. 5G).
Fig. 5. MSC-TR-TK has antitumor effect and its fate can be imaged in vivo.
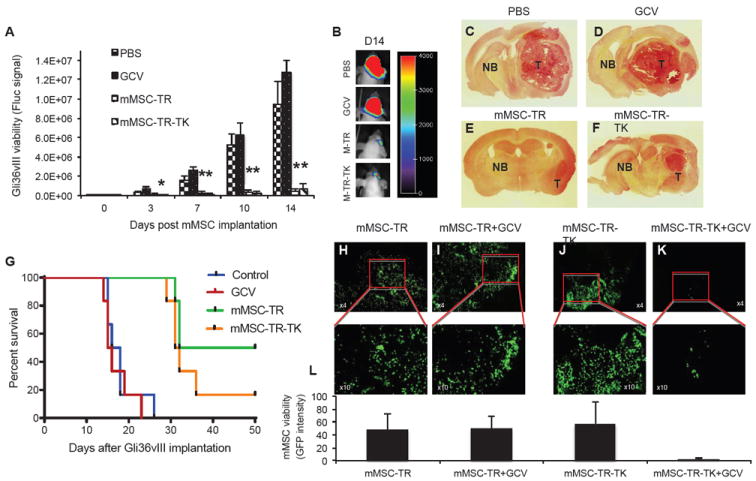
Mice bearing Gli36vIII-FmC were treated intratumorally with mMSC-TR-TK, mMSC-TR, control PBS, or GCV intraperitoneally and imaged for Fluc activity. (A) Plot shows the relative mean bioluminescent signal intensity after quantification of in vivo images. Bars, +SE. (B) One image from a representative mouse of each group at day 14 post mMSC implantation is shown. (C-F) H&E photomicrographs of brain sections of a representative mouse from PBS (C), GCV (D), mMSC-TR (E) or mMSC-TR-TK (F) groups at day 14 post mMSC implantation (original magnification, x2) is shown. (G) Kaplan-Meier survival curves of Gli36vIII-FmC bearing mice treated with PBS, GCV, mMSC-TR or mMSC-TR-TK. (H-K) Gli36vIII-FmC bearing mice were intratumorally implanted with mMSC-TR (H and I) or mMSC-TR-TK (J and K) and fourteen days later treated with GCV or PBS. Photomicrographs (original magnification, x4 upper panel and x10 lower panel) from brain sections of PBS (H and J) or GCV (I and K) groups post 18 days after GCV treatment are shown. Quantification of mMSC viability measured by GFP intensity from the brain section above (L). In all panels, *, p < 0.05 versus controls. Abbreviations: mMSC, primary mouse mesenchymal stem cells; GCV, ganciclovir; T, Tumor tissue; NB, Normal brain; TK, Herpes Simplex Virus Thymidine Kinase; TR, tumor necrosis factor apoptosis-inducing ligand; PBS, phosphate buffered saline.
To determine the fate of therapeutic mMSC post tumor treatment, a set of Gli36vIII-FmC tumor bearing mice were treated with mMSC-TR-TK or mMSC-TR and fourteen days later mice were injected with GCV. Histopathological analysis on brain sections of treated mice revealed the presence of mMSC-TR-TK and mMSC-TR post tumor treatment (Fig. 5H, J and L; supplemental Fig. 3A) and the specific elimination of mMSC-TR-TK (Fig. 5K and L) but not mMSC-TR (Fig. 5I and L) in the brains of mice treated with GCV. These results show that MSCs co-expressing S-TRAIL and HSV-TK or S-TRAIL alone induce tumor regression in a highly aggressive GBM model, thus leading to a significant increase in survival times in mice. Furthermore, therapeutic MSC-TR-TK, unlike mMSC-TR, can be selectively eliminated post tumor treatment to avoid the continuous release of therapeutics from MSCs and to circumvent any malignant transformation of MSCs.
DISCUSSION
In this study, we have explored the possibility of characterizing MSCs co-expressing therapeutic S-TRAIL and HSV-TK to study their fate and therapeutic efficacy in GBM models and using HSV-TK to introduce a safety parameter, which allows the eradication of therapeutic MSC after tumor treatment. Our results show that while MSCs expressing TRAIL and HSV-TK induce tumor cell specific killing, they can be monitored by clinically relevant PET imaging and be selectively eliminated post tumor treatment.
The ability of MSCs to specifically migrate to tumors has led to the use of these cells as a delivery vector for anticancer agents in different tumor models. Human MSCs have been engineered to express and provide targeted delivery of interferonβ (IFNβ) [3], immunomodulatory cytokine IL2 [5], HSV-TK [25] or TRAIL [20, 26] to many types of tumors including GBM models. The administration of therapeutic MSCs has revealed the reduction of tumor growth resulting in increased survival of GBM bearing mice [20]. In this study, we chose S-TRAIL as the therapeutic agent because of its potent anti-tumor effect and its very limited toxicity in non-cancerous tissues. In addition, TRAIL therapy in the form of recombinant protein and monoclonal antibodies to DR4 or DR5 has been used in phase 2 clinical trials with good initial safety and tolerability data [27, 28]. However, the short half-life of recombinant TRAIL protein and the inefficient delivery of monoclonal antibodies, particularly to brain tumors may significantly reduce the efficacy of TRAIL in vivo [29]. To solve that problem, we have previously shown that MSC are resistant to TRAIL mediated apoptosis, and that MSCs expressing S-TRAIL allow a prolonged the secretion of the therapeutic agent in the tumor [20].
To utilize stem cell based therapies for cancer in the clinical settings there are two essential issues: 1) determine the dynamics of MSC migration and tumor distribution in real time with a clinically relevant imaging modality; 2) eliminate therapeutic stem cells post-tumor therapy. MSCs have been administered to many patients without considerable side effects [30, 31]. However, recent reports suggest a need to introduce a safety mechanism in therapeutic stem cells [32-34] especially as the field of stem cell based therapies expands. In this study, the prodrug converting system HSV-TK/GCV was chosen to eliminate therapeutic MSC post tumor treatment. Transduction of MSC with HSV-TK has shown a selective killing of MSC after GCV treatment [25, 35]. Recently, Almeida et al reported the use of another prodrug converting strategy based on an inducible caspase-9 (iCasp9) protein that is activated using a specific chemical inducer of dimerization (CID) to control the survival of MSC [36]. Some of the advantages of using the HSV-TK over the iCasp9 approach is the well-know efficacy of the system, the ability of the prodrug GCV to cross the blood brain barrier and its dual function as a PET imager [37]. In addition, spontaneous dimerization of iCasp9 can cause the early killing of the therapeutic MSCs [36]. On the other hand, one of the potential disadvantages of using HSV-TK is that it induces cell death by a cell cycle dependent mechanism. Other prodrug converting systems non-cell cycle dependent, such as Nitroreductase/CB1954 [38] or Cytochrome P450 2B1/ifosfamide [39] could be used. However, HSV-TK, unlike the other genes, incorporates the ability to image viable HSV-TK expressing cells, which is vital for following MSCs in translational applications, and allows for the eradication of any malignant transformation of MSC, one of the major safety concerns in MSC-based therapies. Several groups have used prodrug converting systems simultaneously as the tumor therapeutic as well as the safety parameter [17-19]. However, one major limitation of using MSC engineered to express prodrug converting genes as antitumor strategy is the immediate elimination of the therapeutic cells post prodrug activation, which allows a very short therapeutic window for tumor cell treatment. In our study the primary therapeutic agent is S-TRAIL and we have utilized HSV-TK as a safety strategy to eliminate therapeutic MSC-S-TRAIL. This is further clarified by our experimental settings in which S-TRAIL secreted from MSC is initially allowed to target tumor cells for 14 days and then mice are injected with the prodrug GCV to eradicate MSC-TR-TK cells. As MSC are resistant to S-TRAIL and not HSV-TK/GCV system, this strategy allows to prolong the secretion of therapeutic agent (S-TRAIL) and utilize GCV as the MSC elimination agent.
Recent studies have shown that hASCs implanted in mice brains can differentiate into endothelial cells and integrate into tumor vessels [40]. In this study, although some therapeutic mMSC-TR-TK localized in the vicinity of tumor vessels (CD31 positive cells) we did not observe any co-localization of MSC with tumor vessels (supplemental Fig. 3B). This result suggests that the mMSC-TR-TK do not differentiate to endothelial cells and may be undifferentiated, thus reinforcing the need to eradicate therapeutic MSC after tumor treatment. The difference in the fate of MSC observed in the studies by Alieva M. et al. [40] and our studies might be due to the difference in the tissue of origin of the MSC (adipose tissue vs bone marrow), species (human vs mouse) as well as the specific tumor environment of the GBMs in mice (U87 vs Gli36vIII).
BLI and optical fluorescent imaging have been employed as non-invasive methods to track MSC migration and monitor therapeutic efficacy in animal models [7]. Although they have become a powerful tool and provided accurate results in pre-clinical studies [22] the clinical utility of those imaging modalities is limited by poor tissue penetration and low spatial resolution, making them impractical for use in patient trials. Magnetic Resonance Imaging (MRI) and PET are commonly used in preclinical and clinical studies for visualization of various tumor and drug interactions and for understanding tumor metabolism with high specificity [29, 42, 43]. Novel imaging contrast agents have emerged and offer possibility of visualizing the fate of stem cells in vivo using MRI. Ferumoxide-protamine sulfate complex (FE-Pro) [44] or superparamagnetic iron oxide (Fe3O4) nanoparticles [45] have been recently used for tracking MSC in a variety of tissues. These particles generate a local magnetic field perturbation that can be detected by MRI. However, one of the limitations of this approach is the possibility of false positive interpretation of the MRI signal, which may be produced by macrophages that have engulfed non-viable labeled MSC or freely-dispersed iron nanoparticles released from non-viable MSC that can be taken up by GBM cells in the brain tissue [46-48]. Furthermore, some studies suggest that iron particles can affect MSC viability [44].
PET scanning using a reporter gene-imaging agent pair has a number of advantages for human translation over other imaging approaches. Positron detection is the most sensitive clinical imaging modality and can detect radiolabeled ligands in concentrations on the order of 10-12 M. PET ligands such as 18F-FHBG [11] 18F-FEAU [12] and 124I-FAIU [13] have been used to image the activity of HSV-TK. PET scanning provides three dimensional images of the concentration of cells in different locations within a tumor. In addition, the trapping of the ligand occurs only in viable cells expressing HSV-TK, so that false positive signal from phagocytized nanoparticles does not occur. Finally, the HSV-TK expression can be repeatedly evaluated over time, and does not decrease with cell division. A limitation of the technique includes spatial resolution on the order of 2-4 mm clinically, which is typically more than sufficient to guide additional interventional therapy. In this study, the use of HSV-TK to both selectively sensitize MSC-TR-TK expressing cells to GCV and to image them, make PET scan the ideal technique for clinical tracking of MSC-TR-TK. Moreover, our in vivo PET imaging observations were cross validated by bioluminescence and fluorescence imaging.
The radiotracer used in the current study was 18F-FHBG, which produces abdominal background signal due to a predominantly hepatobilliary elimination route. For hepatic imaging, alternatives such as 18F-FEAU, which is eliminated by renal clearance, could be used [15]. However, non-specific hepatic uptake of 18F-FHBG does not affect brain or other central nervous system imaging. In order to improve the signal intensity, the use of HSV-sr39TK, a point mutated HSV-TK with high enzymatic activity, could provide greater sensitivity for imaging compared with the wild-type HSV-TK in terms of phosphorylation and selective retention of 18F-FHBG [12].
To our knowledge, this is the first study to report diagnostic, antitumor therapy and safety in stem cell-based therapy approach and it may serve as a foundation for further studies toward potential clinical application of MSC-TR-TK in patients with brain tumors.
CONCLUSION
We demonstrate that stem cells engineered to co-express S-TRAIL and HSV-TK can simultaneously act as therapeutic and diagnostic agents. The stem cells also incorporate a safety component that is essential for the translational application of these therapies. These therapeutic stem cells have an antitumor effect, allow the systematic visualization of the therapeutic cells with clinically relevant PET imaging and provide a mechanism for eventually selectively eliminating the cells after tumor therapy. This study has thus direct implications in translating stem cell based therapies into cancer patients.
Supplementary Material
Acknowledgments
We would like to thank Alicia Leece for the synthesis of 18F-FHBG used in this study. This work was supported by RO1 CA138922 (KS), RO1 NS071197 (KS), R01 CA173077 (KS), American Cancer Society (KS) and James McDonald Foundation (KS).
Footnotes
Author contributions: J.M-Q: conception and design, data collection and assembly, data analysis and interpretation, manuscript writing; D.B. and D.H. data collection and assembly; P.H. and U.M. provision of study material, data collection and analysis; K.S., conception and design, data analysis and interpretation, financial support, manuscript writing.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors indicate no potential conflicts of interest.
References
- 1.Sathornsumetee S, Rich JN. New approaches to primary brain tumor treatment. Anticancer Drugs. 2006;17:1003–1016. doi: 10.1097/01.cad.0000231473.00030.1f. [DOI] [PubMed] [Google Scholar]
- 2.Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev. 2011 doi: 10.1016/j.addr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–3318. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 4.Miletic H, Fischer Y, Litwak S, et al. Bystander killing of malignant glioma by bone marrow-derived tumor-infiltrating progenitor cells expressing a suicide gene. Mol Ther. 2007;15:1373–1381. doi: 10.1038/sj.mt.6300155. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura K, Ito Y, Kawano Y, et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004;11:1155–1164. doi: 10.1038/sj.gt.3302276. [DOI] [PubMed] [Google Scholar]
- 6.Almasan A, Ashkenazi A. Apo2l/trail: Apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev. 2003;14:337–348. doi: 10.1016/s1359-6101(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 7.Shah K, Bureau E, Kim DE, et al. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann Neurol. 2005;57:34–41. doi: 10.1002/ana.20306. [DOI] [PubMed] [Google Scholar]
- 8.Kauer TM, Figueiredo JL, Hingtgen S, et al. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat Neurosci. 2011 doi: 10.1038/nn.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fillat C, Carrio M, Cascante A, et al. Suicide gene therapy mediated by the herpes simplex virus thymidine kinase gene/ganciclovir system: Fifteen years of application. Curr Gene Ther. 2003;3:13–26. doi: 10.2174/1566523033347426. [DOI] [PubMed] [Google Scholar]
- 10.Tjuvajev JG, Doubrovin M, Akhurst T, et al. Comparison of radiolabeled nucleoside probes (fiau, fhbg, and fhpg) for pet imaging of hsv1-tk gene expression. J Nucl Med. 2002;43:1072–1083. [PubMed] [Google Scholar]
- 11.Hung SC, Deng WP, Yang WK, et al. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res. 2005;11:7749–7756. doi: 10.1158/1078-0432.CCR-05-0876. [DOI] [PubMed] [Google Scholar]
- 12.Gambhir SS, Bauer E, Black ME, et al. A mutant herpes simplex virus type 1 thymidine kinase reporter gene shows improved sensitivity for imaging reporter gene expression with positron emission tomography. Proc Natl Acad Sci U S A. 2000;97:2785–2790. doi: 10.1073/pnas.97.6.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tjuvajev JG, Avril N, Oku T, et al. Imaging herpes virus thymidine kinase gene transfer and expression by positron emission tomography. Cancer Res. 1998;58:4333–4341. [PubMed] [Google Scholar]
- 14.Nagano H, Miyamoto A, Wada H, et al. Interferon-alpha and 5-fluorouracil combination therapy after palliative hepatic resection in patients with advanced hepatocellular carcinoma, portal venous tumor thrombus in the major trunk, and multiple nodules. Cancer. 2007;110:2493–2501. doi: 10.1002/cncr.23033. [DOI] [PubMed] [Google Scholar]
- 15.Kucerova L, Altanerova V, Matuskova M, et al. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007;67:6304–6313. doi: 10.1158/0008-5472.CAN-06-4024. [DOI] [PubMed] [Google Scholar]
- 16.Cavarretta IT, Altanerova V, Matuskova M, et al. Adipose tissue-derived mesenchymal stem cells expressing prodrug-converting enzyme inhibit human prostate tumor growth. Mol Ther. 2010;18:223–231. doi: 10.1038/mt.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aboody KS, Brown A, Rainov NG, et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc Natl Acad Sci U S A. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito S, Natsume A, Shimato S, et al. Human neural stem cells transduced with ifn-beta and cytosine deaminase genes intensify bystander effect in experimental glioma. Cancer Gene Ther. 2010;17:299–306. doi: 10.1038/cgt.2009.80. [DOI] [PubMed] [Google Scholar]
- 19.Rath P, Shi H, Maruniak JA, et al. Stem cells as vectors to deliver hsv/tk gene therapy for malignant gliomas. Curr Stem Cell Res Ther. 2009;4:44–49. doi: 10.2174/157488809787169138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasportas LS, Kasmieh R, Wakimoto H, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A. 2009;106:4822–4827. doi: 10.1073/pnas.0806647106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sena-Esteves M, Tebbets JC, Steffens S, et al. Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Methods. 2004;122:131–139. doi: 10.1016/j.jviromet.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 22.Shah K, Hingtgen S, Kasmieh R, et al. Bimodal viral vectors and in vivo imaging reveal the fate of human neural stem cells in experimental glioma model. J Neurosci. 2008;28:4406–4413. doi: 10.1523/JNEUROSCI.0296-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corsten MF, Miranda R, Kasmieh R, et al. Microrna-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered s-trail in human gliomas. Cancer Res. 2007;67:8994–9000. doi: 10.1158/0008-5472.CAN-07-1045. [DOI] [PubMed] [Google Scholar]
- 24.Lee RH, Pulin AA, Seo MJ, et al. Intravenous hmscs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein tsg-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uchibori R, Okada T, Ito T, et al. Retroviral vector-producing mesenchymal stem cells for targeted suicide cancer gene therapy. J Gene Med. 2009;11:373–381. doi: 10.1002/jgm.1313. [DOI] [PubMed] [Google Scholar]
- 26.Kim SM, Lim JY, Park SI, et al. Gene therapy using trail-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008;68:9614–9623. doi: 10.1158/0008-5472.CAN-08-0451. [DOI] [PubMed] [Google Scholar]
- 27.Hotte SJ, Hirte HW, Chen EX, et al. A phase 1 study of mapatumumab (fully human monoclonal antibody to trail-r1) in patients with advanced solid malignancies. Clin Cancer Res. 2008;14:3450–3455. doi: 10.1158/1078-0432.CCR-07-1416. [DOI] [PubMed] [Google Scholar]
- 28.Greco FA, Bonomi P, Crawford J, et al. Phase 2 study of mapatumumab, a fully human agonistic monoclonal antibody which targets and activates the trail receptor-1, in patients with advanced non-small cell lung cancer. Lung Cancer. 2008;61:82–90. doi: 10.1016/j.lungcan.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 29.Asselin MC, O’Connor JP, Boellaard R, et al. Quantifying heterogeneity in human tumours using mri and pet. Eur J Cancer. 2012;48:447–455. doi: 10.1016/j.ejca.2011.12.025. [DOI] [PubMed] [Google Scholar]
- 30.Tyler MA, Ulasov IV, Sonabend AM, et al. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 2009;16:262–278. doi: 10.1038/gt.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed AU, Thaci B, Alexiades NG, et al. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol Ther. 2011;19:1714–1726. doi: 10.1038/mt.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thaci B, Ahmed AU, Ulasov IV, et al. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: Targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 2012;19:431–442. doi: 10.1038/cgt.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 34.Zhu W, Xu W, Jiang R, et al. Mesenchymal stem cells derived from bone marrow favor tumor cell growth in vivo. Exp Mol Pathol. 2006;80:267–274. doi: 10.1016/j.yexmp.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Niess H, Bao Q, Conrad C, et al. Selective targeting of genetically engineered mesenchymal stem cells to tumor stroma microenvironments using tissue-specific suicide gene expression suppresses growth of hepatocellular carcinoma. Ann Surg. 2011;254:767–774. doi: 10.1097/SLA.0b013e3182368c4f. discussion 774-765. [DOI] [PubMed] [Google Scholar]
- 36.Ramos CA, Asgari Z, Liu E, et al. An inducible caspase 9 suicide gene to improve the safety of mesenchymal stromal cell therapies. Stem Cells. 2010;28:1107–1115. doi: 10.1002/stem.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun N, Lee A, Wu JC. Long term non-invasive imaging of embryonic stem cells using reporter genes. Nat Protoc. 2009;4:1192–1201. doi: 10.1038/nprot.2009.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bridgewater JA, Springer CJ, Knox RJ, et al. Expression of the bacterial nitroreductase enzyme in mammalian cells renders them selectively sensitive to killing by the prodrug cb1954. Eur J Cancer. 1995;31A:2362–2370. doi: 10.1016/0959-8049(95)00436-x. [DOI] [PubMed] [Google Scholar]
- 39.Huch M, Abate-Daga D, Roig JM, et al. Targeting the cyp2b 1/cyclophosphamide suicide system to fibroblast growth factor receptors results in a potent antitumoral response in pancreatic cancer models. Hum Gene Ther. 2006;17:1187–1200. doi: 10.1089/hum.2006.17.1187. [DOI] [PubMed] [Google Scholar]
- 40.Alieva M, Bago JR, Aguilar E, et al. Glioblastoma therapy with cytotoxic mesenchymal stromal cells optimized by bioluminescence imaging of tumor and therapeutic cell response. PLoS One. 2012;7:e35148. doi: 10.1371/journal.pone.0035148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bipat S, Niekel MC, Comans EF, et al. Imaging modalities for the staging of patients with colorectal cancer. Neth J Med. 2012;70:26–34. [PubMed] [Google Scholar]
- 43.Morgan B. Opportunities and pitfalls of cancer imaging in clinical trials. Nat Rev Clin Oncol. 2011;8:517–527. doi: 10.1038/nrclinonc.2011.62. [DOI] [PubMed] [Google Scholar]
- 44.Thu MS, Najbauer J, Kendall SE, et al. Iron labeling and pre-clinical mri visualization of therapeutic human neural stem cells in a murine glioma model. PLoS One. 2009;4:e7218. doi: 10.1371/journal.pone.0007218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loebinger MR, Kyrtatos PG, Turmaine M, et al. Magnetic resonance imaging of mesenchymal stem cells homing to pulmonary metastases using biocompatible magnetic nanoparticles. Cancer Res. 2009;69:8862–8867. doi: 10.1158/0008-5472.CAN-09-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pawelczyk E, Arbab AS, Chaudhry A, et al. In vitro model of bromodeoxyuridine or iron oxide nanoparticle uptake by activated macrophages from labeled stem cells: Implications for cellular therapy. Stem Cells. 2008;26:1366–1375. doi: 10.1634/stemcells.2007-0707. [DOI] [PubMed] [Google Scholar]
- 47.Amsalem Y, Mardor Y, Feinberg MS, et al. Iron-oxide labeling and outcome of transplanted mesenchymal stem cells in the infarcted myocardium. Circulation. 2007;116:I38–45. doi: 10.1161/CIRCULATIONAHA.106.680231. [DOI] [PubMed] [Google Scholar]
- 48.Terrovitis J, Stuber M, Youssef A, et al. Magnetic resonance imaging overestimates ferumoxide-labeled stem cell survival after transplantation in the heart. Circulation. 2008;117:1555–1562. doi: 10.1161/CIRCULATIONAHA.107.732073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.