

Abstract
Hepatic ischemia/reperfusion injury (IRI), an exogenous antigen-independent local inflammation response, occurs in multiple clinical settings including liver transplantation, hepatic resection, trauma, and shock. The nervous system maintains extensive crosstalk with the immune system through neuropeptide and peptide hormone networks. This study examines the function and therapeutic potential of VIP (vasoactive intestinal peptide) neuropeptide in a murine model of liver “warm” ischemia (90min) followed by reperfusion. Liver IR triggered an induction of gene expression of intrinsic VIP, peaking by 24h of reperfusion, and coinciding with hepatic self-healing phase. Treatment with the neuropeptide VIP protected livers from IRI, evidenced by diminished serum alanine aminotransferase (sALT) levels, well-preserved tissue architecture, and was associated with elevated intracellular cyclic adenosine monophosphate - protein kinase A (cAMP-PKA) signaling. The hepatocellular protection rendered by VIP was accompanied by diminished neutrophil/macrophage infiltration and activation, reduced hepatocyte necrosis/apoptosis, and increased hepatic IL-10 expression. Strikingly, PKA inhibition restored liver damage in otherwise IR-resistant VIP-treated mice. In vitro, VIP not only diminished macrophage TNF-α/IL-6/IL-12 expression in an PKA-dependent manner, but also prevented necrosis/apoptosis in primary mouse hepatocyte cultures. Conclusion: Our findings document the importance of VIP neuropeptide-mediated cAMP-PKA signaling in hepatic homeostasis and cytoprotection in vivo. As the enhancement of neural modulation differentially regulates local inflammation and prevents hepatocyte death, these results provide the rationale for novel approaches to manage liver IRI in transplant patients.
Keywords: Cyclic Adenosine Monophosphate, Ischemia/Reperfusion Injury, Protein kinase A, Toll-like Receptor 4, Vasoactive intestinal peptide
Introduction
Hepatic ischemia/reperfusion injury (IRI) remains the major challenge in clinical liver transplantation, hepatic resection, trauma, and shock (1). Two phases of IR-triggered hypoxic cellular stress, and inflammation-mediated hepatocellular injury may be distinguished. First, endogenous reactive oxygen species (ROS)-inflicted damage initiates local circulatory disturbances and inflammation, leading to the ultimate hepatocyte death. Second, activation of host innate immune system exacerbates local liver injury. Our group was among the first to document that activation of the sentinel TLR4 signaling is required in the mechanism of liver IRI (2), and that IR-triggered TLR4, primarily on Kupffer cells/macrophages, facilitates downstream “signature” pro-inflammatory cytokine programs, i.e., TNF-α, IFN-β, and CXCL-10 (3,4).
The immune system and the nervous system maintain active communication, and mount integrated responses to danger signals through intricate chemical messengers, neuropeptides, and peptide hormones. The innate immune system acts as the first defensive firewall against invading pathogens, through recognition of pathogen-associated molecular patterns (PAMPs), phagocytosis and releasing pro-inflammatory mediators (5). These immune components convey the peripheral message to the brainstem and preoptic area of the anterior hypothalamus. Then, the regional neural-hormonal–stress response may amplify local inflammation immune cascade to eliminate pathogens (6-9), restore host homeostasis, and return to a resting state (10).
A 28-amino-acid VIP (vasoactive intestinal peptide), was first isolated from the gastrointestinal tract as vasodilator (11). Later, VIP has been recognized as a widely distributed neuroregulator in the central and peripheral nervous system (12). VIP was shown similarities with other gastrointestinal hormones, such as secretin, glucagon and pituitary adenylate cyclase activating polypeptide (PACAP; 68% identity). VIP conveys its immuoregulatory function almost exclusively through two G protein-coupled receptors (GPCRs): VPAC1, constitutively expressed by lymphocytes/macrophages and VPAC2, expressed selectively by activated lymphocytes/macrophages (13). PACAP acts on these same receptors with high affinity, but also interacts with the highly selective PAC1 receptor (14), which is also expressed in macrophages (15). Hepatocytes, on the other hand, constitutively express all three VPAC1, VPAC2 and PAC1 receptors (16).
The widespread distribution of VIP correlates with its involvement in various biological processes. Indeed, VIP peptides inhibit phagocytic activity, production of free radicals and adherence/migration of macrophages and neutrophils (17). VIP peptides inhibit production of pro-inflammatory cytokines/chemokines, downregulate the expression of inducible nitric oxide synthase (iNOS) and subsequent release of nitric oxide, and enhance anti-inflammatory IL-10 produced by activated macrophages, microglia and monocytes (18,19). We recently showed that the PACAP-mediated cAMP-PKA activation provided protection in a model of murine liver IRI (20,21), while the ability of VIP to act in this manner is unknown. Experiments to determine this are important for two reasons: 1) cytoprotective actions of PACAP are thought to be mediated primarily by the PAC1 receptor (14), which is highly selective (1000-fold higher affinity) for PACAP (22), and 2) VIP neuropeptides and long-acting VIP analogues are currently being developed clinically for treatment of chronic inflammatory lung disorders in sarcoidosis (23), bronchial asthma/chronic obstructive pulmonary disease (COPD) (24) and pulmonary arterial hypertension (PAH) (25,26) as well as neuroblastoma and Alzheimer’s disease (27). Although recent studies imply beneficial effects of VIP neuropeptide in liver IRI (28,29), the underling mechanisms remain to be determined.
This study examines putative therapeutic function and mechanisms by which VIP may affect IR-hepatocellular insult and contribute to liver homeostasis. As stress triggers pro- and anti-inflammatory neuropeptides, we first determined endogenous VIP expression in hepatic IRI. We then determined if exogenous VIP could diminish pro-inflammatory response and promote hepatocyte survival in IR-stressed liver, and studied its actions in cultured macrophages and hepatocytes. Finally, we examined the dependence of these in vivo and in vitro actions on the cAMP-PKA signaling pathway.
Experimental Procedures
Animals
C57BL/6 wild-type (WT) mice were purchased from Jackson Laboratory (male, 8-12 weeks old). Animals were housed in the UCLA animal facility under pathogen-free conditions and received humane care according to the criteria outlined in Guide for the Care and Use of Laboratory Animals (prepared by the National Academy of Sciences; NIH publication 86-23, revised 1985).
Mouse warm liver IRI model
We used a mouse model of partial “warm” hepatic IRI (2). In brief, animals were anesthetized, injected with heparin (100U/kg i.v.), and the arterial/portal venous blood supply to the cephalad lobes was interrupted by an atraumatic clip for 90min. Sham-operated mice underwent the same procedure, but without vascular occlusion. In the treatment groups, animals were infused i.v. (penile vein) 1h prior to the onset of liver ischemia with a single dose of VIP neuropeptide (50nmol/mouse, Phoenix Pharmaceuticals, Burlingame, CA) dissolved in PBS. Some recipients were given H-89 (cAMP-PKA inhibitor; 20nmol/mouse i.v., Sigma-Aldrich, St. Louis, MO) dissolved in dimethyl sulfoxide (DMSO). Mice were sacrificed at various time-points of reperfusion; liver and serum samples were collected for analysis.
The hepatocellular damage
Serum alanine aminotransferase (sALT) levels were measured by IDEXX Laboratory (Westbrook, ME). Culture medium ALT levels were measured by ALT kit (Stanbio, Boerne, TX). Untreated hepatocyte lysates were used to determine total ALT level. Cell death was expressed as ALT released from treated cells (percentage of the total ALT).
Histopathology
Liver specimens (4μm), stained with hematoxylin and eosin (H&E), were analyzed blindly. Primary mAb against mouse neutrophils Ly-6G (1A8; BD Biosciences, San Jose, CA) and macrophages CD68 (FA-11; AbD Serotec, Raleigh, NC) were used (30). Liver sections were evaluated blindly by counting labeled cells in 10 high-power fields (HPF).
Myeloperoxidase activity assay
The presence of myeloperoxidase (MPO) was used as an index of neutrophil accumulation in the liver (30). One absorbance unit (U) of MPO activity was defined as the quantity of enzyme degrading 1mol peroxide/min at 25°C/gram of tissue.
Quantitative RT-PCR
Quantitative PCR was performed with platinum SYBR green quantitative PCR kit (Invitrogen, Carlsbad, CA) by the Chromo 4 detector (MJ Research, Waltham, MA). Primers to amplify specific gene fragments were published (21). VIP primer sequences were synthesized by Invitrogen: forward GAAATACCTGAACTCCATCCTGA, and reverse TTCTCCAGCTCTTCAAGAAAGTC. Target gene expressions were calculated by their ratios to the housekeeping gene hypoxanthine-guanine phosphoribosyl transferase (HPRT).
Western blots
Western blots were performed with liver proteins (30μg/sample) and rabbit anti-mouse Bcl-2, Bcl-xl, p-IκBα, p-NF-κB p65, and β-actin mAbs (Cell Signaling Technology, Danvers, MA), as described (30).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay
DNA fragments in liver sections, resulting from oncotic necrosis and apoptosis were detected by TUNEL method (Klenow-FragEL DNA Fragmentation Detection Kit, Calbiochem, La Jolla, CA) (30). TUNEL positive cells were counted in 10 HPF/section under light microscopy (×400).
Caspase-3 activity assay
Caspase-3 activity was performed using Caspase-3 Cellular Activity Assay Kit (Calbiochem). Liver tissue sample and cell lysis were used according to the manufacturer’s instruction.
The cAMP/PKA kinase activity assays
The cAMP levels and PKA activity in tissue samples were measured by cAMP Enzyme Immunoassay and PKA kinase activity kit, respectively (Enzo Life Sciences, Farmingdale, NY).
Cell cultures
Bone marrow-derived macrophages (BMM), separated from femurs/tibias of C57BL/6 mice were cultured (5×106/well) with 10% L929 conditioned medium for 6 days. The cell purity was 94-99% CD11b+. BMM were activated by LPS (10ng/ml, Sigma-Aldrich) in the presence of VIP (10nM), or PBS control, and incubated for 24h. The H-89 (10μM) pretreatment at 1h before LPS was used to block cAMP-PKA pathway. Cell-free supernatants were assayed for cytokine levels by ELISA (eBioscience, San Diego, CA).
Mouse hepatocytes were isolated by in situ two-stage collagenase perfusion, cultured with complete L-15 medium plus 6.25μg/ml insulin, 1μM dexamethasone, and 10% fetal bovine serum for 24h before experiments. Hepatocyte viability was 95-99%. After pretreatment with VIP (10nM), H-89 (10μM) or DMSO for 1h, hepatocyte death was induced by hydrogen peroxide (0.5mM, Sigma-Aldrich) or TNF-α (10ng/ml, R&D Systems, Minneapolis, MN) in combination with actinomycin D (0.4μg/ml, Sigma-Aldrich) during 5h incubation. Cells were processed for flow cytometry/caspase-3 activity, and supernatants were assessed for ALT/LDH levels.
Lactate dehydrogenase (LDH) release assay
Culture medium LDH activity was measured by LDH kit (Stanbio). Untreated hepatocyte lysates were used to determine total LDH activity. Cell death was expressed as LDH activity released from the treated cells as a percentage of total LDH activity.
Flow cytometry
Hepatocytes stained with FITC-ANNEXIN V and 7-AAD (BD Biosciences, Mountain View, CA) were analyzed on a FACS-Calibur cytometer (BD Biosciences). Dead cells were identified as ANNEXIN V+7-AAD+.
Statistical analysis
All values are expressed as the mean ± standard deviation (SD). Data were analyzed with an unpaired, two-tailed Student’s-t test. P<0.05 was considered to be statistically significant.
Results
Treatment with VIP ameliorates liver IRI
First, we determined whether IR triggers endogenous VIP gene expression in mouse livers subjected to 90min of ischemia, followed by reperfusion. Compared with sham controls, VIP mRNA levels transiently dropped after ischemia insult (0.5h), and increased progressively thereafter, peaking by 24h of reperfusion (Fig. 1a: p<0.001). To test the significance of VIP, separate groups of WT mice were pretreated with VIP neuropeptide. Unlike PBS-treated controls, VIP ameliorated liver IRI, evidenced by reduced sALT levels (Fig. 1b: 634±68 vs. 5627±655 U/L; p<0.001); and well-preserved hepatic architecture (Fig. 1c: minimal sinusoidal congestion, no edema, vacuolization or necrosis).
Figure 1.
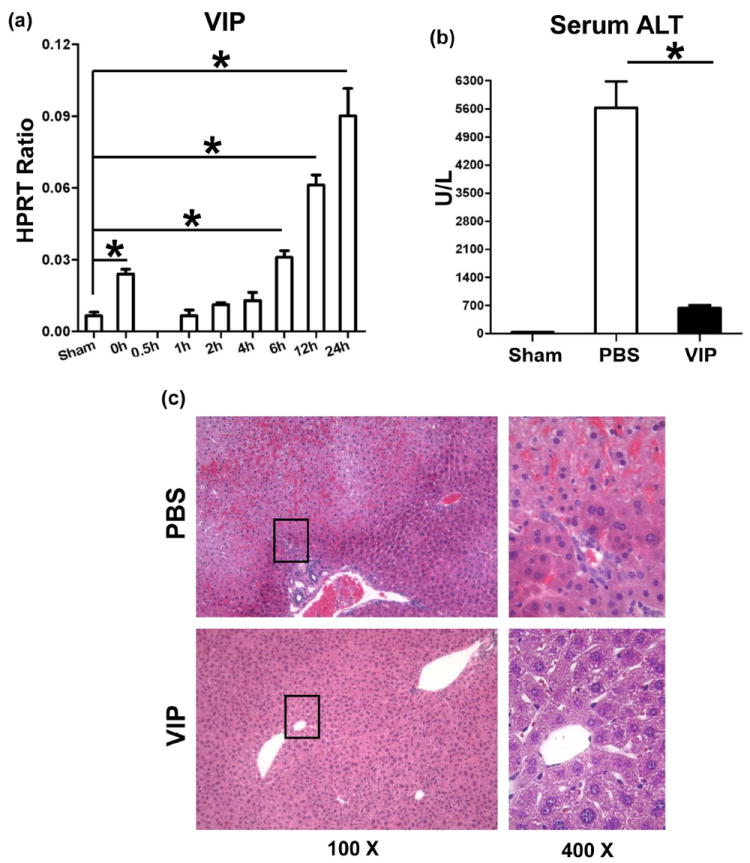
(a) Liver IRI triggers VIP gene expression. Liver samples were harvested from B6 mice that were either sham-operated or subjected to 90min of partial warm ischemia, followed by reperfusion (0.5h – 24h).
Livers in WT mice pretreated with VIP or PBS were subjected to ischemia (90min). At 6h of reperfusion, the hepatocellular function was analyzed by (b) sALT levels, and (c) liver histology (representative H&E staining; magnification ×100 and ×400) (*p<0.001, n=4-5/group).
VIP suppresses neutrophil/macrophage migration to IR-livers
Liver neutrophil activity (MPO assay) was depressed in mice pretreated with VIP, compared with controls (Fig. 2a: 0.22±0.10 vs.1.35±0.35; p<0.001). This correlated with diminished frequency of neutrophils in VIP-treatment group (Fig. 2b: 1.75±0.5 vs. 28.5±7.6; p<0.001). The macrophage recruitment was also decreased in VIP-treated livers (Fig. 2c: 2.5±1.3 vs. 58.0±8.0; p<0.001).
Figure 2.
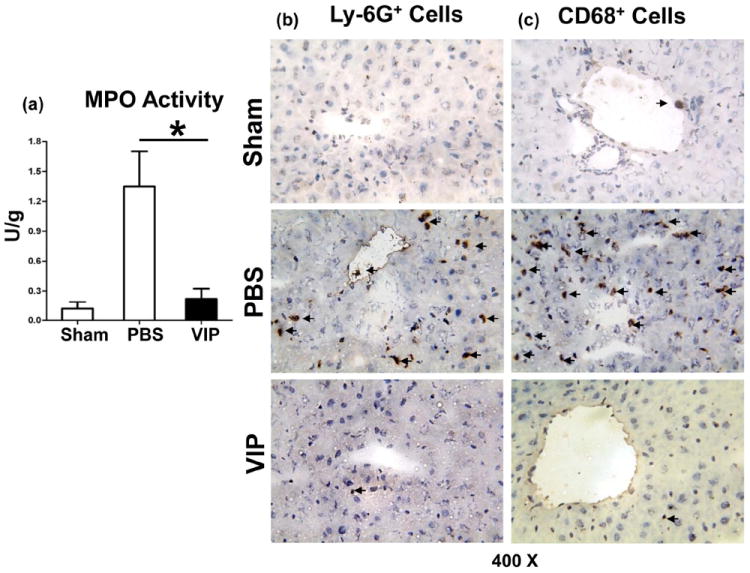
Accumulation of neutrophils and macrophages in IR-livers following administration of VIP neuropeptide (6h of reperfusion after 90min of ischemia). (a) MPO levels; (b) Ly-6G+ neutrophils; (c) CD68+ macrophages in IR-liver lobes (magnification ×400) (*p<0.001, n=4-5/group).
VIP differentially down-regulates IR-induced liver cytokine/chemokine programs
To analyze the immunoregulatory function of VIP neuropeptide, we assessed hepatic chemokine/cytokine gene expression. The neutrophil/monocyte-derived pro-inflammatory chemokine (CXCL-1, CCL-2, and CXCL-10) and cytokine (TNF-α, IFN-β, IL-1β, and IL-6) programs were suppressed in VIP-treated animals, as compared with controls (Fig. 3a,c; p<0.001, p<0.01). In contrast, IL-10 levels were selectively elevated after VIP treatment (Fig. 3b; p<0.001).
Figure 3.
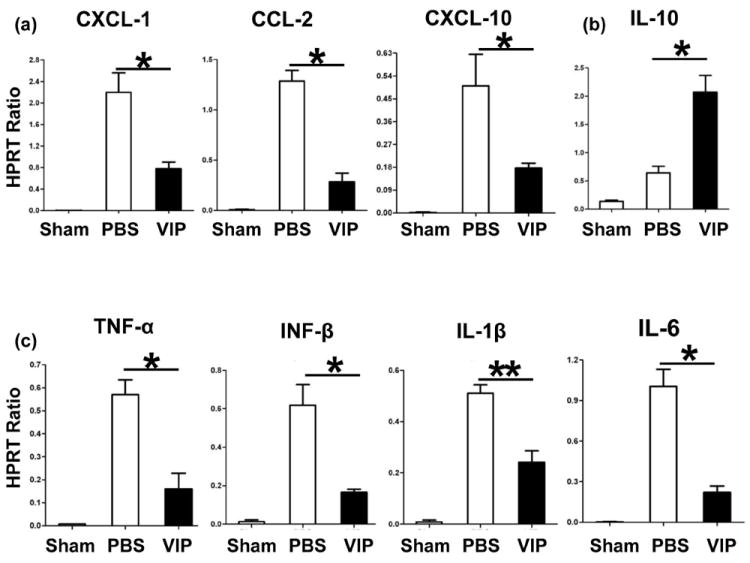
Quantitative RT-PCR-assisted detection of cytokines/chemokines in mouse livers (6h of reperfusion after 90min of ischemia: (a) CXCL-1, CCL-2, CXCL-10; (b) IL-10; and (c) TNF-α, IFN-β, IL-1β, IL-6). Data normalized to HPRT gene expression (**p<0.01, *p<0.001, n=4-5/group).
VIP depresses IR-mediated liver necrosis/apoptosis
We screened for IR-induced hepatic oncotic necrosis and apoptosis. Indeed, VIP treatment diminished otherwise abundant hepatocellular necrosis/apoptosis, evidenced by reduced caspase-3 activity (Fig. 4a: 8.8±2.0 vs. 20.7±2.1 [PBS]; p<0.01) and decreased frequency of TUNEL+ cells (Fig. 4b: 2.0±0.8 vs. 31.3±5.4 [PBS]; p<0.001). Western blot analysis has revealed selectively increased expression of Bcl-2/Bcl-xl, yet suppressed phosphorylation of IκBα/NF-κB p-65 proteins after VIP treatment, as compared with PBS controls (Fig. 4c).
Figure 4.
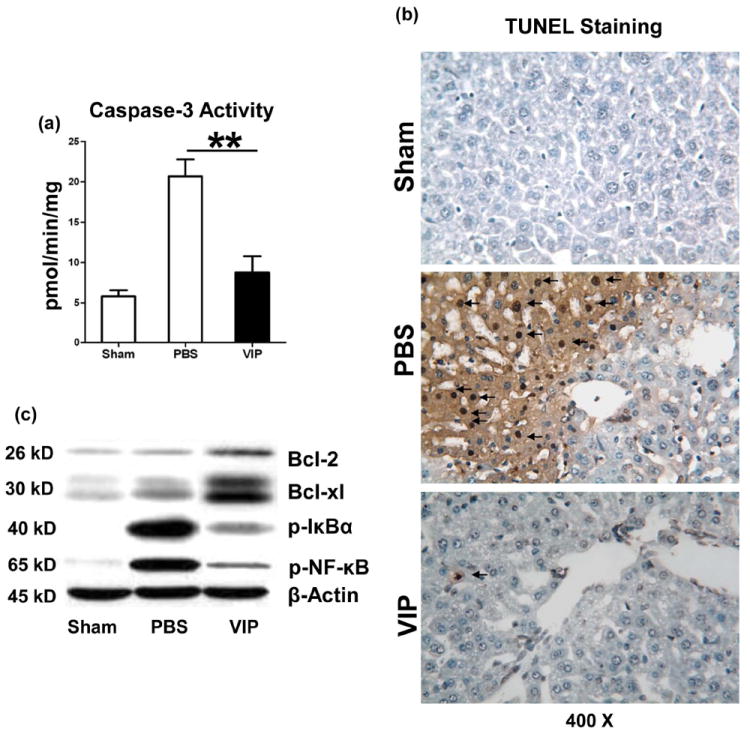
Necrosis/apoptosis in IR-livers (6h of reperfusion after 90min ischemia). (a) caspase-3 activity and (b) TUNEL-assisted detection of hepatic necrosis/apoptosis (dark arrows) in ischemic liver lobes (magnification ×400); (c) Western blot-assisted detection of Bcl-2/Bcl-xl; p-IκBα, and p-NF-κB. (**p<0.01, n=4-5/group).
The cAMP-PKA pathway is essential in VIP modulation of liver IRI
As VIP suppresses macrophage function via cAMP-PKA (17,18), we asked whether VIP may trigger cAMP-PKA signaling in our model. We have recently found that IR may trigger cAMP expression (20). Indeed, administration of VIP increased cAMP levels (Fig. 5a: 895±157 vs. 512±88; umol/g; p<0.01), and PKA activity (Fig. 5a: 10.9±0.7 vs. 4.0±0.3; ng/g; p<0.001), compared with controls.
Figure 5.
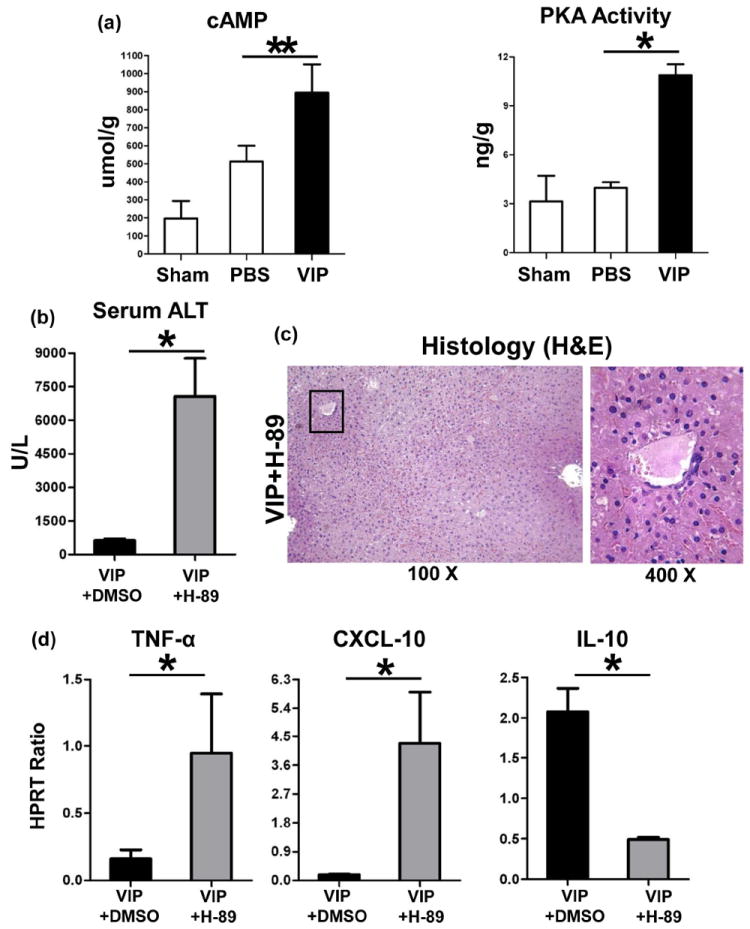
The functional significance of cAMP-PKA in hepatic VIP regulation. Administration of VIP peptides increased liver cAMP levels and elevated PKA activity (a). Adjunctive use of H-89, a PKA inhibitor, restored liver injury in VIP-pretreated groups, evidenced by (b) sALT levels; (c) liver histology (representative H&E; magnification ×100 and ×400); and (d) TNF-α, CXCL-10, and IL-10 expression (**p<0.01, *p<0.001; n=4-5/group).
We used H-89, a specific PKA inhibitor, to study as to whether cAMP-PKA activation is essential for VIP-mediated immunomodulation. Adjunctive inhibition of PKA activity exacerbated liver IRI in VIP-pretreated mice, evidenced by increased sALT levels (Fig. 5b: 7072±1699 [VIP+H-89] vs. 634±68 [VIP] U/L; p<0.001) and hepatocellular damage. Livers after combined VIP and PKA inhibition therapy were characterized by extended zonal/pan-lobular parenchyma necrosis, with widespread sinusoidal congestion and severe edema (Fig. 5c), comparable with PBS controls. Intrahepatic expression of pro-inflammatory TNF-α and CXCL-10 was uniformly heightened, whereas IL-10 levels diminished after VIP plus H-89 treatment (Fig. 5d).
VIP modulates macrophage TLR4 response
TLR4 activation triggers IR-mediated liver inflammation (2). Since macrophages express all three VIP receptors (13-15), TLR4-targeted regulation may contribute to VIP effects in our model. We tested as to whether and how VIP-mediated cAMP-PKA may affect macrophage TLR4 response. BMM cultures were stimulated with LPS (TLR4 ligand) in the absence or presence of VIP; H89 (PKA inhibitor); or DMSO (control). VIP depressed otherwise enhanced LPS-induced TNF-α, IL-6 and IL-12p40 (Fig. 6a,b,c; p<0.001), but increased IL-10 expression (Fig. 6d; p<0.001). In contrast, H-89-mediated PKA inhibition enhanced TNF-α, IL-6, and IL-12p40 levels in VIP groups (Fig. 6a,b,c; p<0.001, p<0.001), as compared with VIP unmodified cultures. Moreover, IL-10 levels decreased in BMM cultures supplemented with VIP plus H-89 (Fig. 6d; p<0.001), compared with VIP alone.
Figure 6.
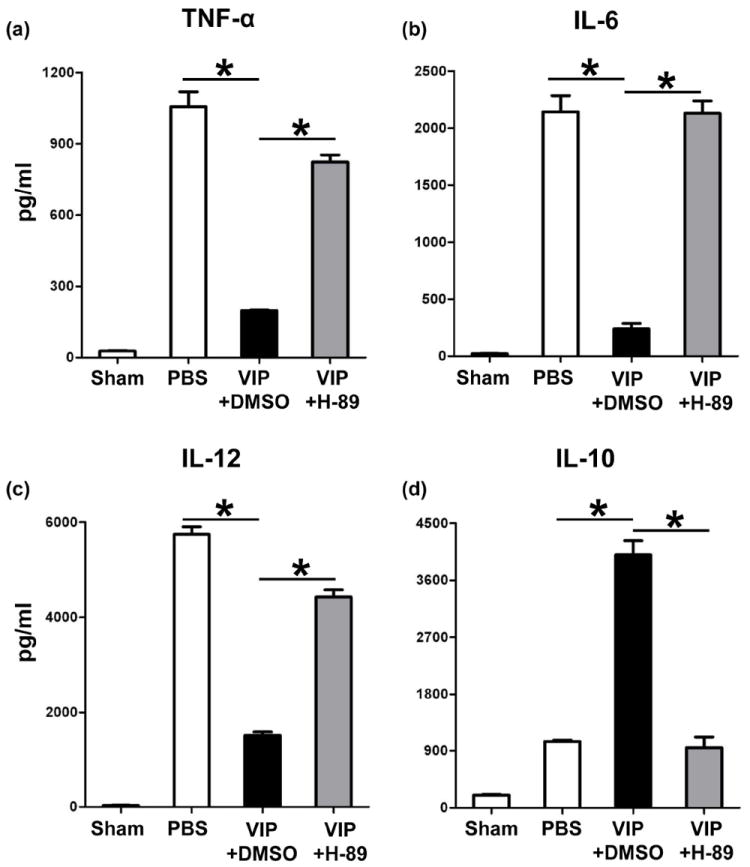
The effects of VIP neuropeptide upon macrophage TLR4 activation in vitro. Bone marrow-derived macrophages were stimulated with LPS in the absence or presence of VIP peptides + H-89 (PKA inhibitor), or DMSO (control). Production of (a) TNF-α, (b) IL-6, (c) IL-12, and (d) IL-10 in culture supernatants. (*p<0.001). Representative of n=3/group.
VIP attenuates hepatocyte death
To assess the immunomodulatory function of VIP-induced cAMP-PKA signaling in hepatocytes, we designed primary hepatocyte culture that mimic liver IR-mediated hepatocellular damage in vivo. Since both necrosis and apoptosis are essential in the pathophysiology of liver IRI, we used hydrogen peroxide (H2O2) to mimic in vivo ROS-triggered hepatocyte necrosis, or TNF-α/actinomycin D (ActD) to induce apoptosis. Native mouse hepatocytes were cultured in the presence of VIP; with H-89 (PKA antagonist); or DMSO (control). Addition of VIP consistently suppressed the hepatocyte death, assessed by FACS-assisted frequency (%) of Annexin V+7-AAD+ cells (Fig. 7a); diminished caspase-3 activity (Fig. 7b); LDH and ALT release (Fig. 7c,d), as compared with controls. In contrast, PKA inhibition enhanced the hepatocyte death (Fig. 7a); and caspase-3 activity (Fig. 7b). In addition, PKA inhibition increased LDH (Fig. 7c) and ALT (Fig. 7d) release in hepatocyte cultures.
Figure 7.
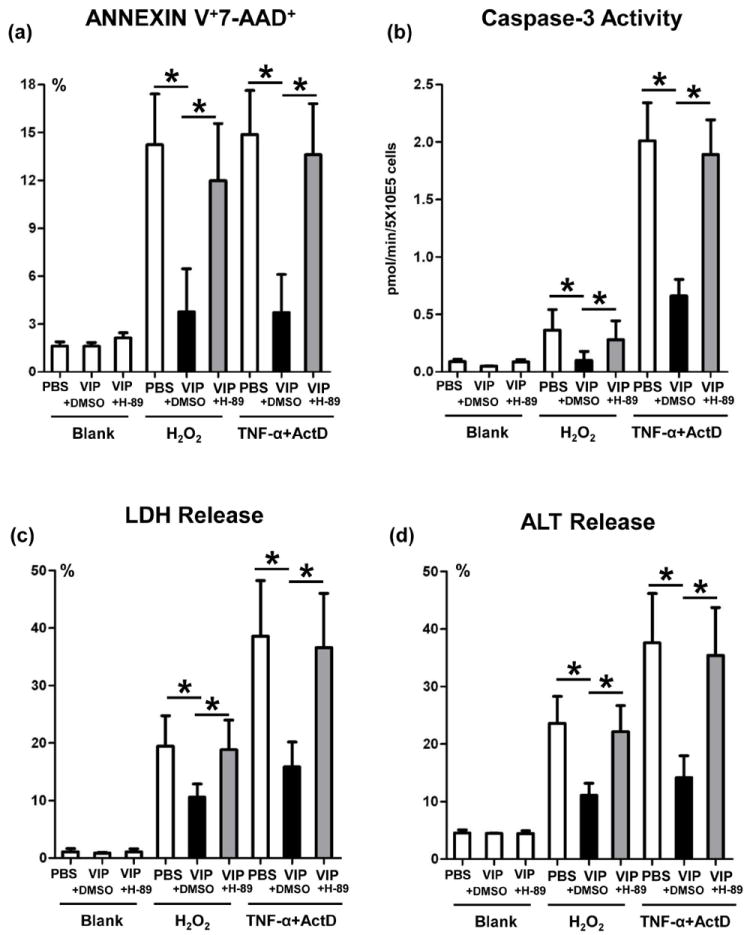
The cytoprotective effects of VIP neuropeptides upon hepatocytes in vitro. Hydrogen peroxide (H2O2), or TNF-α+actinomycin D (ActD) were used to induce primary murine hepatocyte necrosis/apoptosis, in the absence or presence of VIP peptides with H-89 (PKA inhibitor), or DMSO (control). (a) FACS-assisted detection of ANNEXIN V+7-AAD+ dead cells; (b) Hepatocellular caspase-3 activity; (c) LDH release; and (d) ALT levels in supernatants (*p<0.001). Representative of n=3/group.
Discussion
Although VIP neuropeptides regulate macrophage activation and stimulate glucose-induced insulin secretion (18,19,31), their role in innate immunity-driven liver inflammation and IRI remains ill-defined. Here, we show that: i/ VIP was induced in a mouse model of liver warm IR damage; ii/ exogenous VIP protected livers against IRI by inhibiting TLR4 activation and improving the hepatocyte survival; iii/ VIP-mediated cytoprotection was cAMP-PKA dependent. These results are consistent with our findings on PACAP neuropeptides in this model (21).
In the present study, we first found local VIP expression in IR-stressed liver, the levels of which were elevated between 12-24h of reperfusion. This may imply a regulatory role for intrinsic VIP in liver’s self-repair. We then asked whether administration of exogenous VIP may attenuate liver IRI. As IR-induced liver damage peaks at 6h of reperfusion (30), we focused on this time point to demonstrate the modulatory role of VIP neuropeptides. Strikingly, VIP treatment diminished the hepatocellular damage, evidenced by decreased sALT levels and amelioration of cardinal features of liver IRI, i.e., edema, vacuolization and necrosis. These findings are consistent with the ability of VIP to prevent transient ischemic brain damage in a rat model of focal cerebral ischemia (32).
We found increased infiltration by CD68+ macrophages, consistent with preferential pro-inflammatory chemokine gene expression profile in IR-stressed livers (1-4). As VIP therapy suppresses macrophage function (17-19), others have suggested VIP may act as an essential neural immunomodulator in autoimmune diseases (33). We observed reduced macrophage migration and decreased activation/function, along with diminished expression of IRI signature genes, i.e., TNF-α, IL-1β, IL-6, CXCL-10 and CCL-2 (MCP-1). Indeed, CXCL-10, one of the key mediators in type I IFN pathway downstream of TLR4 (3,4), may be directly regulated by VIP. In agreement with our in vivo findings, VIP nearly abolished TLR4-mediated pro-inflammatory cytokine programs in BMM cultures.
PKA pathway in VIP regulation (18,19) may modulate multiple intracellular events (34). We have identified cAMP-PKA activation as a regulator, which halts pathological cell recruitment, prevents destructive immune reactions, and promotes hepatocyte survival (20). This implies PKA activation may raise defensive thresholds to IR-inflammatory response. Indeed, VIP treatment enhanced hepatic cAMP levels and augmented PKA activity, whereas PKA inhibition restored pro-inflammatory profile in VIP-treated BMM cultures. Strikingly, in vivo PKA antagonism rendered otherwise IR-resistant VIP treated hosts restored liver IRI pathology.
TLR4-mediated innate immune activation progresses through the MyD88- and/or TRIF-dependent pathways (35). Our previous studies indicated that signaling via TRIF-IRF3 rather than MyD88, is instrumental for downstream NF-κB activation, IR-inflammation, and hepatocellular damage (2,4). We have shown that cAMP-PKA activation may directly inhibit NF-κB by modulating p65 phosphorylation, stabilizing/elevating IκB, as well as regulating transactivation/stability of NF-κB complexes (20). The cAMP-PKA signaling cascade may also indirectly enhance cAMP response element-bing (CREB) phosphorylation, which has higher affinity for CREB-binding protein (CBP), resulting in competitive sequestration of p65/CBP complexes in IR-livers (20). Here, we show that VIP-induced cAMP-PKA activation decreased phosphorylation/proteolytic degradation of IκB subunit, suppressed phosphorylation of NF-κB p65 and downstream pro-inflammatory programs, yet augmented IL-10, all of which enhance hepatocyte survival. Consistent with in vivo data, we found that PKA activation diminished pro-inflammatory cytokine profile in LPS-activated BMM cultures.
Activated neutrophils (PMNs) generate ROS to promote tissue damage in the second phase of liver IRI (1). Unlike increased neutrophil infiltration/MPO activity in controls, livers in VIP-conditioned mice showed decreased Ly-6G+ neutrophil infiltration, MPO activity and depressed CXCL-1 (KC) levels, the key neutrophil chemoattractant. As neutrophil activity can be enhanced by macrophage-produced cytokines, VIP can also exert its regulatory function during liver IRI through pro-inflammatory cytokine/chemokine networks.
Both necrosis and apoptosis are responsible for IR-hepatocyte damage (36). The death receptor activation, mitochondrial Ca2+ loading, and ROS promote mitochondrial permeability transition, leading to hepatocellular swelling, rupture of the plasma membrane, release of cytochrome C, ultimately resulting in ATP depletion-dependent oncotic necrosis and caspase-dependent apoptosis (1). Hepatocyte oncotic necrosis and apoptosis, which proceed via DNA degradation, can be detected by TUNEL assay (36). Interestingly, VIP treatment inhibited necrosis/apoptosis, evidenced by decreased frequency of TUNEL+ cells and caspase-3 activity in IR-livers. VIP enhanced hepatic expression of Bcl-2/Bcl-xl, suggesting PKA activation-mediated cytoprotection by anti-necrotic/apoptotic proteins. It is plausible that neural immunomodulation prevents the hepatocellular damage by modifying pro-/anti-apoptotic ratio, maintaining mitochondria integrity, or promoting ATP generation. To distinguish between necrosis and apoptosis in hepatocyte cultures, we employed hydrogen peroxide (H2O2) to mimic in vivo ROS-triggered necrosis, or TNF-α to induce apoptosis. VIP supplement diminished hepatocyte death, reduced capase-3 activity, and ameliorated ALT/LDH release in both culture systems. These results, in agreement with our in vivo data, reinforce the immunomodulatory role of VIP to depress NF-κB in nonparenchymal and parenchymal liver compartments, with resultant improvement of the hepatocellular function. Moreover, PKA inhibition exacerbated hepatocyte death, confirming neural regulation at the hepatocyte level is cAMP-PKA dependent.
In conclusion, this study is the first to reveal the mechanisms of exogenous VIP neuropeptide to attenuate liver IRI by depressing macrophage function and improving hepatocyte survival in cAMP-PKA dependent manner. Harnessing immune regulatory and cytoprotective mechanisms by VIP may be essential in the maintenance of hepatic homeostasis in vivo by minimizing local organ damage and promoting IL-10 cytoprotection. As VIP is being developed into a therapeutic principle for humans (23-27), this “very important peptide” should also be considered as a novel therapy to target IR-triggered hepatic inflammation and damage in liver transplant patients.
Acknowledgments
Financial Support: NIH Grants RO1 DK062357 (JWKW) ; The Diann Kim Foundation; The Dumont Research Foundation. HJ is a recipient of American Society of Transplant Surgeons Scientist Scholarship.
List of Abbreviations
- cAMP
cyclic adenosine monophosphate
- CREB
cAMP response element-bing
- IRI
ischemia/reperfusion injury
- LDH
lactate dehydrogenase
- MPO
myeloperoxidase
- PKA
protein kinase A
- ROS
reactive oxygen species
- sALT
serum alanine aminotransferase
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- VIP
vasoactive intestinal peptide
- WT
wild-type
References
- 1.Ji H, Zhai Y, Kupiec-Weglinski JW. Innate-Adaptive Immune Responses in Organ Ischemia/Reperfusion Injury. In: Hartmann G, Wagner H, editors. Innate Immunity: Resistance and Disease-Promoting Principles. Else Kröner-Fresenius Symposia. Vol. 4. Basel: Karger; 2013. pp. 36–41. [Google Scholar]
- 2.Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 3.Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, et al. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47:207–214. doi: 10.1002/hep.21986. [DOI] [PubMed] [Google Scholar]
- 4.Zhai Y, Qiao B, Gao F, Shen X, Vardanian A, Busuttil RW, et al. Type I, but not type II, interferon is critical in liver injury induced after ischemia and reperfusion. Hepatology. 2008;47:199–206. doi: 10.1002/hep.21970. [DOI] [PubMed] [Google Scholar]
- 5.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 6.Goehler LE, Gaykema RP, Hansen MK, Anderson K, Maier SF, Watkins LR. Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton Neurosci. 2000;85:49–59. doi: 10.1016/S1566-0702(00)00219-8. [DOI] [PubMed] [Google Scholar]
- 7.Emch GS, Hermann GE, Rogers RC. TNF-alpha activates solitary nucleus neurons responsive to gastric distension. Am J Physiol Gastrointest Liver Physiol. 2000;279:G582–G586. doi: 10.1152/ajpgi.2000.279.3.G582. [DOI] [PubMed] [Google Scholar]
- 8.Steinman L. Elaborate interactions between the immune and nervous systems. Nat Immunol. 2004;5:575–581. doi: 10.1038/ni1078. [DOI] [PubMed] [Google Scholar]
- 9.Brogden KA, Guthmiller JM, Salzet M, Zasloff M. The nervous system and innate immunity: the neuropeptide connection. Nat Immunol. 2005;6:558–564. doi: 10.1038/ni1209. [DOI] [PubMed] [Google Scholar]
- 10.Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9:418–428. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Said SI, Mutt V. Polypeptide with broad biological activity: isolation from small intestine. Science. 1970;169:1217–1218. doi: 10.1126/science.169.3951.1217. [DOI] [PubMed] [Google Scholar]
- 12.Said SI, Rosenberg RN. Vasoactive intestinal polypeptide: abundant immunoreactivity in neural cell lines and normal nervous tissue. Science. 1976;192:907–908. doi: 10.1126/science.1273576. [DOI] [PubMed] [Google Scholar]
- 13.Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004;56:249–290. doi: 10.1124/pr.56.2.7. [DOI] [PubMed] [Google Scholar]
- 14.Seaborn T, Masmoudi-Kouli O, Fournier A, Vaudry H, Vaudry D. Protective effects of pituitary adenylate cyclase-activating polypeptide (PACAP) against apoptosis. Curr Pharm Des. 2011;17:204–214. doi: 10.2174/138161211795049679. [DOI] [PubMed] [Google Scholar]
- 15.Delgado M, Munoz-Elias EJ, Gomariz RP, Ganea D. VIP and PACAP inhibit IL-12 production in LPS-stimulated macrophages. Subsequent effect on IFN gamma synthesis by T cells. J Neuroimmunol. 1999;96:167–181. doi: 10.1016/s0165-5728(99)00023-5. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen TD, Heintz GG, Wolfe MS. Structural characterization of PACAP receptors on rat liver plasma membranes. Am J Physiol. 1993;265:G811–G818. doi: 10.1152/ajpgi.1993.265.5.G811. [DOI] [PubMed] [Google Scholar]
- 17.Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004;56:249–290. doi: 10.1124/pr.56.2.7. [DOI] [PubMed] [Google Scholar]
- 18.Delgado M, Pozo D, Martinez C, Leceta J, Calvo JR, Ganea D, et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit endotoxin-induced TNF-alpha production by macrophages: in vitro and in vivo studies. J Immunol. 1999;162:2358–2367. [PubMed] [Google Scholar]
- 19.Delgado M, Munoz-Elias EJ, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide enhance IL-10 production by murine macrophages: in vitro and in vivo studies. J Immunol. 1999;162:1707–1716. [PubMed] [Google Scholar]
- 20.Ji H, Shen XD, Zhang Y, Gao F, Huang CY, Chang WW, et al. Activation of cyclic adenosine monophosphate-dependent protein kinase a signaling prevents liver ischemia/reperfusion injury in mice. Liver Transpl. 2012;18:659–670. doi: 10.1002/lt.23399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji H, Zhang Y, Shen XD, Gao F, Huang CY, Abad C, et al. Neuropeptide PACAP in mouse liver ischemia and reperfusion injury: Immunomodulation by the cAMP-PKA pathway. Hepatology. 2013;57:1225–1237. doi: 10.1002/hep.25802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harmar AJ, Fahrenkrug J, Gozes I, Laburthe M, May V, Pisegna JR, et al. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br J Pharmacol. 2012;166:14–17. doi: 10.1111/j.1476-5381.2012.01871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prasse A, Zissel G, Lützen N, Schupp J, Schmiedlin R, Gonzalez-Rey E, et al. Inhaled vasoactive intestinal peptide exerts immunoregulatory effects in sarcoidosis. Am J Respir Crit Care Med. 2010;182:540–548. doi: 10.1164/rccm.200909-1451OC. [DOI] [PubMed] [Google Scholar]
- 24.Onoue S, Yamada S, Yajima T. Bioactive analogues and drug delivery systems of vasoactive intestinal peptide (VIP) for the treatment of asthma/COPD. Peptides. 2007;28:1640–1650. doi: 10.1016/j.peptides.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Hamidi SA, Lin RZ, Szema AM, Lyubsky S, Jiang YP, Said SI. VIP and endothelin receptor antagonist: an effective combination against experimental pulmonary arterial hypertension. Respir Res. 2011;12:141. doi: 10.1186/1465-9921-12-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petkov V, Mosgoeller W, Ziesche R, Raderer M, Stiebellehner L, Vonbank K, et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111:1339–1346. doi: 10.1172/JCI17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Offen D, Sherki Y, Melamed E, Fridkin M, Brenneman DE, Gozes I. Vasoactive intestinal peptide (VIP) prevents neurotoxicity in neuronal cultures: relevance to neuroprotection in Parkinson’s disease. Brain Res. 2000;854:257–262. doi: 10.1016/s0006-8993(99)02375-6. [DOI] [PubMed] [Google Scholar]
- 28.Jiang W, Tang W, Geng Q, Xu X. Inhibition of Toll-like receptor 4 with vasoactive intestinal peptide attenuates liver ischemia-reperfusion injury. Transplant Proc. 2011;43:1462–1467. doi: 10.1016/j.transproceed.2011.01.191. [DOI] [PubMed] [Google Scholar]
- 29.Leister I, Mbachu EM, Post S, Samel ST, Stojanovic T, Gutt CN, et al. Vasoactive intestinal polypeptide and gastrin-releasing peptide attenuate hepatic microvasculatory disturbances following intestinal ischemia and reperfusion. Digestion. 2002;66:186–192. doi: 10.1159/000066761. [DOI] [PubMed] [Google Scholar]
- 30.Ji H, Shen X, Gao F, Ke B, Freitas MC, Uchida Y, et al. Programmed death-1/B7-H1 negative costimulation protects mouse liver against ischemia and reperfusion injury. Hepatology. 2010;52:1380–1389. doi: 10.1002/hep.23843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanlioglu AD, Karacay B, Balci MK, Griffith TS, Sanlioglu S. Therapeutic potential of VIP vs PACAP in diabetes. J Mol Endocrinol. 2012;49:R157–R167. doi: 10.1530/JME-12-0156. [DOI] [PubMed] [Google Scholar]
- 32.Yang J, Song TB, Zhao ZH, Qiu SD, Hu XD, Chang L. Vasoactive intestinal peptide protects against ischemic brain damage induced by focal cerebral ischemia in rats. Brain Res. 2011;1398:94–101. doi: 10.1016/j.brainres.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 33.Delgado M, Abad C, Martinez C, Leceta J, Gomariz RP. Vasoactive intestinal peptide prevents experimental arthritis by downregulating both autoimmune and inflammatory components of the disease. Nat Med. 2001;7:563–568. doi: 10.1038/87887. [DOI] [PubMed] [Google Scholar]
- 34.Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 35.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 36.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]