

Abstract
Carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) is expressed on activated natural killer (NK) cells wherein it inhibits lysis of CEACAM1-bearing tumor cell lines. The mechanism for this is unknown. Here we show that interleukin-2-induced expression of CEACAM1 on both mouse and primary human NK cells impairs the ability of NKG2D to stimulate cytolysis of CEACAM1-bearing cells. This process requires the expression of CEACAM1 on the NK cell and on the tumor cells, which is consistent with the involvement of trans-homophilic interactions between CEACAM1. Mechanistically, co-engagement of NKG2D and CEACAM1 results in a biochemical association between these two surface receptors and the recruitment of Src homology phosphatase 1 (SHP1) by CEACAM1 which leads to dephosphorylation of the guanine nucleotide exchange factor Vav1 and blockade of downstream signaling that is associated with the initiation of cytolysis. Thus, CEACAM1 on activated NK cells functions as an inhibitory receptor for NKG2D-mediated cytolysis, which has important implications for understanding the means by which CEACAM1 expression adversely affects tumor immunity.
Keywords: CEACAM1, cytolysis, interleukin-2, NKG2D, NK cell
Introduction
Natural killer (NK) cells constitute a first line of defense against tumors and viral infections. Accumulating evidence has shown that NK cells may also play an important role in immune tolerance and autoimmune diseases. NK cells exert these roles in immune surveillance via activating NK-cell receptors, which in turn are regulated by inhibitory NK-cell receptors. The inhibitory receptors on NK cells suppress activating receptors by recognizing their ligands, in some cases major histocompatibility complex (MHC) class I molecules expressed on target cells. However, the ligands for most activating NK-cell receptors are not defined. An activating NK-cell receptor that has well-defined ligands is natural killer gene 2 member D (NKG2D, gene name KLRK1). NKG2D is a C-type lectin-like type II transmembrane activating receptor [1]. Each polypeptide of an NKG2D homodimer associates with a homodimer of the adapter molecule DAP10 in human and mouse cells [2–4], or DAP12 exclusively in mouse NK cells [5, 6]. After engagement with its ligands, the NKG2D-DAP10 receptor complex recruits the growth factor receptor binding protein 2 (Grb2), a guanine nucleotide exchange factor Vav, and the p85 subunit of the phosphatidylinositide kinase (PI3K) to transduce signals that endow NKG2D with a pivotal role in NK-cell- and T-cell-mediated immunity against cancers and infections [6–8], as well as in the development of autoimmune diseases [9, 10].
The ligands for NKG2D are usually induced by stress such as during infection and malignancy. The NKG2D ligands (NKG2DL) in humans include MHC class I chain-related gene A and B (MICA, MICB) [11] and the ULBP proteins (ULBP1-6) [12–15], and in mouse, include the retinoic acid early inducible gene-1 (Rae-1), H60, and MULT1 proteins [16–18]. Once engagement of NKG2DL reaches a threshold on target cells, NKG2D on NK cells will surmount the restraint provided by inhibitory NK-cell receptors, resulting in the activation of cytolysis. However, NKG2D-bearing NK cells often are not able to lyse target cells, including tumor cells that have lost MHC class I expression, a ligand for some inhibitory NK-cell receptors [19]. In this context, NK cells may be induced to express new inhibitory molecules within the tumor environment such as carcinoembryonic antigen (CEA)-related cell adhesion molecule 1 (CEACAM1) [19].
CEACAM1 is a member of the CEA-family of immunoglobulin (Ig)-like transmembrane glycoproteins [20, 21]. It is constitutively expressed on a wide range of normal hematopoietic and parenchymal cells and can be observed on tumor cells. Its expression on NK cells and T cells is, however, mainly induced by cytokines and membrane-activating receptor engagement [21–25]. When expressed, CEACAM1 is characterized by significant alternate RNA splicing, leading to 12 isoforms in humans and at least 4 isoforms in mice. These isoforms differ in the length of the cytoplasmic tail (CT) and the number of extracellular Ig-like domains and are named accordingly. The most membrane distal amino terminal Ig domain of CEACAM1, the N-domain, is involved in the homophilic interactions that characterize these proteins. The majority of CEACAM1 isoforms possess either a long (CEACAM1-L) CT or a short (CEACAM1-S) CT. CEACAM1-L isoforms predominate in NK cells and T cells, and contain two immunoreceptor tyrosine-based inhibitory motifs (ITIM) [25–27]. Previous studies have shown that CEACAM1-L isoforms inhibit T-cell receptor (TCR)/CD3 complex, B-cell receptor (BCR), and Toll-like receptor 2 (TLR-2)-mediated immune responses among others [21, 27–31]. In each of these cases, this inhibition is mechanistically related to growth factor receptor tyrosine kinase- or Src kinase-mediated phosphorylation of the CEACAM1-L CT-associated ITIMs, recruitment of Src homology phosphatase 1 (SHP-1) and/or SHP-2, and consequently inhibition of downstream signaling elements [26, 27, 32–37].
Interleukin-2 (IL-2) is an important cytokine for maintaining growth, proliferation and function of T cells and NK cells. IL-2 also functions as an important homeostasis factor in immune responses by inducing T-cell death. We and others have previously reported that IL-2 induces T cells to express CEACAM1, which in turn attenuates T-cell-mediated immunity [27]. Moreover, expression of CEACAM1 on the cell surface of NK cells is associated with inhibition of cytolysis of CEACAM1-bearing NK susceptible targets in a mechanism that has yet to be defined. Here we show that IL-2 can induce NK cells to express the long CT isoform of CEACAM1, which in turn associates with and inhibits NKG2D cytolytic function and signaling. Taken together, our findings suggest that induction of CEACAM1 expression on NK cells is an alternative pathway to attenuate immune responses induced by IL-2.
Results
IL-2 induces CEACAM1 expression on NK cells without altering the expression of NK-cell receptors
When expressed on NK cells, CEACAM1 inhibits NK-cell-mediated cytolysis of CEACAM1-bearing tumor cells but not CEACAM1-null tumor cells of the same type, indicating that trans-homophilic interactions between CEACAM1 on NK cells and tumor cells engage and induce inhibitory signaling to impede the cytolytic function of activating NK-cell receptors [38]. However, the activating NK-cell receptors targeted by such an inhibitory mechanism associated with CEACAM1 have not been documented. Based upon our previous studies demonstrating that tumor cells expressing CEACAM1 escape NK-cell-mediated-immune surveillance by reducing the abundance of NKG2D ligands on the tumor in a cell autonomous manner [39] and previous reports by others that NK cells of melanoma patients exhibit decreased expression of the activating NK-cell receptor, NKp46 [40], we hypothesized that CEACAM1 on NK cells might also inhibit NKG2D expression.
To test this hypothesis, we took advantage of Ceacam1−/− mice (KO) mice. We isolated primary NK cells from the spleens of KO and wildtype (WT) littermate mice and stimulated them in vitro with IL-2-containing media for various periods of time. Figure 1A shows that after stimulation with IL-2 all NK cells of WT mice exhibited CEACAM1 expression on day 8 with additional expression after 12 days of culture in IL-2. As expected, no CEACAM1 was expressed on NK cells obtained from KO mice. Notably, the levels of NKG2D on the NK cells of both WT and KO mice were virtually identical (Figure 1A), indicating that CEACAM1 induction does not inhibit NKG2D expression on the cell surface of NK cells. We also observed that the expression of CEACAM1 on NK cells did not influence the expression of the activating NKp46 or inhibitory Ly-49 receptors (Figure 1B), suggesting that CEACAM1 impedes NK-cell-mediated immunity by directly inhibiting cytolytic function rather than by altering the expression of other activating or inhibitory NK-cell receptors.
Figure 1.

IL-2-induced CEACAM1 expression in mouse NK cells. (A) Flow cytometry analysis of primary NK cells from Ceacam1−/− (KO) and WT mice that were cultured with IL-2 for the indicated times. (B) Flow cytometry analysis for surface expression of NKp46 and Ly-49 receptors C/I/F/H on NK cells from CEACAM1 KO and WT mice. Histograms are gated on the CD3-negative, NK1.1-positive cell population. All results are representative of three independent experiments.
IL-2-induced CEACAM1 in NK cells inhibits NKG2D-mediated cytolysis of tumor cells
We therefore examined whether CEACAM1 expression on NK cells inhibits the activating NK-cell receptor NKG2D. To test this, we first performed cytotoxicity assays using freshly isolated primary NK cells, which express NKG2D but have not yet upregulated CEACAM1, from the spleens of either WT or KO mice as effectors and the target cells indicated in Figure 2A. We found that naïve NK cells from KO and WT mice exhibited the same ability to lyse different tumor cells. Mechanistically, we observed that CEACAM1 was not expressed on the cell surface of the WT NK cells during the time period associated with the cytotoxicity assay (data not shown). These results indicate that genetic disruption of CEACAM1 expression per se does not affect the cytolytic function of naïve, NKG2D-bearing, but CEACAM1-deficient NK cells.
Figure 2.
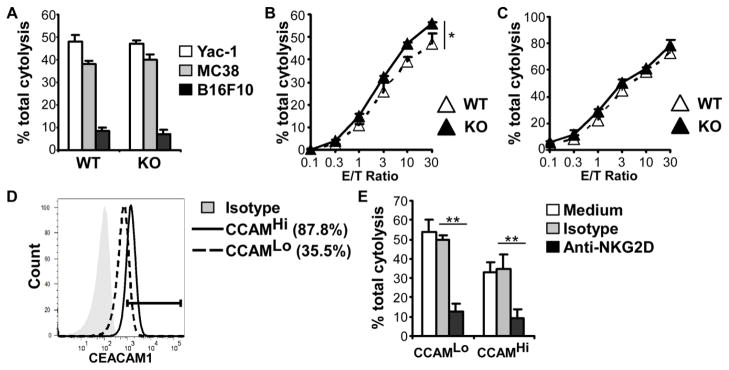
Downregulation of NKG2D function by IL-2 via induction of CEACAM1 expression on NK cells. (A) 51Chromium release cytotoxicity assays for comparison of the cytolytic potential of NK cells from CEACAM1 WT and KO mice. Effector NK cells were freshly isolated from mouse spleens; target cells as indicated. (B) 51Chromium release cytotoxicity assays for comparison of the cytolytic potential of NK cells from CEACAM1 WT and KO mice. Effector NK cells: IL-2-cultured NK cells for 8 days, target cell: CEACAM1-non-silenced MC38 cells. (C) 51Chromium release cytotoxicity assays for comparison of the cytolytic potential of NK cells from CEACAM1 WT and KO mice. Effector NK cells: IL-2-cultured NK cells for 8 days; target cells: CEACAM1-silenced MC38 cells. (D) Flow cytometry analysis of CEACAM1 surface expression on CEACAM1-silenced MC38 (CCAM1Lo) and CEACAM1-non-silenced MC38 cells (CCAM1Hi). (E) Anti-NKG2D blocking assay. NK cells were pretreated with anti-NKG2D (5 μg/ml) and analyzed by 51Chromium release cytotoxicity assays at an effector/target (E/T) ratio = 30:1. Effector NK cells: WT mouse NK cells cultured with IL-2 for 8 days; target cells: CEACAM1-silenced MC38 (CCAM1Lo) and CEACAM1-non-silenced MC38 cells (CCAM1Hi). (A–D) Data are shown as mean + SEM of triplicate cultures and are from one experiment representative of three experiments performed. *P < 0.05; **P < 0.01; Student’s t test.
We examined the effects of CEACAM1 induction on the cell surface of NK cells after IL-2 stimulation. As noted, NK cells from spleens of WT mice express significant levels of CEACAM1 on the cell surface after 8 days of stimulation with IL-2 as shown in Figure 1. When these activated NK cells were used as effector cells and the mouse colon cancer cell line, MC38, which expresses both CEACAM1 and Rae-1, were used as target cells in a cytotoxicity assay, we observed that the IL-2-stimulated NK cells from KO mice exhibited enhanced cytolytic activity than that observed with IL-2 stimulated NK cells from WT mice (Figure 2B). This indicates that CEACAM1 on NK cells of WT mice impedes the cytolytic function of NK cells, likely through homophilic interactions with CEACAM1 on the tumor cells. To confirm this, we silenced the expression of CEACAM1 on MC38 cells and used these as target cells in a cytotoxicity assay with IL-2-stimulated WT NK cells as effectors. As expected, the cytolytic activity of the IL-2-stimulated WT NK cells was similar to the levels observed with IL-2-stimulated NK cells from KO mice (Figure 2C). Thus, loss of CEACAM1 expression on the target cell reversed the inhibitory effects of CEACAM1 expression on the activated NK cell.
These observations with WT IL-2-stimulated NK cells was consistent with previous studies showing that CEACAM1-bearing NK cells are disabled in their ability to lyse tumor cells that express CEACAM1 in comparison with their ability to destroy CEACAM1-null tumor cells of the same type [38, 41]. To further determine whether this decreased efficiency of CEACAM1-bearing NK cells to lyse tumor cells that express CEACAM1 was due to the inhibition of NKG2D cytolytic function by CEACAM1, we performed an anti-NKG2D mAb blocking assay with CEACAM1- silenced (CEACAM1Lo) or non-silenced (CEACAM1Hi) MC38 cells (Figure 2D). Although the lysis of CEACAM1Hi cells was less than that of CEACAM1-silenced CEACAM1Lo MC38 cells, mouse NKG2D blocking antibody almost completely prevented WT NK cell-mediated cytotoxicity of both types of MC38 tumor cells (Figure 2E).
To confirm and extend these findings, we turned our attention to human NK cells and took advantage of the human NK-cell line NK92. NK92 cells are maintained in medium containing IL-2 and consequently express NKG2D and CEACAM1 (Figure 3A), which is predominantly derived from long CT-containing isoforms based upon PCR data (Figure 3B). We observed that NK92 cells were able to lyse the EBV-transformed human B cell line 721.221 transfected with MICA at an effector/target (E/T) ratio = 0.7 or higher (data not shown). To confirm that the lysis of these MICA-transfected 721.221 cells by NK92 cells was mediated by NKG2D, we performed a blocking assay. An anti-human NKG2D blocking antibody was shown to inhibit NK92-elicited lysis of MICA-transfected 721.221 cells in a dose dependent manner at an E/T ratio = 0.7 (Figure 3C). This indicates that the augmented lysis of MICA-transfected 721.221 by NK92 cells is mainly mediated by NKG2D. Since it has been previously reported that 721.221 parental cells do not express sufficient levels of CEACAM1 to inhibit CEACAM1-bearing NK cell-mediated cytolysis [38, 41], and to demonstrate that the CEACAM1 that is expressed on NK92 cells is indeed inhibitory to NKG2D-mediated lysis, we furthermore engaged the CEACAM1 expressed on NK92 cells with a human CEACAM1 antibody (5F4) that is weakly agonistic at high antibody concentrations under cross-linking conditions and previously demonstrated to elicit inhibitory signaling by human cytolytic T cells under these conditions [27]. The 5F4 anti-CEACAM1 antibody was observed to inhibit NKG2D-mediated lysis of the MICA-transfected 721.221 cell line at an E/T ratio of 0.7 in a dose-dependent fashion (Figure 3C). We also evaluated human primary NK cell cytotoxicity using an antibody-redirected lysis assay in order to confirm that direct co-ligation of CEACAM1 and NKG2D was required for inhibition of cytotoxicity. CEACAM1 expression on the primary human NK cells was induced by IL-2 stimulation for 12 days, similar to our observation with mouse NK cells (Figure 3D). Reverse transcription PCR (RT-PCR) showed that these IL-2-stimulated human NK cells expressed CEACAM1-L isoforms (data not shown). Primary NK cells stimulated with IL-2 for 12 days were examined in an antibody-redirected lysis assay using Fc receptor-bearing P815 cells as target cells in the presence of anti-NKG2D antibodies with or without concomitant anti-CEACAM1 antibodies. These studies confirmed that anti-NKG2D induced cytotoxicity was inhibited by CEACAM1 co-ligation with a mouse anti-human CEACAM1 antibody (5F4) in a dose-dependent fashion (Figure 3E). We further observed that cross-linking of NKG2D-induced IFN-γ production by human NK cells treated with IL-2 for 12 days in the presence of a non-binding IgG1 control antibody (Figure 3F, left panel). In addition, NKGD-mediated stimulation of IFN-γ production was only observed under conditions of cross-linking using a mouse specific F(ab′)2 antibody (Figure 3E, left panel), but not with an irrelevant rabbit specific F(ab′)2 antibody (Figure 3E, right panel). Moreover, anti-NKG2D antibody-induced IFN-γ was inhibited by CEACAM1 co-ligation with the 5F4 antibody under conditions conducive to cross-linking (Figure 3E, left panel) but not conditions that were unable to induce cross-linking (Figure 3E, right panel). These data confirm that direct co-ligation of CEACAM1 and NKG2D in cis is indeed required for CEACAM1-mediated inhibition of NKG2D.
Figure 3.

Engagement of CEACAM1 on human NK cells attenuates NKG2D-mediated cytotoxicity. (A) Flow cytometry analysis shows the expression of CEACAM1 and NKG2D on NK92 cells. (B) RT-PCR results shows that NK92 cells predominantly express CEACAM1-L. CEACAM1-L and -S: corresponding cDNA plasmids for positive controls; NK92: NK92 RNA. (C) 51Chromium release cytotoxicity assay to reveal that engagement of CEACAM1 by anti-human CEACAM1 antibody (5F4) under previously described cross-linking conditions with goat anti-mouse IgG [28] decreases NKG2D-mediated cytotoxicity. Effector: NK92 cells, target: MICA-transfected 721.221 cells. Data are shown as mean + SEM of triplicate cultures. Effector/target ratio was 0.7:1. (A–C) Data shown are representative of four independent experiments. (D) Flow cytometric analysis of primary human NK cells which were cultured with IL-2 for the indicated times. Filled histogram = isotype control. Solid line = CEACAM1. (E) Antibody-redirected cytotoxicity assay in the presence of the anti-human NKG2D antibody (2 μg/ml) with anti-human CEACAM1 antibody or control mouse IgG1 (indicated concentrations). Data are shown as mean + SEM of triplicate cultures. Effector: primary human NK cells treated with IL-2 for 12 days, target: P815 cells. E/T ratio was 20:1. (F) Anti-human CEACAM1 antibody inhibits IFN-γ production by human NK cells in response to IL-2 and anti-human NKG2D antibody stimulation. The IL-2 treated primary human NK cells were stimulated with mouse anti-human NKG2D antibody (4 μg/ml) and either mouse anti-human CEACAM1 antibody or control mouse IgG1 (indicated concentrations) in the presence of F(ab′)2 fragments (8 μg/ml) from goat anti-mouse Ig or control goat anti-rabbit F(ab′)2 fragments for 24 hours. IFN-γ levels in culture supernatants were measured by ELISA and are shown as mean + SEM of triplicate wells. (D–F) Data shown are representative of two independent experiments performed. *P<0.05; **P<0.01, paired, two-tailed Student’s t test.
NKG2D stimulation induces association of NKG2D with CEACAM1
It has been previously shown that CEACAM1, which is expressed on T and B cells, can translocate into the TCR and BCR immune synapse [27, 30]. We therefore examined whether CEACAM1 expressed on IL-2-activated NK cells could associate with NKG2D leading to an impairment of the consequent cytolytic activity of the NK cells. To test this hypothesis, we initially used two methodological approaches to demonstrate an association between NKG2D and CEACAM1 upon NKG2D stimulation. In the first approach, we co-engaged NK92 cells with an anti-human NKG2D agonistic antibody (1D11) together with either an anti-human CEACAM1 antibody (5F4) or antibody directed at CD45, a tyrosine phosphatase, fixed the cells, and then detected the localization of NKG2D, CEACAM1, and CD45 by confocal microscopy analysis. We observed the co-localization of NKG2D with CEACAM1 but not with CD45, as shown by the presence of punctate immunofluorescence at the membrane surface and intracellularly within vesicular structures (Figure 4A). These results are consistent with the co-localization of NKG2D with CEACAM1, but not CD45, upon NKG2D ligation. In an alternative approach, we further confirmed the association between NKG2D and CEACAM1 by co-immunoprecipitation assays. In this case, we stimulated NK92 cells with an anti-NKG2D antibody (1D11), removed the free anti-NKG2D antibody and immunoprecipitated CEACAM1 with different antibodies as indicated. Our results showed that CEACAM1 could only be immunoprecipitated by anti-human CEACAM1 and anti-NKG2D antibodies but not by mouse IgG control or anti-human HLA-A, B, C class I antibodies. This indicates that activation of NKG2D specifically promotes its association with CEACAM1 (Figure 4B) and thus induces a localized rather than diffusible association further supporting the observations above in an antibody-redirected cytolysis assay.
Figure 4.
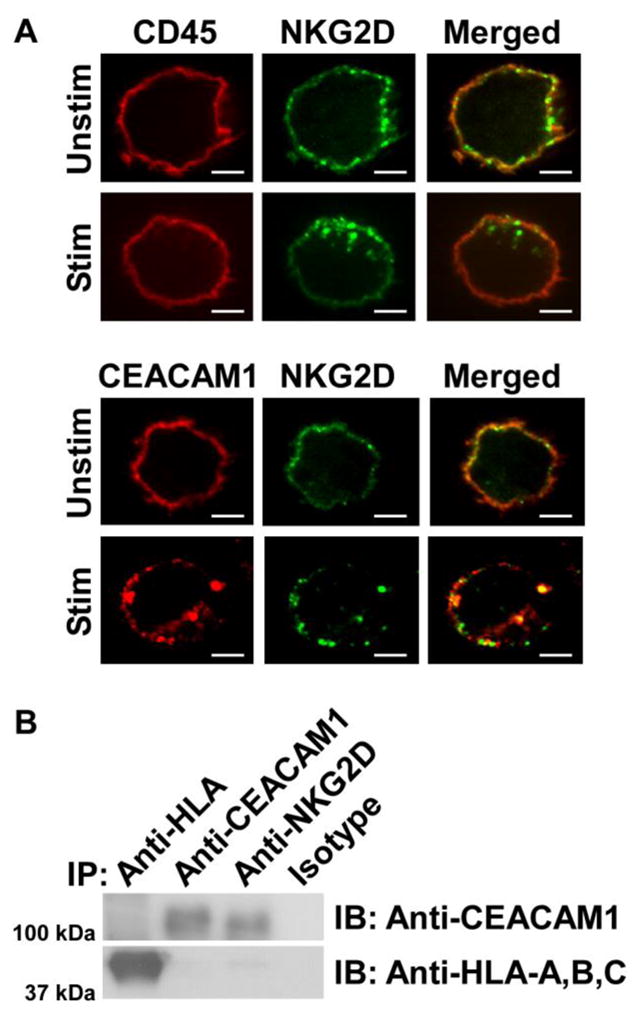
NKG2D association with CEACAM1 in NK92 cells. (A) Confocal microscopy analysis shows that after stimulation NKG2D co-localizes and co-internalizes with CEACAM1, but not CD45, (as demonstrated by yellow staining) in NK92 cells. Scale bar: 5 μm. (B) Immunoblot (IB) results show that NKG2D associates with CEACAM1, but not HLA-A, B, C. IP: immunoprecipitation. All results shown are representative of three experiments performed.
CEACAM1 attenuates NKG2D cytolytic signaling through SHP-1
NK cells constitutively express inhibitory receptors that can associate with SHP1 and inhibit the cytolytic function of activating NK-cell receptors in the presence of MHC class I expression on target cells [42]. However, certain tumor cells are characterized by the loss of MHC class I expression by a variety of mechanisms yet remain insensitive to NK cell-mediated cytolysis [43]. It has been proposed that CEACAM1 may replace MHC class I-inhibitory receptors in this scenario [21, 44]. Thus, in the following set of studies we sought to determine whether CEACAM1 on NK cells associates with SHP1 and uses this phosphatase to inhibit NK-cell cytolytic function. Since we and others have found that the association between CEACAM1 and SHP1 requires stimulation of activating receptors on T and B cells, we tested whether stimulation of NKG2D induces the association between CEACAM1-L and SHP-1 in human NK92. As shown in Figure 5A, upon NKG2D stimulation, CEACAM1 was observed to associate with SHP-1. Our next group of studies therefore focused on the effects of SHP1 recruitment by CEACAM1 on the activity and function of Vav1. Vav1 is one of the most proximal elements associated with NKG2D-mediated cytolytic signaling which is shared in mouse and human cells [7]. As shown in Figure 5B, the CEACAM1-SHP-1 immune complex that was specifically immunoprecipitated by an anti-CEACAM1 monoclonal antibody (5F4), was capable of dephosphorylating phosphorylated-Vav1 obtained from NK92 cells by immunoprecipitation with an anti-Vav1 antibody. This indicates that the tyrosine phosphorylated Vav1 in NK cells induced by NKG2D stimulation is a target of SHP1 phosphatase activity. Consistent with this, we further demonstrated that engagement of NK92 cells with an anti-NKG2D antibody in the presence of an anti-CEACAM1 antibody (5F4), but not control Ig, significantly decreased the phosphorylation not only of Vav1 but also of JNK, which is downstream of NKG2D-activated Vav1, as shown by an evaluation of their phosphorylated forms with individual phospho-specific antibodies (Figure 5C).
Figure 5.

CEACAM1 recruits SHP1 to attenuate NKG2D-dependent cytolytic signaling. (A) Immunoblot (IB) results show that anti-human CEACAM1 antibody, but not control Ig, co-immunoprecipitates (IP) SHP-1 from anti-human NKG2D-stimulated NK92 cell lysates. (B) CEACAM1-associated SHP1 dephosphorylates Vav1. The immunoblot (IB) results show that anti-human CEACAM1 but not control Ig immune complexes from the lysates of anti-human NKG2D-stimulated NK92 cells dephosphorylate Vav1 immunoprecipitated by anti-Vav specific antibody. (C) Co-engagement of CEACAM1 and NKG2D for 5 min inhibits phosphorylation of Vav1 as well as downstream elements. The immunoblot (IB) shows that co-engagement of NK92 by anti-human CEACAM1 monoclonal antibody, but not control Ig, inhibits anti-human NKG2D-induced phosphorylation of Vav1 and JNK as evaluated by corresponding phospho-specific antibodies. (A–C) Results shown are representative of four experiments performed.
Discussion
We here show that IL-2-induced CEACAM1 expression on NK cells dampens the cytolytic function of NK cells induced by NKG2D ligation by NKG2DL. Our data specifically show that NKG2D activation leads to a physical association with CEACAM1 resulting in the recruitment of SHP1 leading to the deactivation of the proximal elements of NKG2D-initiated cytolytic signaling and consequently reduced lysis of NKG2DL-expressing target cells such as tumor cells. We also demonstrate that CEACAM1 expressed on NK cells does not reduce the protein expression of NKG2D or of either Ly-49 inhibitory or NKp46 activating receptors. Thus, CEACAM1-mediated inhibition of IL-2-induced NK-cell activation occurs directly through its effects on NKG2D-initiated signaling of cytolytic function in a pathway that requires trans-homophilic interactions with CEACAM1. This mechanism is distinct from our previous studies showing that CEACAM1 on tumor cells retains NKG2DL intracellularly in a cell autonomous manner, which diminishes the sensitivity of tumors to NKG2D-mediated lysis [39]. Thus, our studies reveal distinct mechanistic pathways associated with expression of CEACAM1 on tumor cells and NK cells that lead to diminished NK-cell-dependent immune surveillance which is capable of targeting a wide variety of cell types. In the first instance, this is associated with diminished sensitivity of the target cell to lysis. This is a cell autonomous process that is independent of trans-homophilic interactions and associated with CEACAM1-mediated retention of NKG2DL as previously described [39]. The second mechanism is due to direct interactions of CEACAM1 with NKG2D and inhibition of downstream signaling associated with cytolysis. This latter mechanism is a trans-homophilic-dependent process and requires CEACAM1 on the target (tumor) cell as previously proposed [41]. Overall, this study provides a mechanistic basis for previous findings demonstrating that CEACAM1-bearing NK cells are not able to efficiently lyse CEACAM1-positive tumor cells [29]. This study also provides new clues to explain the observation that cancer patients are refractory to the treatment of IL-2 and/or adoptively transferred IL-2-activated LAK (lymphokine-activated killer) cells [45].
Human NK cells only express the long isoform of NKG2D that exclusively associates with the adaptor molecule, DAP10, which lacks an ITAM [1]. However, a YXXM motif in the CT of DAP10 can mediate association with GRB2, Vav1, and PI3K, which transmit intracellular signals that endow NKG2D with a crucial function in NK cell-mediated immunity [1, 4, 7]. Using a well-known human NK-cell line, we were able to demonstrate that CEACAM1 functions to recruit SHP1 into the vicinity of NKG2D and down-regulates the activity of Vav1 and its downstream signaling associated with cytolysis. However, activated mouse NK cells are also capable of expressing a short isoform of NKG2D, which can associate with the adaptor molecule, DAP12, which contains an ITAM [1]. ITAM-containing adaptor molecules can activate the SYK kinase-dependent signaling pathways [1]. We and others have previously found that CEACAM1 in T cells can inhibit TCR-mediated activation of ZAP-70, a member of SYK tyrosine kinase family [27]. In these other studies, we have further demonstrated that engagement of the TCR/CD3 complex induces CEACAM1 association with and recruitment of SHP1 to the TCR/CD3 complex wherein SHP1 dephosphorylates CD3-ζ and ZAP-70. Thus, in activated mouse NK cells, CEACAM1-mediated inhibitory signaling is also likely to lead to decreased SYK kinase-related cytolytic signaling that is mediated by the NKG2D/DAP12 complex and worthy of further investigation in future studies. This notion is indirectly confirmed in this study by showing that NK cells from WT mice exhibited decreased NKG2D-mediated cytolysis of CEACAM1-bearing tumor cells by NKG2D in a pathway that depended upon IL-2 induced CEACAM1 expression on NK cells; a process that was not observed when NK cells were obtained from CEACAM1-deficient mice. These studies are consistent with a model in which trans-homophilic interactions between CEACAM1 expressed on tumor cells and NK cells is crucial for the ability of CEACAM1 on NK cells to trigger inhibitory signaling as a consequence of NKG2D ligation by NKG2DL on target cells. Thus, this work elucidates the molecular mechanisms that are operative in our previously published studies on NK-cell-mediated tumor cytotoxicity mediated by CEACAM1 [39] and studies by others which demonstrate the ability of CEACAM1 to inhibit NK cytotoxicity [38, 40, 41].
These studies significantly add to the mechanisms identified that are associated with inhibition of NKG2D function. Immunoglobulin-like inhibitory NK-cell receptors not only employ SHP1 to negatively regulate NK-cell activation [42] but also down-regulate expression of activating NK-cell receptors on NK and/or CD8+ T cells [46]. Based upon our observations, CEACAM1 utilizes the first mechanism to disable NKG2D-mediated signaling.
Another aspect of our study deserves comment. Notably, SHP1 is normally excluded from membrane lipid rafts [47, 48] where an immune synapse complex localizes [49]. Previous studies by our lab and others have demonstrated that CEACAM1 is able to shuttle between lipid rafts and non-lipid raft membranes [27, 50]. Given our observations, and specifically the fact that CEACAM1 becomes associated with the NKG2D receptor complex upon co-engagement of NKG2D and CEACAM1, suggests that CEACAM1 facilitates entry of SHP1 into the vicinity of the NKG2D complex signalosome, which is likely enriched in lipid rafts. Further studies of the details of this mechanism, including whether there is any role played by disruption of target cell-effector conjugate formation, will be required to fully understand this process.
Finally, it should be noted that CEACAM1 can associate with IL-2 receptor in human T cells and lead to a reduction of IL-2 receptor expression [50]. However, CEACAM1, as shown here, did not affect the cell-surface expression of NKG2D and other NK-cell receptors in NK cells. Consistent with our observations, CEACAM1 can associate with Toll-like receptor 2 [31], granulocyte colony-stimulating factor receptor (G-CSFR) [51], and TCR [27, 50] without decreasing their cell surface expression.
In summary, we provide a mechanistic basis for previous observations that have shown CEACAM1 expression on NK cells inhibits their cytolytic function when they are exposed to CEACAM1-bearing targets such as tumor cell lines [38, 41, 52]. Specifically, we demonstrate that this involves inhibition of NKG2D-mediated signaling that leads to cytolysis of target cells. This pathway is enabled by IL-2 through its ability to induce CEACAM1 expression on the NK cell, which allows for trans-homophilic interactions with CEACAM1 on the target cell [38] and involves NKG2D-stimulated association between CEACAM1 and NKG2D. CEACAM1 can thus be brought into the immediate vicinity of the NKG2D signalosome, leading to inactivation of proximal signaling events downstream of NKG2D such as Vav1 and its subsequent effector elements as exemplified by JNK. These studies add to burgeoning information on the importance of CEACAM1 as a negative regulator of immune surveillance against tumors and potentially other diseases that require NK-cell and CD8+ T cell immunity such as viral infections.
Materials and methods
Mice
Wild-type (WT) C57BL/6 (B6) mice were purchased from Charles River Laboratories (Wilmington, MA) or Taconic Farms Inc. (Germantown, NY). Ceacam1−/− (KO) mice on the C57BL/6 background have previously been described [43]. All mice were used between 8 and 12 weeks of age. Mice were maintained under specific pathogen-free conditions at the Harvard Center for Comparative Medicine at Harvard Medical School. All animal experimentation was performed in accordance with the Institutional Animal Care and Use Committee (IACUC) at Harvard Medical School, which granted permission for this study.
Cell lines
MC38, B16F10, Yac-1, and P815 cells were from the ATCC (Manassas, VA). MICA*0019-expressing 721.221 cells were constructed as previously described [53] and MC38-silenced cells have been previously reported [39]. All cells were cultured in RPMI-1640 medium supplemented with 10% FBS, ampicillin, streptomycin, 10 mM HEPES, and 3 μM β–ME.
NK cells and cytotoxicity assays
Mouse primary NK cells were isolated from mouse spleens and human primary NK cells were purified from leukopacks (Kraft Family Blood Donor Center of the Dana-Farber Cancer Institute and Brigham and Women’s Hospital) using the corresponding NK-cell isolation kits (Miltenyi, Auburn, CA) according to the manufacturer’s instruction. The human NK-cell line NK92 (kindly provided by Dr. Jack Strominger, Harvard Medical School) and human primary NK cells were cultured in RPMI-1640 supplemented with 10% FBS and 200 units/mL IL-2 (NIH). In some experiments, the effector cells were pretreated with anti-NKG2D (Clone 191004 for mouse and Clone 149810 for human, R&D Systems, Minneapolis, MN) blocking antibodies or anti-human CEACAM1 antibody (5F4) [54], as indicated, for 30 minutes at 4°C. Antibody-redirected cytotoxicity assays were performed as previously described [27, 54]. A standard 4-hour 51Cr (PerkinElmer, Waltham, MA) release cytotoxicity assay and calculation of percentage of specific cytolysis were performed as described previously [27].
Immunoprecipitation and western blotting
Immunoprecipitation (IP) and western blotting were performed as previously described by using specific antibodies as indicated [27]. Anti-HLA-A, B, C antibody (Clone W6/32, BioLegend) was used as control. For Vav dephosphorylation, NK92 cells were stimulated with 2 μg/5×106 cells of anti-NKG2D antibodies (1D11, eBioscience, San Diego, CA) followed by cross-linking with a F(ab)2 of goat anti-mouse Ig (Jackson ImmunoResearch, PA) for 2 minutes. To remove antibodies, cells were washed twice with 100 mM glycine containing 50 mM NaCl (pH 3) and subsequently washed in PBS. The cell lysates were immunoprecipitated either with anti-Vav1 (Cell Signaling Technology, Danvers, MA) or with anti-human CEACAM1 (5F4) antibodies or control mouse IgG of the relevant isotype. Anti-Vav immunoprecipitates were mixed with either anti-human CEACAM1 or mouse IgG immunoprecipitates in phosphatase buffer for 30 minutes. Samples were analyzed by immunoblotting with an anti-phospho-Vav1 specific antibody (Invitrogen, Carlsbad, CA).
RNA silencing, detection
The methods for silencing expression of CEACAM1 have been previously reported [39]. CEACAM1 isoforms were identified as previously described [18] by RT-PCR.
Antibody cross-linking assay
IL-2 stimulated human primary NK cells were stimulated with anti-human NKG2D antibody (1D11, 2 μg/ml) and either anti-human CEACAM1 antibody (5F4) or control mouse IgG1 (MOPC) as indicated. These antibodies were cross-linked using F(ab)2 fragments (8 μg/ml) from goat anti-mouse Ig (Invitrogen) or control goat anti-rabbit Ig (Invitrogen). After 24 hours, the quantity of human IFN-γ in culture supernatants was measured by ELISA (BD Biosciences) according to the manufacturer’s instructions.
Confocal microscopy
NK92 cells were incubated with FITC-conjugated anti-human NKG2D (eBioscience) with either biotin-labeled anti-human CEACAM1 or anti-CD45 (eBioscience) antibodies on ice for 1 hour. After removal of free antibodies by washing three times with PBS the cells were transferred to 37°C for 30 minutes, fixed with 4% paraformaldehyde, permeabilized with 0.2% saponin, and then stained with rhodamine-conjugated streptavidin. The slides were analyzed on the workstation of a Nikon TE2000-E inverted microscope coupled to a Perkin-Elmer spinning disk confocal unit and an Orca AG cooled CCD camera (from Hamamatsu) with a Plexiglas chamber enclosing the stage for control of the sample environment (temperature and humidity).
Flow cytometry
For CEACAM1 expression analysis, cells were incubated with mouse anti-mouse CEACAM1 monoclonal antibody (CC1) [55], (a gift kindly provided by Dr. K. Holmes, University of Colorado, Denver) or mouse anti-human CEACAM1 monoclonal antibody (5F4) for 20 minutes followed by FITC-conjugated rat anti-mouse IgG1 (Jackson ImmunoResearch). For detection of NKp46 and Ly-49 surface expression, cells were stained with anti-NKp46 (BD Pharmingen), Ly-49 C/I/F/H (eBioscience) antibodies, and anti-NK1.1 antibody (BD Pharmingen) for 30 minutes.
Statistics
A student’s t test (paired, two tailed) was used to determine significance. A P value of less than 0.05 was considered as significant.
Acknowledgments
The authors appreciate the technical assistance of Jessica Wagner for confocal microscopy analysis and Jennifer Cusick for technical support. We thank Dr. K. Holmes for providing the anti-mouse CEACAM1 specific antibody (CC1). RSB was supported by NIH DK051362, DK044319, DK053056, DK088199, Harvard Digestive Diseases Center (NIH DK034854) and the High Pointe Foundation. RSB and OM were supported by Israel-U.S. Binational Research Award. KB and NB were supported by the Canadian Institutes of Health Research. LLL is an American Cancer Society Professor and is supported by NIH AI066897. YHH and LC were supported by a Research Fellowship Award from the Crohn’s & Colitis Foundation of America. SZ was supported by theDeutsche Forschungsgemeinschaft (Ze 814/1-1) and the Crohn’s and Colitis Foundation of America. TO was supported by the Deutsche Forschungsgemeinschaft (OL 424/1-1). The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: human IL-2 from Dr. Maurice Gately, Hoffmann-La Roche Inc.
Abbreviations
- CEACAM1
Carcinoembryonic antigen-related cell adhesion molecule 1
- CEACAM1-L
CEACAM1 long cytoplasmic tail isoform
- CEACAM1-S
CEACAM1 short cytoplasmic tail isoform
- CT
cytoplasmic tail
- E/T ratio
effector/target ratio
- IB
immunoblot
- MICA
MHC class I chain-related gene A
- NKG2D
natural killer gene 2 group D
- NKG2DL
NKG2D ligand
- Rae1
retinoic acid early inducible gene-1
- shRNA
short hairpin RNA
- Vav
guanine nucleotide exchange factor Vav
Footnotes
Conflict of interest
The authors declare no financial or commercial conflict of interest.
References
- 1.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diefenbach A, Tomasello E, Lucas M, Jamieson AM, Hsia JK, Vivier E, Raulet DH. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol. 2002;3:1142–1149. doi: 10.1038/ni858. [DOI] [PubMed] [Google Scholar]
- 3.Garrity D, Call ME, Feng J, Wucherpfennig KW. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc Natl Acad Sci U S A. 2005;102:7641–7646. doi: 10.1073/pnas.0502439102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilfillan S, Ho EL, Cella M, Yokoyama WM, Colonna M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat Immunol. 2002;3:1150–1155. doi: 10.1038/ni857. [DOI] [PubMed] [Google Scholar]
- 5.Rosen DB, Araki M, Hamerman JA, Chen T, Yamamura T, Lanier LL. A Structural basis for the association of DAP12 with mouse, but not human, NKG2D. J Immunol. 2004;173:2470–2478. doi: 10.4049/jimmunol.173.4.2470. [DOI] [PubMed] [Google Scholar]
- 6.Zompi S, Hamerman JA, Ogasawara K, Schweighoffer E, Tybulewicz VL, Di Santo JP, Lanier LL, et al. NKG2D triggers cytotoxicity in mouse NK cells lacking DAP12 or Syk family kinases. Nat Immunol. 2003;4:565–572. doi: 10.1038/ni930. [DOI] [PubMed] [Google Scholar]
- 7.Upshaw JL, Arneson LN, Schoon RA, Dick CJ, Billadeau DD, Leibson PJ. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat Immunol. 2006;7:524–532. doi: 10.1038/ni1325. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, Phillips JH. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 1999;285:730–732. doi: 10.1126/science.285.5428.730. [DOI] [PubMed] [Google Scholar]
- 9.Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2003;100:9452–9457. doi: 10.1073/pnas.1632807100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, Lanier LL. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20:757–767. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 12.Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14:123–133. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- 13.Chalupny NJ, Sutherland CL, Lawrence WA, Rein-Weston A, Cosman D. ULBP4 is a novel ligand for human NKG2D. Biochem Biophys Res Commun. 2003;305:129–135. doi: 10.1016/s0006-291x(03)00714-9. [DOI] [PubMed] [Google Scholar]
- 14.Eagle RA, Traherne JA, Hair JR, Jafferji I, Trowsdale J. ULBP6/RAET1L is an additional human NKG2D ligand. Eur J Immunol. 2009;39:3207–3216. doi: 10.1002/eji.200939502. [DOI] [PubMed] [Google Scholar]
- 15.Eagle RA, Flack G, Warford A, Martínez-Borra J, Jafferji I, Traherne JA, Ohashi M, et al. Cellular Expression, Trafficking, and Function of Two Isoforms of Human ULBP5/RAET1G. PLoS ONE. 2009;4:e4503. doi: 10.1371/journal.pone.0004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerwenka A, Bakker AB, McClanahan T, Wagner J, Wu J, Phillips JH, Lanier LL. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12:721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 17.Malarkannan S, Shih PP, Eden PA, Horng T, Zuberi AR, Christianson G, Roopenian D, et al. The molecular and functional characterization of a dominant minor H antigen, H60. J Immunol. 1998;161:3501–3509. [PubMed] [Google Scholar]
- 18.Carayannopoulos LN, Naidenko OV, Fremont DH, Yokoyama WM. Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J Immunol. 2002;169:4079–4083. doi: 10.4049/jimmunol.169.8.4079. [DOI] [PubMed] [Google Scholar]
- 19.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 20.Beauchemin N, Draber P, Dveksler G, Gold P, Gray-Owen S, Grunert F, Hammarstrom S, et al. Redefined nomenclature for members of the carcinoembryonic antigen family. Exp Cell Res. 1999;252:243–249. doi: 10.1006/excr.1999.4610. [DOI] [PubMed] [Google Scholar]
- 21.Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat Rev Immunol. 2006;6:433–446. doi: 10.1038/nri1864. [DOI] [PubMed] [Google Scholar]
- 22.Azuz-Lieberman N, Markel G, Mizrahi S, Gazit R, Hanna J, Achdout H, Gruda R, et al. The involvement of NK cells in ankylosing spondylitis. Int Immunol. 2005;17:837–845. doi: 10.1093/intimm/dxh270. [DOI] [PubMed] [Google Scholar]
- 23.Moller MJ, Kammerer R, Grunert F, von Kleist S. Biliary glycoprotein (BGP) expression on T cells and on a natural-killer-cell sub-population. Int J Cancer. 1996;65:740–745. doi: 10.1002/(SICI)1097-0215(19960315)65:6<740::AID-IJC5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 24.Nakajima A, Iijima H, Neurath MF, Nagaishi T, Nieuwenhuis EE, Raychowdhury R, Glickman J, et al. Activation-induced expression of carcinoembryonic antigen-cell adhesion molecule 1 regulates mouse T lymphocyte function. J Immunol. 2002;168:1028–1035. doi: 10.4049/jimmunol.168.3.1028. [DOI] [PubMed] [Google Scholar]
- 25.Singer BB, Scheffrahn I, Heymann R, Sigmundsson K, Kammerer R, Obrink B. Carcinoembryonic antigen-related cell adhesion molecule 1 expression and signaling in human, mouse, and rat leukocytes: evidence for replacement of the short cytoplasmic domain isoform by glycosylphosphatidylinositol-linked proteins in human leukocytes. J Immunol. 2002;168:5139–5146. doi: 10.4049/jimmunol.168.10.5139. [DOI] [PubMed] [Google Scholar]
- 26.Beauchemin N, Kunath T, Robitaille J, Chow B, Turbide C, Daniels E, Veillette A. Association of biliary glycoprotein with protein tyrosine phosphatase SHP-1 in malignant colon epithelial cells. Oncogene. 1997;14:783–790. doi: 10.1038/sj.onc.1200888. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, Chen L, Qiao SW, Nagaishi T, Blumberg RS. Carcinoembryonic antigen-related cell adhesion molecule 1 inhibits proximal TCR signaling by targeting ZAP-70. J Immunol. 2008;180:6085–6093. doi: 10.4049/jimmunol.180.9.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boulton IC, Gray-Owen SD. Neisserial binding to CEACAM1 arrests the activation and proliferation of CD4+ T lymphocytes. Nat Immunol. 2002;3:229–236. doi: 10.1038/ni769. [DOI] [PubMed] [Google Scholar]
- 29.Chen Z, Chen L, Blumberg RS. Editorial: CEACAM1: fine-tuned for fine-tuning. J Leukoc Biol. 2009;86:195–197. doi: 10.1189/jlb.0309122. [DOI] [PubMed] [Google Scholar]
- 30.Lobo EO, Zhang Z, Shively JE. Pivotal advance: CEACAM1 is a negative coreceptor for the B cell receptor and promotes CD19-mediated adhesion of B cells in a PI3K-dependent manner. J Leukoc Biol. 2009;86:205–218. doi: 10.1189/jlb.0109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slevogt H, Zabel S, Opitz B, Hocke A, Eitel J, N’Guessan PD, Lucka L, et al. CEACAM1 inhibits Toll-like receptor 2-triggered antibacterial responses of human pulmonary epithelial cells. Nat Immunol. 2008;9:1270–1278. doi: 10.1038/ni.1661. [DOI] [PubMed] [Google Scholar]
- 32.Abou-Rjaily GA, Lee SJ, May D, Al-Share QY, Deangelis AM, Ruch RJ, Neumaier M, et al. CEACAM1 modulates epidermal growth factor receptor--mediated cell proliferation. J Clin Invest. 2004;114:944–952. doi: 10.1172/JCI21786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huber M, Izzi L, Grondin P, Houde C, Kunath T, Veillette A, Beauchemin N. The carboxyl-terminal region of biliary glycoprotein controls its tyrosine phosphorylation and association with protein-tyrosine phosphatases SHP-1 and SHP-2 in epithelial cells. J Biol Chem. 1999;274:335–344. doi: 10.1074/jbc.274.1.335. [DOI] [PubMed] [Google Scholar]
- 34.Izzi L, Turbide C, Houde C, Kunath T, Beauchemin N. cis-Determinants in the cytoplasmic domain of CEACAM1 responsible for its tumor inhibitory function. Oncogene. 1999;18:5563–5572. doi: 10.1038/sj.onc.1202935. [DOI] [PubMed] [Google Scholar]
- 35.Klaile E, Muller MM, Kannicht C, Singer BB, Lucka L. CEACAM1 functionally interacts with filamin A and exerts a dual role in the regulation of cell migration. J Cell Sci. 2005;118:5513–5524. doi: 10.1242/jcs.02660. [DOI] [PubMed] [Google Scholar]
- 36.Muller MM, Klaile E, Vorontsova O, Singer BB, Obrink B. Homophilic adhesion and CEACAM1-S regulate dimerization of CEACAM1-L and recruitment of SHP-2 and c-Src. J Cell Biol. 2009;187:569–581. doi: 10.1083/jcb.200904150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Najjar SM. Regulation of insulin action by CEACAM1. Trends Endocrinol Metab. 2002;13:240–245. doi: 10.1016/s1043-2760(02)00608-2. [DOI] [PubMed] [Google Scholar]
- 38.Markel G, Lieberman N, Katz G, Arnon TI, Lotem M, Drize O, Blumberg RS, et al. CD66a interactions between human melanoma and NK cells: a novel class I MHC-independent inhibitory mechanism of cytotoxicity. J Immunol. 2002;168:2803–2810. doi: 10.4049/jimmunol.168.6.2803. [DOI] [PubMed] [Google Scholar]
- 39.Chen Z, Chen L, Baker K, Olszak T, Zeissig S, Huang YH, Kuo TT, et al. CEACAM1 dampens antitumor immunity by down-regulating NKG2D ligand expression on tumor cells. J Exp Med. 2011;208:2633–2640. doi: 10.1084/jem.20102575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markel G, Ortenberg R, Seidman R, Sapoznik S, Koren-Morag N, Besser MJ, Bar J, et al. Systemic dysregulation of CEACAM1 in melanoma patients. Cancer Immunol Immunother. 2010;59:215–230. doi: 10.1007/s00262-009-0740-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Markel G, Seidman R, Stern N, Cohen-Sinai T, Izhaki O, Katz G, Besser M, et al. Inhibition of human tumor-infiltrating lymphocyte effector functions by the homophilic carcinoembryonic cell adhesion molecule 1 interactions. J Immunol. 2006;177:6062–6071. doi: 10.4049/jimmunol.177.9.6062. [DOI] [PubMed] [Google Scholar]
- 42.Orr MT, Lanier LL. Natural killer cell licensing during viral infection. Adv Exp Med Biol. 2011;780:37–44. doi: 10.1007/978-1-4419-5632-3_4. [DOI] [PubMed] [Google Scholar]
- 43.Le Maux Chansac B, Moretta A, Vergnon I, Opolon P, Lecluse Y, Grunenwald D, Kubin M, et al. NK cells infiltrating a MHC class I-deficient lung adenocarcinoma display impaired cytotoxic activity toward autologous tumor cells associated with altered NK cell-triggering receptors. J Immunol. 2005;175:5790–5798. doi: 10.4049/jimmunol.175.9.5790. [DOI] [PubMed] [Google Scholar]
- 44.Markel G, Mussaffi H, Ling KL, Salio M, Gadola S, Steuer G, Blau H, et al. The mechanisms controlling NK cell autoreactivity in TAP2-deficient patients. Blood. 2004;103:1770–1778. doi: 10.1182/blood-2003-06-2114. [DOI] [PubMed] [Google Scholar]
- 45.Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol. 2008;9:486–494. doi: 10.1038/ni1580. [DOI] [PubMed] [Google Scholar]
- 46.Huard B, Karlsson L, Triebel F. KIR downregulation on NK cells is associated with downregulation of activating receptors and NK cell inactivation. Eur J Immunol. 2001;31:1728–1735. doi: 10.1002/1521-4141(200106)31:6<1728::aid-immu1728>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 47.Kosugi A, Sakakura J, Yasuda K, Ogata M, Hamaoka T. Involvement of SHP-1 tyrosine phosphatase in TCR-mediated signaling pathways in lipid rafts. Immunity. 2001;14:669–680. doi: 10.1016/s1074-7613(01)00146-7. [DOI] [PubMed] [Google Scholar]
- 48.Su MW, Yu CL, Burakoff SJ, Jin YJ. Targeting Src homology 2 domain-containing tyrosine phosphatase (SHP-1) into lipid rafts inhibits CD3-induced T cell activation. J Immunol. 2001;166:3975–3982. doi: 10.4049/jimmunol.166.6.3975. [DOI] [PubMed] [Google Scholar]
- 49.Harder T. Lipid raft domains and protein networks in T-cell receptor signal transduction. Curr Opin Immunol. 2004;16:353–359. doi: 10.1016/j.coi.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 50.Chen CJ, Shively JE. The cell-cell adhesion molecule carcinoembryonic antigen-related cellular adhesion molecule 1 inhibits IL-2 production and proliferation in human T cells by association with Src homology protein-1 and down-regulates IL-2 receptor. J Immunol. 2004;172:3544–3552. doi: 10.4049/jimmunol.172.6.3544. [DOI] [PubMed] [Google Scholar]
- 51.Pan H, Shively JE. Carcinoembryonic Antigen-Related Cell Adhesion Molecule-1 Regulates Granulopoiesis by Inhibition of Granulocyte Colony-Stimulating Factor Receptor. Immunity. 2010;33:620–631. doi: 10.1016/j.immuni.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Markel G, Wolf D, Hanna J, Gazit R, Goldman-Wohl D, Lavy Y, Yagel S, et al. Pivotal role of CEACAM1 protein in the inhibition of activated decidual lymphocyte functions. J Clin Invest. 2002;110:943–953. doi: 10.1172/JCI15643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ehrlich LI, Ogasawara K, Hamerman JA, Takaki R, Zingoni A, Allison JP, Lanier LL. Engagement of NKG2D by cognate ligand or antibody alone is insufficient to mediate costimulation of human and mouse CD8+ T cells. J Immunol. 2005;174:1922–1931. doi: 10.4049/jimmunol.174.4.1922. [DOI] [PubMed] [Google Scholar]
- 54.Morales VM, Christ A, Watt SM, Kim HS, Johnson KW, Utku N, Texieira AM, et al. Regulation of human intestinal intraepithelial lymphocyte cytolytic function by biliary glycoprotein (CD66a) J Immunol. 1999;163:1363–1370. [PubMed] [Google Scholar]
- 55.Devlin M, Alsop D, Clapp A, Cottingham D, Fischer M, Gundersen J, Holmes W, et al. Preliminary results from the third flight of the Millimeter Anisotropy Experiment (MAX) Proc Natl Acad Sci U S A. 1993;90:4774–4776. doi: 10.1073/pnas.90.11.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]