

Abstract
The p38 mitogen-activated protein kinase (MAPK) isoforms are phosphorylated by a variety of stress stimuli in neurodegenerative disease and act as upstream activators of myriad pathogenic processes. Thus, p38 MAPK inhibitors are of growing interest as possible therapeutic interventions. Axonal dysfunction is an early component of most neurodegenerative disorders, including the most prevalent optic neuropathy, glaucoma. Sensitivity to intraocular pressure at an early stage disrupts anterograde transport along retinal ganglion cell (RGC) axons to projection targets in the brain with subsequent degeneration of the axons themselves; RGC body loss is much later. Here we show that elevated ocular pressure in rats increases p38 MAPK activation in retina, especially in RGC bodies. Topical eye-drop application of a potent and selective inhibitor of the p38 MAPK catalytic domain (Ro3206145) prevented both the degradation of anterograde transport to the brain and degeneration of axons in the optic nerve. Ro3206145 reduced in the retina phosphorylation of tau and heat-shock protein 27, both down-stream targets of p38 MAPK activation implicated in glaucoma, as well as well as expression of two inflammatory responses. We also observed increased p38 MAPK activation in mouse models. Thus, inhibition of p38 MAPK signaling in the retina may represent a therapeutic target for preventing early pathogenesis in optic neuropathies.
Keywords: p38 MAPK, axonopathy, retinal ganglion cell, glaucoma, neurodegeneration, heat shock protein
Introduction
The p38 MAPK signaling pathways play a diverse and important role in cellular responses to external stimuli, including stress (Mielke and Herdegen, 2000). The four highly homologous isoforms of p38 MAPK (α, β, γ, δ) are activated via dual phosphorylation at tyrosine and threonine residues, activating in turn a multitude of downstream cytoplasmic and transcriptional substrates (Ashwell, 2006; Zarubin and Han, 2005). In neurons, p38 MAPK is activated by a variety of stress-related stimuli, including osmotic shock, excitotoxicity, growth factors, proinflammatory cytokines, and oxidative injury (Bendotti et al., 2005; Harper and LoGrasso, 2001; Mielke and Herdegen, 2000). As well, many p38 MAPK downstream effects are implicated in neurodegenerative disorders. These include phosphorylation of transcription factors involved in release of inflammatory cytokines, such as ATF2 (activating transcription factor 2), upregulation of pro-apoptotic caspases, and hyperphosphorylation of tau neurofilaments (Ackerley et al., 2004; Munoz and Ammit, 2010). Thus, p38 MAPK has become increasingly relevant as a potential therapeutic target in neurodegenerative disease (Bendotti et al., 2005).
Many chronic neurodegenerative disorders demonstrate loss of axonal function as an early hallmark, which is increasingly becoming a target for novel therapeutic interventions (Coleman and Perry, 2002; Coleman, 2005; Whitmore et al., 2005). Early axonopathy is also observed in glaucoma (Calkins, 2012; Nickells et al., 2012), the most common optic neuropathy and a leading cause of irreversible blindness worldwide (Quigley and Broman, 2006). The disease in its most common form involves sensitivity to intraocular pressure (IOP) that results in early degeneration of the retinal ganglion cell (RGC) projection to the brain and later loss of RGC bodies in the retina (Calkins, 2012; Howell et al., 2012). The only modifiable risk factor for glaucoma is IOP, which is treated by either daily topical hypotensive drugs or surgical intervention when topical drugs fail (reviewed in Shih and Calkins, 2012). Like other neurodegenerative diseases, one of the earliest signs of axonal dysfunction in animal models of glaucoma is disruption of axonal transport. We have shown using both chronic and inducible models that failure of anterograde transport from the retina to the superior colliculus (SC), the primary projection for RGCs in rodents, occurs early and prior to outright degeneration of the optic projection, which in turn is earlier than RGC body loss in the retina (Calkins, 2012; Crish et al., 2010; Lambert et al., 2011). Experimental interventions that target upstream effectors of early transport dysfunction are highly effective at ameliorating RGC axonal degeneration and subsequent loss of RGC bodies (Bosco et al., 2008; Howell et al., 2012; Lambert et al., 2011).
Numerous studies have implicated activated p38 MAPK in RGC injury, especially in pathways relevant to glaucoma (El-Remessy et al., 2008; Harada et al., 2006; Kikuchi et al., 2000; Levkovitch-Verbin et al., 2007; Manabe and Lipton 2003; Tezel et al., 2003). Here we tested whether a highly selective and potent inhibitor of the p38 MAPK catalytic domain (Ro3206145) could prevent degeneration with elevated IOP in an inducible model of glaucomatous optic neuropathy. We found that daily topical application of Ro3206145 to the eye prevented loss of axonal transport to the SC and axon degeneration in the optic nerve independent of IOP. Phosphorylated p38 MAPK increased with elevated IOP in RGCs, driving the activation of multiple downstream targets including phosphorylated tau; this activation was prevented by the p38 MAPK inhibitor. Similarly, treatment with Ro3206145 also was effective at reducing some inflammatory responses downstream of p38 MAPK activation. Thus, p38 MAPK appears to be a key regulator in the progression of RGC degeneration. Inhibiting its activation of downstream targets may represent a viable therapeutic target for preventing injury due to ocular stress in glaucoma or other optic neuropathies.
Materials and Methods
Animal Model and Treatments
All experimental procedures were approved by The Vanderbilt University Institutional Animal Care and Use Committee. Adult (6–8 month) Brown Norway rats were obtained from Charles River Laboratories (Wilmington, MA), while C57BL/6 (3 month) and DBA/2J mice were obtained from Jackson Laboratories (Bar Harbor, ME). Animals were maintained in a 12-hour light-dark cycle with standard rodent chow available ad libitum. For rats and C57 mice, we elevated IOP unilaterally via microbead occlusion of aqueous flow, with the opposing eye receiving an equivalent injection of saline as described (Crish et al., 2010; Sappington et al., 2010). We monitored IOP at least once daily using tonometry (TonoPen XL, Medtronic Solan, Jacksonville, FL), as previously described (Inman et al., 2006; Sappington et al., 2010).
Prior to injections, IOP was measured bilaterally for 2–3 days; the measurements from each eye were averaged to obtain a baseline value (day 0). To elevate IOP (day 1), we injected 5 μl of 15 μm polystyrene microbeads (Molecular Probes) into the aqueous chamber of the eye, while the control eye received 5 μl of sterile saline (Fisher Scientific). For C57 mice, volume was reduced to 1 μl. Only a single injection was made in all eyes. After injection, antibiotic drops (0.5% moxifloxacin hydrochloride ophthalmic solution; Alcon) were applied topically to each eye, and the animal was allowed to recover for 24 hours prior to resumption of IOP measurements. For use in rats, we obtained a 1% topical drop saline formulation of the the 4-azaindole Ro3206145 (1-{4-[3-(4-Fluoro-phenyl)-1H-pyrrolo[3,2-b]pyridin-2-yl]-pyridin-2-ylamino}-propan-2-ol), a highly selective p38 MAPK inhibitor (Peifer et al., 2006;Trejo et al. 2003; Wagner and Laufer, 2006; courtesy of Alcon Research and Novartis, Fort Worth, TX). This formulation corresponds to an effective concentration of about 30 μM. For the treatment cohort, we administered bilaterally 10 μl Ro3206145 topically twice daily; the vehicle cohort received phosphate-buffered saline. Animals were assigned to either the treatment or vehicle cohort blindly and were not re-identified until the experiments were completed. Treatment began two days after microbead-induced IOP elevation and continued until sacrifice. Animals were perfused transcardially with 4% paraformaldehyde.
Retinal Explants and p38 MAPK Assay
We removed retinas from the eyes of retired breeder Sprague Dawley rats (~8 month; Charles River Laboratories). These were placed on organotypic culture insert (Millipore, Temecula, CA), grown in six-well culture plates in modified Neurobasal A media (2% B27 and 1% N2 supplements, 2mM L-glutamine, 100uM inosine, 0.1% gentamicin, 50ng/mL BDNF, 20ng/mL CNTF, and 10 ng/mL bFGF), and maintained in an incubator at 37°C with 5% CO2. To induce p38 MAPK activation, explants were UV-irradiated for 30 seconds at room temperature (CL-1000M lamp, UVP, Upland, CA) before re-incubation at 37°C for another hour (Kabuyama et al., 2002). Ro3206145 was dissolved in 0.5% DMSO diluted into media to a series of final concentrations (0, 0.1, 1, 10, 50, 100, 500, 1000, or 5000 μM). Explants were processed with a nonradioactive p38 MAPK assay kit to assess specific activation of the kinase and to demonstrate specificity of Ro3206145 in our hands (Cell Signaling Technology, Beverly, MA: Hsieh and Papaconstantinou, 2006; Ding et al., 2009). For this cell-free assay, phosphorylated p38 MAPK was immuno-precipitated, and an ATP-driven kinase reaction was performed using activating transcription factor 2 (ATF2) protein as a substrate. ATF2 phosphorylation was then detected by western blotting and imaged using the IRDye 800CW secondary antibody (Li-Cor, Lincoln, NE) on the Odyssey infrared imaging system (Li-Cor). Densitometric analysis of western blots was performed using ImageJ 1.44p (National Institutes of Health). The intensity of the phosphorylated ATF2 band represents the relative p38 MAPK kinase activity precipitated by the antibody against phosphorylated p38 MAPK. All experiments were replicated at least in triplicate.
Immunocytochemistry and Quantification
Retinas from perfused animals were dissected, paraffin-embedded and vertically sectioned. Immunolabeling of retinal sections was performed as described previously and at identical conditions between sets (Sappington et al., 2009). To probe for p38 MAPK pathway components, we used polyclonal antibodies against p38 MAPK itself (1:250; Cell Signaling Technology, Beverly, MA), phosphorylated p38 MAPK (1:1000; Cell Signaling Technology), phosphorylated Hsp27 (1:25; Cell Signaling Technology), and phosphorylated Tau (1:500; Santa Cruz Biotechnology, Dallas, TX). As markers for RGCs and astrocytes, we used polyclonal antibodies against tubulin or α-detyrosinated tubulin (1:500; Millipore, Billerica, MA) and against glial fibrillary acidic protein (GFAP,1:500: Millipore), respectively. Imaging was done using a Zeiss FV1000 inverted confocal microscope through the Vanderbilt University Medical Center Cell Imaging Shared Resource with a slice thickness of 0.31 μm. We used identical microscope settings to acquire images for signal quantification, which was done by a naïve observer using custom routines in either ImagePro (Media Cybernetics; Bethesda, MD) or MatLab (MathWorks; Natick, MA).
Anterograde Transport and Axon Count Measurements
Forty eight hours before sacrifice, rats were anesthetized with 2.5% isoflurane using a table top anesthesia system (VetEquip, Inc., Pleasanton, CA) and received an intravitreal injection of 1 μl solution of 0.5 mg Cholera toxin subunit β (CTB) conjugated to Alexa Fluor-488 (Invitrogen, Carlsbad, CA) according to our established protocol (Crish et al., 2010; 2013). After the 2 day period, animals were deeply anesthetized with an overdose of Nembutal (200 mg/kg, Henry Schein, Indianapolis, IN) and perfused intracardially with phosphate buffered saline (PBS) followed by 4% paraformaldehyde in PBS. Brains were removed and cryoprotected in 30% sucrose/PBS overnight and 50 μm coronal slices were taken on a freezing sliding microtome. CTB signal in serial coronal SC sections (50 μm) was digitally photographed on an Olympus AX-70 microscope and intensity was quantified using custom algorithms for ImagePro (Media Cybernetics, Bethesda, MD), as previously described (Crish et al., 2010; 2013). Briefly, CTB signal was normalized with respect to background, and intensity calculations from alternate sections were combined as a colorimetric representation across the retinotopic collicular map. The percentage of intact transport was determined as the region with CTB signal ≥ 70% of the maximum signal for that colliculus. We obtained counts of RGC axons in cross-sections of optic nerve (2–3 mm proximal to the globe) as previously described (Inman et al., 2006; Sappington et al., 2010; Lambert et al., 2011).
Quantitative Real-time Polymerase Chain Reaction (qRT-PCR)
We extracted RNA from retina as previously described for lightly fixed tissue (Hanna and Calkins, 2006). Briefly, tissues were incubated overnight in lysis buffer (10mM Tris/HCl, pH 8.0; 0.1mM EDTA, pH8.0; 2% SDS, pH 7.3; and 500 ug/ml Proteinase K; Clontech Labs, Mountain View, CA). RNA was extracted using Trizol reagent (Invitrogen) with 10 ug of glycogen added as an RNA carrier prior to precipitation. RNA concentration and purity were determined using a NanoDrop 8000 (Thermo Scientific, Wilmington, DE). Samples (1 ug) were DNase-treated (Invitrogen) prior to cDNA synthesis (Applied Biosystems reagents, Foster City, CA). We performed qRT-PCR using an ABI PRISM 7300 Real-Time PCR System and FAM dye-labeled gene-specific probes against sequences encoding ceruloplasmin and tissue inhibitor of metalloproteinases-3 (Applied Biosystems, catalog numbers Rn00561049_m1 and Rn00441826_m1, respectively). Cycling conditions and cycle threshold values were automatically determined by the supplied ABI software (SDS v1.2). Relative product quantities for each transcript were performed minimally in triplicate, normalized to 18s rRNA or β-actin mRNA as endogenous controls, and determined using the 2ΔΔCt analysis method (Livak and Schmittgen, 2001).
Results
Ro3206145 inhibition of kinase activity
The 4-azaindole Ro3206145 is a highly selective p38 MAPK inhibitor that competes with ATP to bind the catalytic domain and reduce phosphorylation of downstream pathways; it is roughly 50x more potent in binding p38 MAPK and several thousand-fold more selective over other MAP kinases than the more commonly used inhibitor, SB203580 (Peifer et al., 2006; Trejo et al. 2003; Wagner and Laufer, 2006). To demonstrate its efficacy in retinal tissue, we used ultra-violet radiation induce phosphorylation of p38 MAPK in retinal explants (Kabuyama et al., 2002), which we then immune-precipitated using a selective antibody provided in a commercial kinase assay (Hsieh and Papaconstantinou, 2006; Ding et al., 2009). Using this assay, we measured how Ro3206145 affected p38 MAPK phosphorylation of the transcription factor ATF2, an established and selective downstream target (Munoz and Ammit, 2010). Exposure to UV light elicited a nearly three-fold increase in p38 MAPK-induced ATF2 phosphorylation for retinal explants maintained ex vivo (Figure 1). Increasing concentrations of Ro3206145 were progressively more effective at inhibiting ATF2 phosphorylation in retinal explants, reaching significance at 10 μM compared to UV exposure without treatment (Figure 1B).
Figure 1. Ro3206145 inhibits p38 MAPK activity ex vivo.
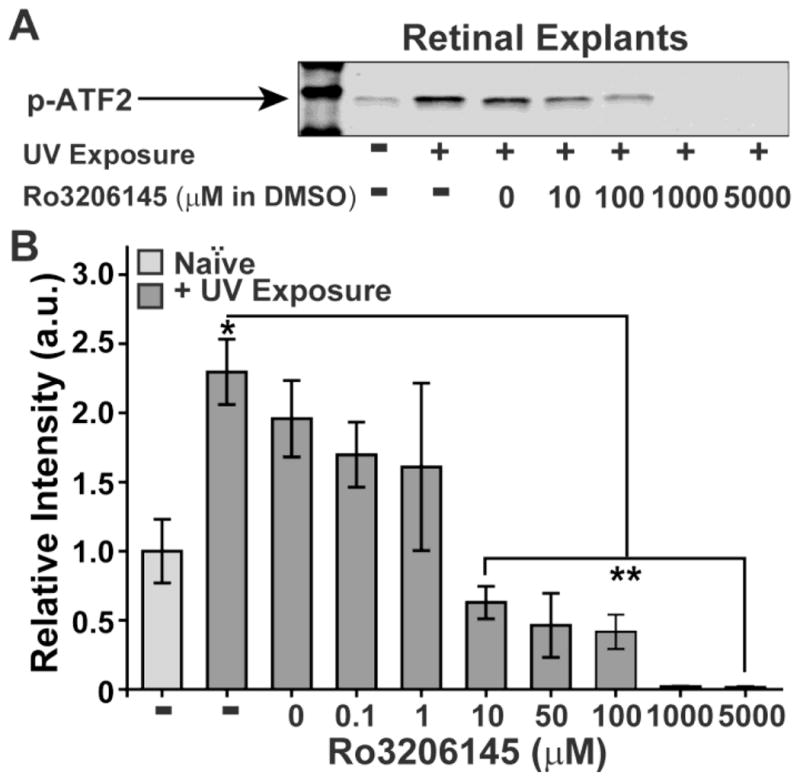
(A) Example western blot of phosphorylated activating transcription factor 2 (p-ATF2) following a kinase reaction with phosphorylated p38 MAPK that was immuno-precipitated from rat retinal explants. Explants were exposed to ultra-violet (UV) light to activate p38 MAPK. Increasing concentrations of Ro3206145 were effective at inhibiting p38 MAPK activation of ATF2. (B) Densitometer quantification (of the ATF2 assay (n≥3 for each condition) shows significant increase in p38 MAPK activity with UV exposure relative to naïve controls (* p = 0.01). Ro3206145 (in DMSO) reduces activity, reaching significance compared to UV exposure alone for 10 μM and higher (** p ≤ 0.003).
Application of Ro3206145 does not affect IOP or activated p38 MAPK
We elevated IOP in two rat cohorts using microbead occlusion of aqueous fluid flow in the anterior chamber of the eye (Chen et al., 2011; Cone et al., 2010; Crish et al., 2010; Sappington et al., 2010). We monitored over a six week period the effects of Ro3206145 or vehicle on IOP with twice-daily topical application, which is a typical regimen in clinical glaucoma. For both cohorts, IOP in the saline-injected control eye remained at about 20 mmHG for the experimental period, while IOP in the microbead eye rose 1–2 days post-injection and remained elevated by 25–30% (Figure 2A). Drug treatment had no significant effect on IOP compared to vehicle for either the saline or microbead eye (Figure 2B; p ≥ 0.14).
Figure 2. Ro3206145 does not affect microbead-induced elevations in IOP.
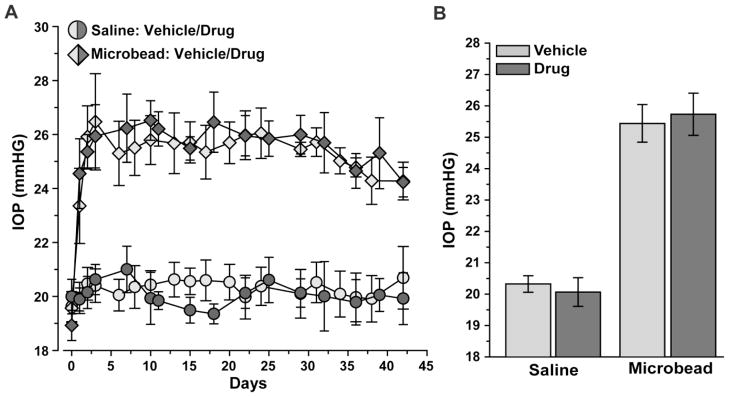
A) Longitudinal IOP measurements after a single unilateral microbead or saline injection (5 μl) in rats receiving twice-daily topical application of either vehicle or Ro3206145 (n = 8 each). IOP post-injection (day 1) was the same in the vehicle vs. drug cohorts for both the saline-injected eye (20.06 ± 0.41 vs. 20.27 ± 0.30 mmHg; p=0.87) and the microbead eye (25.35 ± 0.76 vs. 25.42 ± 1.09 mmHg; p = 0.45). B) IOP during the treatment period was also similar in vehicle vs. drug cohorts for both the saline (20.32 ± 0.27 vs. 20.07 vs. 0.46 mmHG; p=0.14) and microbead (25.44 ± 0.60 vs. 25.73 ± 0.67; p=0.15) eyes.
Microbead-induced elevated IOP increased immuno-labeling for phosphorylated p38 MAPK throughout the retina compared to retina from the saline eye (Figure 3A, left panel). Treatment with Ro3206145 did not affect this increase (Figure 3A, right panel), which was expected given the drug targets the catalytic domain of activated p38 MAPK but not p38 MAPK activation itself (Peifer et al., 2006; Trejo et al., 2003). Across retinal layers, phosphorylated p38 MAPK increased by 2- to 3-fold in the vehicle cohort with elevated IOP (Figure 3B). Interestingly, treatment with Ro3206145 tended to increase baseline levels of phosphorylated p38 MAPK in retinas from the saline eye, especially in the outer retina (Figure 3B). Levels in the microbead retinas were accordingly higher. This could reflect a compensatory mechanism to counter reduced downstream activity in the drug cohort. Elevated IOP increased label particularly in RGC nuclei (Figure 3C), which is consistent with the known role of activated p38 MAPK as a modulator of downstream transcription factors (Zarubin and Han, 2005).
Figure 3. Ro3206145 does not prevent p38 MAPK activation, as expected.

A) Immuno-labeling for phosphorylated p38 MAPK (p-p38) in retinal sections from either saline (top row) or microbead (bottom row) eye from study cohorts. Six weeks of microbead-induced elevated IOP increased signal, especially near RGCs labeled by uptake of cholera toxin B (CTB, arrows), which was injected into the eye prior to sacrifice; this is not affected by Ro3206145 treatment. Abbreviations: ONL (outer nuclear layer), OPL (outer plexiform layer), INL (inner nuclear layer), IPL (inner plexiform layer), GCL (ganglion cell layer), and NFL (nerve fiber layer). B) Quantification of phosphorylated p38 MAPK signal across retinal layers in arbitrary units (a.u.; n≥7 sections per cohort). Elevated IOP increased signal most significantly near RGCs and their axons (NFL-GCL) and in the OPL. Drug treatment tended to exacerbate this increase. * p ≤ 0.05 or ** p ≤ 0.01 for microbead vs. saline retina (red) and drug vs. vehicle cohort (blue brackets). C) High magnification micrographs show increased phosphorylated p38 MAPK with elevated IOP in the nucleus of CTB-labeled RGCs (arrows) for both vehicle and drug cohorts. Scale = 20 μm (A), 5 μm (C).
Inhibition of p38 MAPK prevents RGC distal axonopathy
Failure of anterograde transport from the retina to the SC is an early characteristic of axonopathy in glaucoma and occurs prior to outright degeneration of the optic projection (Calkins, 2012; Crish et al., 2010; Lambert et al., 2011). Following intravitreal injection of CTB, sections of SC from the microbead-injected eye in the vehicle cohort showed diminished anterograde transport from the retina compared to sections of the SC from the saline-injected eye (Figure 4A, top row). The corresponding retinotopic maps reconstructed from serial SC sections demonstrated significant sectors of lost transport compared to control (Figure 4A, bottom row). This pattern is consistent with our previous findings in both chronic and inducible models (Crish et al., 2010; Lambert et al., 2011). In contrast to the vehicle cohort, SC sections from the Ro3206145 cohort were similar for both the saline and microbead eyes with little or no sign of diminished transport with elevated IOP; the corresponding retinotoptic maps were intact for both eyes as well (Figure 4B). On average, for the vehicle cohort, elevated IOP with microbead injection induced a 34% reduction in SC transport compared to the control eye (p=0.01); this was prevented by Ro3206145 treatment (p=0.03; Figure 4C).
Figure 4. p38 MAPK inhibition prevents RGC transport loss with elevated IOP.
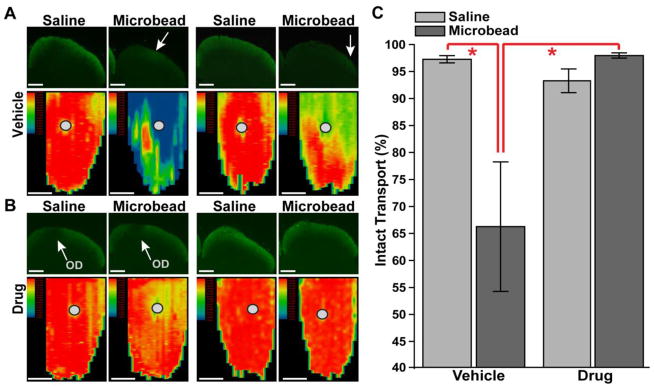
A) Top row: single sagittal sections through the superior colliculus (SC) from saline-and microbead-injected eyes from two vehicle cohort rats. Each section shows anterogradely transported CTB (green) following intravitreal injection; microbead SC sections have only residual signal (arrows). Bottom row: corresponding retinotopic maps of CTB signal reconstructed from serial sections through SC with representation of optic disc gap indicated (circles). Signal density ranges from 100% (red) to 50% (green) to 0% (blue), with SC from microbead eyes demonstrating considerable loss of CTB transport. B). SC sections (top row) and corresponding retinotopic maps (bottom row) show little difference in CTB signal between saline and microbead eyes in Ro3206145 cohort. C) Fraction of intact SC retinotopic map (≥ 70% CTB signal density) is significantly reduced with microbead-induced elevations in IOP for the vehicle group. This reduction is prevented by Ro3206145 treatment. * p ≤ 0.05. Scale = 500 μm.
Degeneration of RGC axons in the optic nerve, while preceding RGC body loss in the retina, is slower than depletion of active transport in most models of glaucoma (Calkins, 2012). Accordingly, cross-sections through optic nerves from the vehicle cohort demonstrated early signs of axonal degeneration with microbead-induced elevated IOP, including increased gliosis and diminished axon packing density (Figure 5A, top row). In contrast, sections of nerves from the Ro3206145 cohort appeared similar for both the microbead and saline eyes, with fascicles of axons that remained largely intact and without increased gliosis (Figure 5A, bottom row). On average, elevated IOP induced a nearly 30% reduction in axon density compared to the control nerve for the vehicle cohort (Figure 5B), while density for the two nerves in the drug cohort did not differ.
Figure 5. p38 MAPK inhibition prevents RGC axon degeneration in the optic nerve.
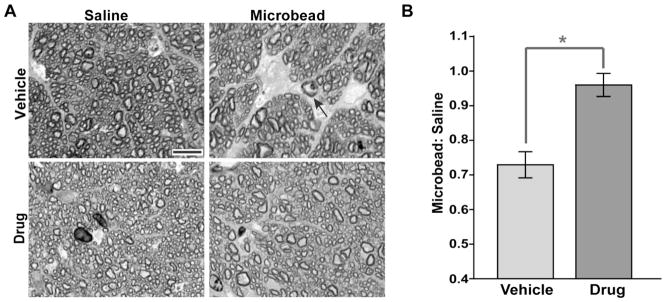
A) Cross-section through optic nerve of microbead-injected eye in the vehicle cohort (top row) shows diminished axon density with intraaxonal inclusions (arrow) and increased gliosis compared to nerve from the saline-injected eye. For the drug cohort, bundles of axons remained intact, and nerves from the saline and microbead eye appeared similar. Scale = 10 μm. B) The ratio of axon density (axons/mm2) in microbead nerve to density in the saline nerve. For the vehicle cohort, the ratio is reduced by 27.1 ± 3.8% (mean ± sd) compared to a ratio of unity (p=0.007). For the drug cohort, axon density is comparable for the two eyes (p=0.58), yielding a ratio that is significantly different than that for the vehicle cohort (* p = 0.015). n ≥ 8 sets of nerves per cohort.
Previously we have shown that depletion of anterograde transport of CTB along the optic projection to the SC is not due to reduced uptake by RGCs (Crish et al., 2010). Here we demonstrate similar patterns of CTB uptake in RGCs for both saline and microbead retina (Figure 6A), consistent with the finding that RGC body loss in our model is later than axon degeneration in the nerve (Calkins, 2012; Chen et al., 2011). This is so for both the vehicle and drug cohorts. Quantification of RGC density in the retina indicated similar numbers of CTB-labeled RGCs between cohorts, for both saline and microbead retina (Figure 6B). Thus, the rescue of anterograde transport to the SC we demonstrate in Figure 4 for the drug cohort is not due to differences in CTB uptake by RGCs.
Figure 6. Rescue of axon transport by p38 MAPK inhibition is not due to changes in CTB uptake.

A) Confocal micrographs through the nerve fiber and ganglion cell layers of whole-mounted retina from vehicle and drug cohorts showing CTB-labeled RGCs and their axons. As in Figure 3, retina from microbead-injected eyes demonstrate increased phosphorylated p38 MAPK (p-p38) in both cohorts; arrows indicate particular examples of CTB-labeled RGCs with p-p38 localizing in the nucleus. Scale = 20 μm. B) Quantification indicates similar numbers of CTB-labeled RGCs between cohorts, for both saline and microbead retina. Thus, differences in anterograde transport to the SC as shown in Figure 4 are not due to differences in CTB uptake by RGCs. n ≥ 3 sets of retina per cohort.
Ro3206145 reduces downstream targets in the retina
Activated p38 MAPK phosphorylates a myriad of downstream targets implicated in neurodegenerative conditions, including the microtubule-stabilizing protein Tau and stress-related heat shock protein 27 (Hsp27; Ackerley et al., 2004; Ferrer et al., 2005; Kostenko and Moens, 2009; Mielke and Herdegen, 2000; Reynolds et al., 2000). Both Tau and Hsp27 have been implicated in neurodegeneration in glaucoma (Gasparini et al. 2011; Gupta et al. 2008; Huang et al., 2007; Nucci et al. 2010; Wax et al., 2008).
We found that microbead-induced elevated IOP increased immuno-labeling for phosphorylated Tau throughout the retina, including in RGCs, compared to retina from the saline eye in the vehicle cohort (Figure 7A, left panel). Treatment with Ro3206145 prevented this increase completely (Figure 7A, right panel); the 10-fold increase with elevated IOP in the vehicle group was reduced to saline-eye levels (Figure 7B). Similarly, label for phosphorylated HSP27 in the retina also increased with elevated IOP in the vehicle but not drug cohort compared to retina from the saline eye (Figure 7C); like Tau, these changes too were significant when quantified (Figure 7D). Thus, topical treatment with Ro3206145 was effective in preventing phosphorylation of downstream targets with microbead-induced elevations in IOP.
Figure 7. Ro3206145 reduces downstream target activation.

A) Immuno-labeling for phosphorylated Tau (p-Tau) in retinal sections from either saline (top row) or microbead (bottom row) eye from study cohorts. Signal is increased throughout the retina in the vehicle cohort with microbead-induced elevated IOP; drug treatment prevents this. CTB label in RGCs included for reference. B) In the vehicle group, intensity of phosphorylated Tau signal in microbead retina was increased 10-fold (10.9 ± 3.5 vs. 0.4 ± 0.1 for saline retinas; mean ± sd). C) Immuno-labeling for phosphorylated Hsp27 (p-Hsp27) also shows increased levels in retinal sections with elevated IOP in the vehicle cohort, especially near RGCs (arrows). Increase was prevented in the drug cohort. D) For retinas from the vehicle group, elevated IOP increased phosphorylated Hsp27 three-fold (11.2 ± 2.8 vs. 4.3 ± 1.2 for saline eye). n ≥ 7 images per cohort; * p ≤ 0.05, ** p ≤ 0.01. Abbreviations as in Figure 3. Scale = 15 μm.
Increased expression of heat-shock protein 60 (Hsp60) is implicated in glaucomatous neurodegeneration (Wax et al., 2008). Consistent with this, elevated IOP elicited a modest increase in Hsp60 label throughout the retina in the vehicle cohort (Figure 8). However, Hsp60 is not – to our knowledge - a direct downstream target of phosphorylated p38 MAPK, unlike Hsp27 (see above). Accordingly, treatment with Ro3206145 had little effect on Hsp60, and levels in microbead retinas did not differ between the vehicle and drug cohorts (p=0.23).
Figure 8. Ro3206145 does not affect Hsp60 levels.

A) Immuno-labeling for Hsp60 in retinal sections from either saline (top row) or microbead (bottom row) eye from study cohorts. Signal increased modestly in retina for the vehicle cohort with elevated IOP, including in CTB-labeled RGCs, and slightly less so in the drug cohort. B) In the vehicle group, intensity of Hsp60 signal (arbitrary units) in microbead retina was increased significantly (12.7 ± 3.4 % vs. 6.4 ± 2.4 for saline retinas; mean ± sd; * p = 0.004). No other comparison was significant. n ≥ 7 images per cohort. Abbreviations as in Figures 3 and 7. Scale = 20 μm.
Activation of p38 MAPK in neuronal systems is associated with several downstream inflammatory signaling cascades (Bendotti et al., 2005). We have begun to link some of these complex cascades to p38 MAPK in our glaucoma model and to determine whether the protective effects of Ro3206145 could be ascribed to them. Evidence suggests that both ceruloplasmin and TIMP3 are important components of tumor necrosis factor α (TNFα) inflammatory signaling downstream of p38 MAPK activation (Lee et al., 2007; Yang et al., 2011; Xu et al., 2012). We found that expression of the mRNA encoding ceruloplasmin in the retina rose significantly with elevated IOP in the vehicle cohort (Figure 9A, left), consistent with findings from human glaucoma and other animal models (Stasi et al., 2007). Ro3206145 dramatically reduced expression for both eyes. Similarly, drug treatment significantly reduced expression of tissue inhibitor of metalloproteinases-3 (TIMP3) in microbead retina (Figure 9A, right). Thus, in these cases, Ro3206145 treatment was effective at interrupting two diverse inflammatory responses at the transcriptional level. Activation of p38 MAPK can also lead to phosphorylation of MAPK-activated protein kinase 2 (MK2), which is implicated in production of TNFα (Ronkina et al., 2007). Interestingly, we found that constitutive levels of phosphorylated MK2 in the retina diminished with microbead-induced elevations in IOP (Figure 9B). This decrease was significant for both vehicle and drug cohorts (Figure 9C), and we observed no other difference in MK2 levels between the two groups. Thus, MK2 represents one example of a p38 MAPK downstream inflammatory target not affected by Ro3206145 at the protein level.
Figure 9. Ro3206145 modulation of inflammatory pathways.

A) Quantitative RT-PCR measurements of mRNA encoding ceruloplasmin (left) and tissue inhibitor of metalloproteinases-3 (TIMP3, right) from retina of vehicle and drug cohorts. All measurements normalized to expression the saline retina of the vehicle cohort. Though variability was high, ceruloplasmin expression increased significantly in microbead retina in the vehicle cohort and was dramatically reduced in both retina with drug treatment. With elevated IOP, expression of TIMP3 increased modestly in the vehicle cohort (p=0.07) and was significantly reduced in the drug cohort. * p = 0.05, ** p ≤ 0.002; n ≥ 5 retina per cohort. B) Immuno-labeling for phosphorylated MAPK-activated protein kinase 2 (p-MK2) in retinal sections from either saline (top row) or microbead (bottom row) eye from study cohorts. Some label is detected in CTB-labeled RGCs (arrows). Signal diminished modestly with elevated IOP. C) In both cohorts, intensity of phospho-MK2 signal (arbitrary units) in microbead retina was decreased significantly (* p ≤ 0.02; n ≥ 3 images per cohort). Scale = 20 μm. Abbreviations as before.
p38 MAPK in mouse retina
Finally, we sought to determine whether elevated IOP in mice could induce activation of p38 MAPK in the retina similar to the pattern of rats (Figure 10). For a cohort of C57 mice, a microbead elevation of 28% over 4 weeks induced a pattern of increased phosphorylated p38 MAPK compared to the saline eye similar to that in rats (Figure 10A). Quantification of the signal indicated a nearly 10-fold increase with elevated IOP near RGCs (Figure 10B). Like the rat, in mouse retina activated p38 MAPK also increased significantly in other retinal layers as well (Figure 10B). In the DBA/2J mouse model of glaucoma, elevated IOP also increased phosphorylated p38 MAPK throughout the retina, including in RGCs. This was true for both 3 mo mice (Figure 10C) and 10 mo mice (Figure 10D). Quantification of western blots showed that retinal levels of activated p38 MAPK increased most significantly in older DBA/2J due to elevated IOP, with a nearly a 3-fold increase in 10 mo mice compared to 3 mo (Figure 10E). We did not find detectable differences in activated p38 MAPK localization across naïve C57 retina with increasing age (not shown).
Figure 10. p38 MAPK activation in mouse models.

A) Immuno-labeling for phosphorylated p38 MAPK in C57 mouse retinal sections from either saline (left) or microbead (right) eye shows an increase with elevated IOP, particularly in RGCs outlined with label against tubulin (arrows). IOP over 4 weeks was 15.15 ± 0.42 mmHg for the saline eye and 19.44 ± 0.47 mmHg for the microbead eye (p< 0.001, n=3 mice). B) Signal intensity in arbitrary units for phosphorylated p38 MAPK was significantly higher throughout retinal layers (n≥3 sections per eye per animal). C) In retina from 3 mo DBA/2J mouse with low IOP (14.3 mmHG, left panel), phosphorylated p38 MAPK is visible in RGCs labeled for α-detyrosinated tubulin (arrows) and throughout the INL. Astrocytes labeled against GFAP are also visible. D) In a 3 mo DBA/2J with higher IOP (15.5 mmHG, right panel), phosphorylated p38 MAPK is increased in RGCs (arrows) as well as other cells. E) Western blots from total retinal protein lysates show a dramatic increase in phosphorylated p38 MAPK with elevated IOP in 10 mo DBA/2J when normalized relative to 3 mo and compared to p38 MAPK in the same retina. Lysates pooled across retinas for each group (n=3). IOP = 17.98 mmHG (10 mo low); 25.90 mmHG (10 mo high). * p ≤ 0.05; ** p ≤ 0.01. Abbreviations as in previous figures; PR= photoreceptors. Scale = 10 μm (A); 15 μm (C,D).
Discussion
The activation of p38 MAPK via phosphorylation is relevant to pathogenesis in a variety of neurodegenerative diseases (Bendotti et al., 2006; Harper and LoGrasso, 2001; Mielke and Herdegen, 2000). For example, in Alzheimer’s disease, p38 MAPK activation occurs early during disease progression and is associated with neuritic plaques, neuropil threads, and neurofibrillary tangles (Hensley et al., 1999; Sun et al., 2003). Once activated, p38 MAPK phosphorylates a variety of downstream effectors of progression, including pro-apoptotic, inflammatory and cytoskeletal mediators as well as numerous transcription factors (Ackerley et al., 2004; Munoz and Ammit, 2010). Accordingly, inhibitors of one or more p38 MAPK isoforms are of increasing interest as possible therapeutic interventions in age-related neurodegenerative disorders (Corrêa and Eales, 2012). For example, in animal models of Alzheimer’s disease, small-molecule p38 MAPK inhibitors were effective at reducing pro-inflammatory cytokine production and synapse loss while improving behavioral performance (Hensley et al., 1999; Munoz et al., 2007; Munoz and Ammit 2010; Sun et al., 2003). The same class of drug may hold promise as a neuroprotective therapy for Parkinson’s disease, ischemic stroke, and Amyotrophic lateral sclerosis (Barone et al., 2001, Legos et al., 2001; Malemud, 2007).
Similarly, p38 MAPK activation has been implicated as a mediator of RGC injury in a variety of models relevant to the pathogenesis of glaucoma, including optic nerve axotomy, elevated IOP (ocular hypertension), and excitotoxic injury (El-Remessy et al., 2008; Harada et al., 2006; Kikuchi et al., 2000; Levkovitch-Verbin et al., 2007; Manabe and Lipton 2003;). Increased p38 MAPK signaling has also been documented in human glaucomatous tissue (Tezel et al., 2003). Here we demonstrated that inhibiting the p38 MAPK catalytic domain prevented RGC axonopathy due to elevated IOP in an inducible rat model. The 4-azaindole Ro3206145 selectively inhibits p38 MAPK downstream targets by competing with ATP for catalytic binding; the drug was developed as an anti-cytokine and anti-arthritic agent for treatment of inflammatory disorders (Wagner and Laufer, 2006). It is highly selective for p38 MAPK, with some evidence suggesting selective binding of the α and β isoforms (Peifer et al., 2006; Trejo et al, 2003; Wagner and Laufer, 2006). It has a p38 MAPK IC50 of 1.29 nM, compared to the most common p38 MAPK inhibitor SB 203580, which has an IC50 of 0.07 μM. Moreover, the drug is nearly 8000-fold more likely to bind p38 MAPK than other stress-related kinases, for which it has an IC50 > 10 μM (Wagner and Laufer, 2006). We used twice-daily topical application via eye-drop to the eye, which is the most common way in which glaucoma is treated; what the steady-state concentration of the drug was in the retina during our experiments is unknown.
Using a kinase assay and retinal explants (Figure 1), we showed that Ro3206145 was highly effective at inhibiting phosphorylation of the transcription factor ATF2, an established p38 MAPK downstream target that has been implicated in glaucoma (Levkovitch-Verbin et al., 2007; Munoz and Ammit, 2010). Daily topical application of Ro3206145 did not influence IOP in our inducible microbead occlusion model (Figure 2), nor did it affect the IOP-induced increase in phosphorylated p38 MAP (Figure 3). This was expected, since the drug does not prevent activation of p38 MAPK only its ATP-dependent catalysis of downstream targets. However, the drug was remarkably effective at eliminating IOP-induced degradation of anterograde axonal transport the SC and RGC axon degeneration in the optic nerve (Figures 4 and 5). Both of these outcomes are early hallmarks of RGC degeneration in glaucoma, preceding by far RGC body loss in the retina across many different animal models (Calkins, 2012). These results were not due to enhanced uptake of our anterograde transport marker, CTB; no difference in the numbers of RGC bodies labeled by CTB were noted (Figure 6).
We found that elevated IOP increased levels of phosphorylated tau and phosphorylated Hsp27 in the retina, including in RGCs (Figure 7). Both are known downstream targets of activated p38 MAPK and have been implicated in RGC degeneration in glaucoma (Ferrer, 2004; Gasparini et al. 2011; Gupta et al. 2008; Huang et al., 2007; Kostenko and Moens, 2009; Nucci et al. 2010; Wax et al., 2008). We found that Ro3206145 was also effective at reducing levels of downstream p38 MAPK products in the retina, including phosphorylated tau and Hsp27 (Figure 7). Indeed p38 MAPK has been shown to phosphorylate neurofilaments in vitro (Ackerley et al., 2004; Reynolds et al., 2000), so inhibition of its downstream activity could prevent axonal transport loss to the SC directly. In contrast, as expected treatment had no effect in levels of Hsp60 – another glaucoma target which is not known to be downstream of p38 MAPK (Wax et al., 2008).
While both phosphorylated tau and Hsp27 are associated directly with RGC neurodegeneration, we also found that Ro3206145 was effective at reducing at least some downstream inflammatory targets. Treatment reduced in the retina the expression of mRNA encoding ceruloplasmin and TIMP3 (Figure 9A), both important components of TNFα inflammatory signaling and downstream targets of p38 MAPK (Lee et al., 2007; Stasi et al., 2007; Yang et al., 2011; Xu et al., 2012). TNFα is a potent inflammatory cytokine implicated in glaucomatous neurodegeneration in both the retina and optic nerve (Almasieh et al., 2012). Interestingly, we found that levels of phosphorylated MK2 in the retina were reduced with microbead-mediated elevations in IOP (Figure 9B). This was unexpected since activation of MK2 is also upstream of TNFα production and downstream of p38 MAPK (Ronkina et al., 2007). Our result could indicate an unknown compensatory mechanism to push back against p38-mediated pathogenic pathways. In any case, Ro3206145 had no effect on levels of phosphorylated MK2. While our exploration of potential inflammatory mediators involved in p38 MAPK signaling has only begun, these early results indicate that the protective effects of Ro3206145 are likely to involve pathways both intrinsic to RGCs directly and those involving a generalized inflammatory response. Since the IOP-induced increase phosphorylated p38 MAPK in the rodent retina was not confined to RGCs (Figures 3 and 10), we cannot make the argument that either p38 MAPK-dependent pathogenesis or efficacy of p38 MAPK inhibition is specific to RGCs. Since we observed increased phosphorylated p38 MAP also in two distinct mouse models (Figure 10), these pathways could be probed in more detail using transgenic tools.
Distal axonopathy is a common feature of most neurodegenerative diseases, involving loss of function along axonal projections with subsequent structural degeneration (Coleman, 2005; Coleman and Perry, 2002; Whitmore et al., 2005). This is so also in experimental models of glaucoma (Calkins, 2012; reviewed in Nickells et al., 2012). Deficits in axonal transport between the retina and central targets in the brain precede outright degeneration in the optic projection, both of RGC axons and axonal terminals and of their post-synaptic structures (Crish et al., 2010; Lambert et al., 2011). RGC bodies themselves persist until much later (Buckingham et al., 2008; Lambert et al., 2011). Progression is similar across a variety of inducible models, including the microbead model used here, and in the popular DBA/2J mouse model of pigmentary glaucoma (Calkins, 2012). Furthermore, abating early axonal dysfunction pharmacologically has proven to be a powerful tool for preventing subsequent degeneration and may be a relevant intervention in the human disease (Bosco et al., 2008; Howell et al., 2012; Lambert et al., 2011). Certainly our results here support further investigation of p38 MAPK inhibitors as possible interventions.
As in other neurodegenerative diseases, myriad intracellular cascades, transcription factors and extracellular signaling pathways (including inflammatory cytokines) contribute to pathogenesis in glaucoma (Almasieh et al., 2012). Many of these are implicated in p38 MAPK signaling (Cuadrado and Nebreda, 2010). While it appears that p38 MAPK signaling is activated by elevated pressure, it may not be the only stress-activated cell signaling kinase pathway involved in glaucomatous neurodegeneration. The p38 MAPK isoforms are important members of the family of stress-activated protein kinases, as are c-Jun N-terminal kinases (JNKs) and extracellular signal-regulated kinases (ERKs; Mielke and Herdegen, 2000). Doubtlessly these too contribute to RGC degeneration. For example, NDMA-induced retinal toxicity involves both JNK and p38 MAPK, and inhibitors of each are protective in this model (Munemasa et al., 2005). Similarly, JNK inhibition was protective against RGC loss in another ocular hypertensive model in dose-dependent fashion (Sun et al., 2011). However, in this study we used Ro3206145 due to its selectivity of p38 MAPK over both ERK and JNK (Trejo et al., 2003). Combined, these studies suggest that an upstream activator common to the stress pathways, like ASK1 (apoptosis signal-regulating kinase 1), could be a primary step in RGC degeneration in glaucoma. If so, it is possible that an early insult stems from mitochondrial dysfunction induced by oxidative stress (Nguyen et al. 2011; Osborne 2010), which is sufficient to activate the ASK1-p38 MAPK pathway in the retina (Harada 2006). Interestingly, ASK1 is predominantly expressed in RGCs and has been linked to optic nerve degeneration (Harada and Namekata, 2010). While it is currently impossible for us to predict the most relevant pathway towards p38 MAPK activation, our study does provide strong evidence that inhibiting downstream targets prevents axon degeneration and loss of functional transport in the SC, an early pathogenic event in progression (Calkins, 2012; Crish et al., 2010).
Highlights.
Disrupted axon transport and axon degradation are early in glaucoma
Elevated pressure increases activation of p38 MAPK in the retina
An inhibitor of the p38 MAP catalytic domain prevents axonopathy
p38 MAPK inhibition reduced downstream targets implicated in glaucoma
Acknowledgments
Support
The authors thank Dr. Mark Hellberg of Novartis for providing Ro3206145. Funding provided by the Alcon Laboratories, Inc. Discovery Research Program, the Melza M. and Frank Theodore Barr Foundation through the Glaucoma Research Foundation (DJC), a Departmental Unrestricted Award and Senior Scientific Investigator Award from Research to Prevent Blindness, Inc. (DJC). Imaging supported through the Vanderbilt University Medical Center Cell Imaging Shared Resource core facility (CTSA grant UL1 RR024975 from NCRR/NIH) and the Vanderbilt Vision Research Center (P30EY008126).
Abbreviatons
- ATF2
activating transcription factor 2
- ASK1
apoptosis signal-regulating kinase 1
- CTB
cholera toxin subunit β
- IOP
intraocular pressure
- MAPK
mitogen-activated protein kinase
- RGC
retinal ganglion cell
- SC
superior colliculus
- 1-{4-[3-(4-Fluoro-phenyl)-1H-pyrrolo[3,2-b]pyridin-2-yl]-pyridin-2-ylamino}-propan-2-ol
Ro3206145
- Hsp27,Hsp60
heat-shock protein 27 and 60
- CP
ceruloplasmin
- TIMP3
tissue inhibitor of metalloproteinases-3
- α-DeTub
α-detyrosinated tubulin
- GFAP
glial acidic fibrillary protein
Footnotes
Disclosure Statement
At the inception of this work, an author was an employee of Alcon Laboratories, Inc (IHP).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jason D. Dapper, Email: jasondapper@hotmail.com.
Samuel D. Crish, Email: scrish@neomed.edu.
Iok-Hou Pang, Email: Iok-Hou.Pang@unthsc.edu.
David J. Calkins, Email: david.j.calkins@vanderbilt.edu.
References
- Ackerley S, Grierson AJ, Banner S, Perkinton MS, Brownlees J, Byers HL, Ward M, Thornhill P, Hussain K, Waby JS, Anderton BH, Cooper JD, Dingwall C, Leigh PN, Shaw CE, Miller CC. p38alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol Cell Neurosci. 2004;26(2):354–64. doi: 10.1016/j.mcn.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 2012 doi: 10.1016/j.preteyeres.2011.11.002. [epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Ashwell JD. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol. 2006;6(7):532–40. doi: 10.1038/nri1865. [DOI] [PubMed] [Google Scholar]
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, Legos JJ, Erhardt JA, Ohlstein EH, Hunter AJ, Harrison DC, Phipott K, Smith BR, Adams JL, Parsons AA. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev. 2001;21(2):129–45. doi: 10.1002/1098-1128(200103)21:2<129::aid-med1003>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Bao Cutrona M, Cheroni C, Grignaschi G, Lo Coco D, Peviani M, Tortarolo M, Veglianese P, Zennaro E. Inter- and intracellular signaling in amyotrophic lateral sclerosis: role of p38 mitogen-activated protein kinase. Neurodegener Dis. 2005;2(3–4):128–34. doi: 10.1159/000089617. [DOI] [PubMed] [Google Scholar]
- Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, Hubbard WC, Calkins DJ, Horner PJ, Vetter ML. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci 2008. 2008 Apr;49(4):1437–46. doi: 10.1167/iovs.07-1337. [DOI] [PubMed] [Google Scholar]
- Buckingham BP, Inman DM, Lambert W, Oglesby E, Calkins DJ, Steele MR, Vetter ML, Marsh-Armstrong N, Horner PJ. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008;28:2735–2744. doi: 10.1523/JNEUROSCI.4443-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins DJ. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res. 2012;31(6):702–19. doi: 10.1016/j.preteyeres.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wei X, Cho KS, Chen G, Sappington R, Calkins DJ, Chen DF. Optic neuropathy due to microbead-induced elevated intraocular pressure in the mouse. Invest Ophthalmol Vis Sci. 2011;52(1):36–44. doi: 10.1167/iovs.09-5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;11:889–98. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Perry VH. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 2002;25(10):532–7. doi: 10.1016/s0166-2236(02)02255-5. [DOI] [PubMed] [Google Scholar]
- Cone FE, Gelman SE, Son JL, Pease ME, Quigley HA. Differential susceptibility to experimental glaucoma among 3 mouse strains using bead and viscoelastic injection. Exp Eye Res. 2010;91(3):415–24. doi: 10.1016/j.exer.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa SA, Eales KL. The Role of p38 MAPK and its substrates in neuronal plasticity and deurodegenerative disease. J Signal Transduct. 2012;2012:649079. doi: 10.1155/2012/649079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci USA. 2010;107(11):5196–201. doi: 10.1073/pnas.0913141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crish SD, Dapper JD, MacNamee SE, Balaram P, Sidorova TN, Lambert WS, Calkins DJ. Failure of axonal transport induces a spatially coincident increase in astrocyte BDNF prior to synapse loss in a central target. Neuroscience. 2013 Jan 15;229:55–70. doi: 10.1016/j.neuroscience.2012.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signaling. Biochem J. 2010;429(3):403–17. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Ding H, Gabali AM, Jenson SD, Lim MS, Elenitoba-Johnson KS. P38 mitogen activated protein kinase expression and regulation by interleukin-4 in human B cell non-Hodgkin lymphomas. J Hematop. 2009 Oct 21;2(4):195–204. doi: 10.1007/s12308-009-0049-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Remessy AB, Tang Y, Zhu G, Matragoon S, Khalifa Y, Liu EK, Liu JY, Hanson E, Mian S, Fatteh N, Liou GI. Neuroprotective effects of cannabidiol in endotoxin-induced uveitis: critical role of p38 MAPK activation. Mol Vis. 2008;14:2190–203. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, Avila J. Current advances in different kinases involved in tau phosphorylation, and implication in Alzheimer’s disease and tauopathies. Curr Alzheimer Res. 2005;2(1):3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- Ferrer I. Stress kinases involved in tau phosphorylation in Alzheimer’s disease, tauopathies and APP transgenic mice. Neurotox Res. 2004;6(6):469–75. doi: 10.1007/BF03033283. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Crowther RA, Martin KR, Berg N, Coleman M, Goedert M, Spillantini MG. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiol Aging. 2011;32(3):419–33. doi: 10.1016/j.neurobiolaging.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Gupta N, Fong J, Ang LC, Yucel YH. Retinal tau pathology in human glaucomas. Can J Ophthalmol. 2008;43(1):53–60. doi: 10.3129/i07-185. [DOI] [PubMed] [Google Scholar]
- Harada C, Nakamura K, Namekata K, Okumura A, Mitamura Y, Iizuka Y, Kashiwagi K, Yoshida K, Ohno S, Matsuzawa A, Tanaka K, Ichijo H, Harada T. Role of apoptosis signal-regulating kinase 1 in stress-induced neural cell apoptosis in vivo. Am J Pathol. 2006 Jan;168(1):261–9. doi: 10.2353/ajpath.2006.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada C, Namekata K, Guo X, Yoshida H, Mitamura Y, Matsumoto Y, Tanaka K, Ichijo H, Harada T. ASK1 deficiency attenuates neural cell death in GLAST-deficient mice, a model of normal tension glaucoma. Cell Death Differ. 2010;17(11):1751–9. doi: 10.1038/cdd.2010.62. [DOI] [PubMed] [Google Scholar]
- Harper SJ, LoGrasso P. Signalling for survival and death in neurons: the role of stress-activated kinases, JNK and p38. Cell Signal. 2001;13(5):299–310. doi: 10.1016/s0898-6568(01)00148-6. [DOI] [PubMed] [Google Scholar]
- Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye QN, Stewart CA, Geddes J, Markesbery WR, Patel E, Johnson GV, Bing G. p38 kinase is activated in the Alzheimer’s disease brain. J Neurochem. 1999;72(5):2053–8. doi: 10.1046/j.1471-4159.1999.0722053.x. [DOI] [PubMed] [Google Scholar]
- Howell GR, Soto I, Zhu X, Ryan M, Macalinao DG, Sousa GL, Caddle LB, Macnicoll KH, Barbay JM, Porciatti V, Anderson MG, Smith RS, Clark AF, Libby RT, John SW. Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma. J Clin Invest. 2012;122:1246–1261. doi: 10.1172/JCI61135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CC, Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. 2006;20(2):259–68. doi: 10.1096/fj.05-4376com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Fileta JB, Filippopoulos T, Ray A, Dobberfuhl A, Grosskreutz CL. Hsp27 phosphorylation in experimental glaucoma. Invest Ophthalmol Vis Sci. 2007;48(9):4129–35. doi: 10.1167/iovs.06-0606. [DOI] [PubMed] [Google Scholar]
- Inman DM, Sappington RS, Horner PJ, Calkins DJ. Quantitative correlation of optic nerve pathology with ocular pressure and corneal thickness in the DBA/2 mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2006;47:986–96. doi: 10.1167/iovs.05-0925. [DOI] [PubMed] [Google Scholar]
- Kabuyama Y, Homma MK, Kurosaki T, Homma Y. Early signaling events induced by 280-nm UV irradiation. Eur J Biochem. 2002 Jan;269(2):664–70. doi: 10.1046/j.0014-2956.2001.02698.x. [DOI] [PubMed] [Google Scholar]
- Kikuchi M, Tenneti L, Lipton SA. Role of p38 mitogen-activated protein kinase in axotomy-induced apoptosis of rat retinal ganglion cells. J Neurosci. 2000;20(13):5037–44. doi: 10.1523/JNEUROSCI.20-13-05037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci. 2009;66(20): 3289–307. doi: 10.1007/s00018-009-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert WS, Ruiz L, Crish SD, Wheeler LA, Calkins DJ. Brimonidine prevents axonal and somatic degeneration of retinal ganglion cell neurons. Mol Neurodegener. 2011;6(1):4. doi: 10.1186/1750-1326-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Yun SJ, Nam KN, Gho YS, Lee EH. Activation of microglial cells by ceruloplasmin. Brain Res. 2007;1171:1–8. doi: 10.1016/j.brainres.2007.07.053. [DOI] [PubMed] [Google Scholar]
- Legos JJ, Erhardt JA, White RF, Lenhard SC, Chandra S, Parsons AA, Tuma RF, Barone FC. SB 239063, a novel p38 inhibitor, attenuates early neuronal injury following ischemia. Brain Res. 2001;892(1):70–7. doi: 10.1016/s0006-8993(00)03228-5. [DOI] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Harizman N, Dardik R, Nisgav Y, Vander S, Melamed S. Regulation of cell death and survival pathways in experimental glaucoma. Exp Eye Res. 2007;85(2):250–8. doi: 10.1016/j.exer.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Malemud CJ. Inhibitors of stress-activated protein/mitogen-activated protein kinase pathways. Curr Opin Pharmacol. 2007;7(3):339–43. doi: 10.1016/j.coph.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Manabe S, Lipton SA. Divergent NMDA signals leading to proapoptotic and antiapoptotic pathways in the rat retina. Invest Ophthalmol Vis Sci. 2003;44(1):385–92. doi: 10.1167/iovs.02-0187. [DOI] [PubMed] [Google Scholar]
- Mielke K, Herdegen T. JNK and p38 stress kinases degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol. 2000;61(1):45–60. doi: 10.1016/s0301-0082(99)00042-8. [DOI] [PubMed] [Google Scholar]
- Munemasa Y, Ohtani-Kaneko R, Kitaoka Y, Kuribayashi K, Isenoumi K, Kogo J, Yamashita K, Kumai T, Kobayashi S, Hirata K, Ueno S. Contribution of mitogen-activated protein kinases to NMDA-induced neurotoxicity in the rat retina. Brain Res. 2005;1044(2):227–40. doi: 10.1016/j.brainres.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Munoz L, Ralay Ranaivo H, Roy SM, Hu W, Craft JM, McNamara LK, Chico LW, Van Eldik LJ, Watternson DM. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology. 2010;58(3):561–8. doi: 10.1016/j.neuropharm.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. 2012;35:153–79. doi: 10.1146/annurev.neuro.051508.135728. [DOI] [PubMed] [Google Scholar]
- Nucci C, Martucci A, Martorana A, Sancesario GM, Cerulli L. Glaucoma progression associated with altered cerebral spinal fluid levels of amyloid-beta and tau proteins. Clin Experiment Ophthalmol. 2010 doi: 10.1111/j.1442-9071.2010.02452.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Nguyen D, Alavi MV, Kim KY, Kang T, Scott RT, Noh YH, Lindsey JD, Wissinger B, Ellisman MH, Weinreb RN, Perkins GA, Ju WK. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011;2:e240. doi: 10.1038/cddis.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN. Mitochondria: Their role in ganglion cell death and survival in primary open angle glaucoma. Exp Eye Res. 2010;90(6):750–757. doi: 10.1016/j.exer.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Peifer C, Wagner G, Laufer S. New approaches to the treatment of inflammatory disorders small molecule inhibitors of p38 MAP kinase. Curr Top Med Chem. 2006;6(2):113–49. doi: 10.2174/156802606775270323. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90(3):262–7. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J Neurochem. 2000;74(4): 1587–95. doi: 10.1046/j.1471-4159.2000.0741587.x. [DOI] [PubMed] [Google Scholar]
- Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel M, Telliez JB. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol. 2007;27(1):170–81. doi: 10.1128/MCB.01456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappington RM, Sidorova T, Long DJ, Calkins DJ. TRPV1: contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest Ophthalmol Vis Sci. 2009;50:717–728. doi: 10.1167/iovs.08-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappington RM, Carlson BJ, Crish SD, Calkins DJ. The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci. 2010;51(1):207–16. doi: 10.1167/iovs.09-3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih GC, Calkins DJ. Secondary neuroprotective effects of hypotensive drugs and potential mechanisms of action. Expert Rev Ophthalmol. 2012;7(2):161–175. doi: 10.1586/eop.12.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasi K, Nagel D, Yang X, Ren L, Mittag T, Danias J. Ceruloplasmin upregulation in retina of murine and human glaucomatous eyes. Invest Ophthalmol Vis Sci. 2007;48(2):727–32. doi: 10.1167/iovs.06-0497. [DOI] [PubMed] [Google Scholar]
- Sun A, Liu M, Nguyen XV, Bing G. p38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp Neurol. 2003;183(2):394–405. doi: 10.1016/s0014-4886(03)00180-8. [DOI] [PubMed] [Google Scholar]
- Sun H, Wang Y, Pang IH, Shen J, Tang X, Li Y, Liu C, Li B. Protective effect of a JNK inhibitor against retinal ganglion cell loss induced by acute moderate ocular hypertension. Mol Vis. 2011;17:864–75. [PMC free article] [PubMed] [Google Scholar]
- Tezel G, Chauhan BC, LeBlanc RP, Wax MB. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest Ophthalmol Vis Sci. 2003;44(7):3025–33. doi: 10.1167/iovs.02-1136. [DOI] [PubMed] [Google Scholar]
- Trejo A, Arzeno H, Browner M, Chanda S, Cheng S, Comer DD, Dalrymple SA, Dunten P, Lafarque J, Lovejoy B, Freire-Moar J, Lim J, Mcintosh J, Miller J, Papp E, Reuter D, Roberts R, Sanpablo F, Saunders J, Song K, Villasenor A, Warren SD, Welch M, Weller P, Whiteley PE, Zeng L, Goldstein DM. Design and synthesis of 4-azaindoles as inhibitors of p38 MAP kinase. J Med Chem. 2003;46(22):4702–13. doi: 10.1021/jm0301787. [DOI] [PubMed] [Google Scholar]
- Wagner G, Laufer S. Small molecular anti-cytokine agents. Med Res Rev 2006. 2006 Jan;26(1):1–62. doi: 10.1002/med.20042. [DOI] [PubMed] [Google Scholar]
- Wax MB, Tezel G, Yang J, Peng G, Patil RV, Agarwal N, Sappington RM, Calkins DJ. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell derived fas-ligand. J Neurosci. 2008;28(46):12085–96. doi: 10.1523/JNEUROSCI.3200-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore A, Libby RT, John SW. Glaucoma: thinking in new ways-a role for autonomous axonal self-destruction and other compartmentalised processes? Prog Retin Eye Res. 2005;24:639–662. doi: 10.1016/j.preteyeres.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Yang Y, Jalal FY, Thompson JF, Walker EJ, Candelario-Jalil E, Li L, Reichard RR, Ben C, Sang QX, Cunningham LA, Rosenberg GA. Tissue inhibitor of metalloproteinases-3 mediates the death of immature oligodendrocytes via TNF-α/TACE in focal cerebral ischemia in mice. Sci Signal. 2012;5(222) doi: 10.1186/1742-2094-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Liu J, Sakaki-Yumoto M, Derynck R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. J Neuroinflammation. 2011;8:108. doi: 10.1126/scisignal.2002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15(1):11–8. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]