

Summary
Retroviral vectors derived from the murine leukemia virus (MuLV) are widely used as the starting material in the development of vectors for gene therapy and critical in answering questions relating to viral pathogenesis. The p30 capsid (CA) is the major viral core protein and an internal group antigen in MuLV. In this study, an enzyme-linked immunosorbent assay (ELISA) was developed for quantitation of MuLV infectious particles with p30 CA core antigen protein. The ELISA was developed using several goat-polyclonal serum against MuLV p30 generated by the NCI as primary antibody and a rat-monoclonal antibody to CA available from ATCC. The MuLV p30 CA antigen was standardized against recombinant MuLV p30 CA expressed from bacteria. The assay is sensitive, accurate and linear within a defined concentration range of CA. Comparison with different MuLV quantitative methods including reporter gene transfer, reverse transcriptase activity assay, and viral RNA quantitative PCR, showed this ELISA protocol to be highly quantifiable within defined ranges, which can be correlated with infectious viral titer.
Keywords: MuLV CA ELISA, p30 CA, Virus normalization
The use of ELISAs against viral proteins has become standard for quantifying viral particles for many viruses, including HIV-1 (p24 ELISA). For MuLV, one of the initial assays for normalizing viral particles was based on quantifying the level of reverse transcriptase within viral particles (Goff, Traktman, and Baltimore, 1981). For studies involving mutations in the pol gene product that can affect packaging of Gag-Pol in particles, normalizing based on RT is not optimal. An ELISA for p30 was only recently available (Cell Biolabs, San Diego CA USA), at a high expense ($595/96 assays). Antibodies to the CA protein had been generated through the NCI, and have been utilized by many laboratories in the field. In this study, an ELISA was developed based on anti-CA antibodies widely distributed among the retroviral community. The basic assay uses the monoclonal antibody CRL-1912 (Chesebro et al., 1983) distributed by ATCC (Manassa VA USA) to capture the antigen, conjugates the bound CA using goat-anti CA polyclonal antibodies generated by the NCI, and quantifies the results with an HRP modified anti-goat antibody.
The M-MuLV CA protein was expressed and purified from bacteria and used as the protein standard for the ELISA. pTYB1 pLysS CA is a pETDuet based vector that has an Mxe intein in frame at the CA C-terminus followed by a chitin binding domain. The region encoding CA was PCR amplified using 2.5 pmoles forward primer–5′ GGTGGTCATATGCCCCTCCGCGCAGGAAAC 3′ (NdeI site underlined), 2.5 pmoles reverse primer 5′ CGGGGTACCCTTGGCAAAGCACAATAGCTTGCTCATCTCTCT 3′ (KpnI site underlined), 2.5 U Pfu DNA polymerase (Stratagene, La Jolla CA USA), 0.2 mM each dNTPs, 1X Pfu polymerase buffer and pNCA-C template DNA (Felkner and Roth, 1992) in a 100 μL reaction mix. CA was cloned in pTYB1 pLysS using the NdeI and KpnI (New England Biolabs, Ipswich MA USA) restriction sites and introduced into competent E.coli BL21 (DE3) cells. Protein expression was induced from a 1 liter culture at an O.D.600 of 0.7 for 24 hours at 15°C. Cells were harvested and lysed in ice cold buffer containing 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 10 mM CHAPS, and complete EDTA protease inhibitor tablets (1 tablet per 50 ml. of buffer) (Roche, Indianapolis, IN USA). Resuspended cells were dounced to homogeneity followed by sonication using a sonic dismembrator model 100 (Fisher Scientific, Rockford IL USA) at setting 6 with 5 pulses of 30 seconds each. The cell suspension was centrifuged in a high speed Sorvall centrifuge at 16,500 rpm for 1 hour at 4°C using a SS-34 rotor. The soluble fusion protein fraction was purified using chitin beads (New England Biolabs, Ipswich MA USA) as per the manufacturer’s recommendations. CA was released from the fusion protein by incubating for 48 hours at 4°C with 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 10% glycerol, 50 mM dithiotreitol (Fluka, St. Louis MO USA), and 0.1 mM EDTA. CA was then separated from the chitin binding domain and the uncleaved fusion protein as per manufacturer’s instructions. Greater than 90% of the CA protein was separated from the chitin tag (see western blot, Fig. S1). Purified protein was then concentrated using Ultracel 10K concentrator (Millipore) to about 1 mg/ml. The yield of CA protein was 3.7 mg/L bacterial culture.
For the ELISA, the anti-MuLV p30 monoclonal antibody was purified from the monoclonal hybridoma cell line (ATCC Number: CRL-1912, Manassas VA USA) culture medium using Protein-G agarose column chromatography (Åkerström and Björck, 1986). The hybridoma cells were cultured in RPMI 1640 containing 10% ultra-low IgG FBS (Gibco), 1 mM sodium pyruvate (Gibco), and antibacterials & antimycotics (Gibco, Grand Island NY USA). Medium was harvested every 48 hr and stored at 4°C. 50 ml of culture supernatant was applied to Protein G-Agarose gel (P4691, Sigma, St. Louis MO USA, 1 ml) equilibrated with cold PBS. The column was washed with 25 ml cold PBS solution, and the antibody was eluted with 1 ml 0.2M glycine buffer, pH 2.7, into tubes containing 0.25 ml neutralizing buffer (0.2M Na2HPO4, pH 8.9). The antibody concentration was determined by A280 on a Nanodrop 2000 (Thermo Scientific, Rockford IL USA).
The MuLV CA ELISA was performed according to the following protocol. Assays were performed in 96 well flat bottom MaxiSorp Immuno plates (Thermo Scientific, Rockford IL USA 439454). The plates were coated with purified anti-MuLV p30 monoclonal antibody in 100 μl/well of coating buffer (0.1 M sodium bicarbonate buffer, pH 9.6) for 24 hr at 4°C and rinsed twice with 200 μl/well PBS (see Fig. 1 for titration). The plates were blocked with 5% BSA in PBS (100 μl/well) for 1hr at 37°C. Dilutions of purified recombinant p30 CA standard were prepared from 20 ng/ml to 0.312 ng/ml in lysis buffer (0.5% TritonX-100 in PBS). Samples to be assayed were prepared in 1X lysis buffer (0.5% TritonX-100 in PBS) with the following four dilutions of 1X, 1/10X, 1/100X, 1/1000X and then incubated at 37°C, 30min. Into each well of the ELISA plate, 100 μl of sample or 100 μl MuLV p30 standards were added and incubated for 1hr at 37°C. Each sample was analyzed in duplicate or triplicate. The samples were then washed 5 times with PBST buffer (0.05% Tween 20 in PBS). 100 μl/well of goat-anti p30 polyclonal antibody (1:3000) in gelatin-buffer (0.25% gelatin, 0.15M NaCl, 5mM EDTA, 0.05% Tween 20 in 50mM Tris-HCl, pH 8.0) was added and incubated at RT for 1 hr on a shaker. Samples were washed 6 times with 200 μl PBST buffer. 100 μl/well of the bovine anti-goat polyclonal antibody, diluted 1:5000 in gelatin-buffer was added and incubated at RT for 1 hr on a shaker. The samples were washed 6 times with 200 μl PBST buffer. 75 μl/well of 1-step TM Ultra TMB-ELISA substrate (Thermo Scientific Rockford IL USA, 34028) was added and incubated at RT on the shaker for 3–20 min. The reactions were stopped by adding 75 μl of 2N H2SO4 solution when the standard 20 ng/ml p30 well changed to blue. The optical density was read at O.D.450 nm immediately.
Figure 1. Non-specific interaction between HRP-Conjugating 2nd antibody and monocolonal anti-p30 antibody.
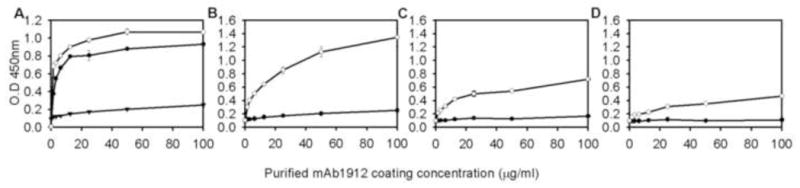
(A) HRP conjugated anti-goat 2nd antibodies were tested with varying concentrations of CRL-1912 monoclonal antibody coating the ELISA trays. Filled circles, rabbit anti-goat antibody; open circle, donkey anti-goat antibody; filled triangles, bovine anti-goat antibody. (B–D). Dilution of bovine anti-goat antibody in the presence of CRL-1912 monoclonal antibody and MuLV p30 recombinant protein. Panel B, 1:5000 dilution; Panel C, 1:10,000 dilution; Panel D, 1:20,000 dilution of bovine anti-goat antibody. Closed circles, absence of CA; open circles, 20 ng/nl CA.
Optimization of the ELISA involved decreasing the background non-specific signals while maximizing the experimental signal. Although the HRP conjugated anti-goat secondary antibody should not bind to the primary rat monoclonal antibody (CRL-1912), varying levels of cross-reaction were observed. Three commercially available anti-goat-HRP antibodies including rabbit anti-goat antibody (31402, Pierce, Rockford IL USA, 0.8 mg/ml), donkey anti-goat antibody, (sc-2020, Santa Cruz Biotechnology, Dallas TX USA, 0.4 mg/ml) and bovine anti-goat antibody (805-035-180, Jackson ImmunoResearch Laboratories, West Grove PA USA, 0.8 mg/ml) were tested for their background binding to CRL1912, in the absence of p30 protein (Fig. 1). The level of CRL-1912 coating the plates was varied between 1 to 100 μg/ml and the HRP-conjugated antibodies were held constant at 1:5000 dilution. The background from rabbit anti-goat antibody and donkey anti-goat antibody increased with the level of bound CRL-1912 antibody. The background signal was fivefold higher than bovine anti-goat antibody at levels of CRL-1912 greater than 20 μg/ml. The assay was therefore developed using the bovine anti-goat HRP conjugated antibody.
In the presence of 20 ng/ml purified p30 protein and goat anti-CA 81S-263 (1:3000), the level of coating monoclonal antibody CRL-1912 and bovine anti-goat HRP conjugated antibody were then optimized. Figs. 1B–D show the titration of monoclonal antibody CRL-1912 between 0–100 μg/ml at 1:5000, 1:10,000, and 1:20,000 dilutions, respectively, of bovine anti-goat HRP antibody. Maximal signal required coating the plates at concentrations greater than 25 μg/ml and using dilutions of HRP conjugated bovine anti-goat antibody at 1:5000. Dilutions of bovine anti-goat-HRP conjugated 2nd antibody greater than 1:5000 reduced the signal for 20 ng/ml of p30 CA protein.
Four different goat anti-p30 serums generated at the NCI-BCB Repository (distributed by Microbiological Associates, Farmington Hills MI USA) were tested. These serums were made against the p30 protein from Rauscher murine leukemia virus (R-MuLV). The p30 CA from Moloney murine leukemia virus (M-MuLV) has 95% protein sequence identity (250/263) with R-MuLV. MuLV viral particles pseudotyped with VSV-G were collected from supernatant of viral D17 canine producer cells and maintained as previously described (Schneider et al., 2012). MuLV particles were concentrated by centrifugation at 21000 x g for 30 min at 4°C and analyzed by western blotting on PVDF membranes after SDS-PAGE. Immunoblots were developed using anti-p30 goat antiserum (74S-428, 74S-454, 81S-263, and 75S-287) from NCI-BCB Repository, a peroxidase-conjugated anti-goat antibody and an ECL detection kit (Pierce, Rockford IL USA). Viral particles produced in the presence of 10 nM of the protease inhibitor Amprenavir (A634400, Toronto Research Chemicals, Toronto, Ontario Canada) were enriched for the precursor Pr65Gag protein. All four sera recognized both the M-MuLV p30 and precursor Pr65 proteins by western blot (Fig. 2A). Cross-reactivity to other MuLV proteins was observed, in particular with 71S-454, where a protein of <20 kDa was observed to be recognized by the antibody in addition to the CA protein. In addition, the monoclonal CRL1912 antibody recognized both the Pr65 precursor protein (in the presence of Amprenavir) and the processed p30 CA by western blot using a goat anti-rat HRP conjugated secondary antibody. Significant to the development of the ELISA, the different goat anti-p30 sera varied in their slopes and R square-value when titrated against increasing concentration of CA. In Fig. 2B–E, three of the serums showed a linear relationship of CA concentration over 0.3–20 ng/ml to OD450 with R square values of ~0.9. The serum 81S-263 showed the largest signal response, displaying the highest slope value. Although serum 75S-287 showed high specificity to CA on a western blot (Fig. 2A), within the ELISA it displayed a low slope and R-square value. Presumably the high OD450 background observed with 75S-287 serum in this ELISA could be due to its reacting with the coating monoclonal antibody.
Figure 2. Analysis of different goat anti-p30 sera.
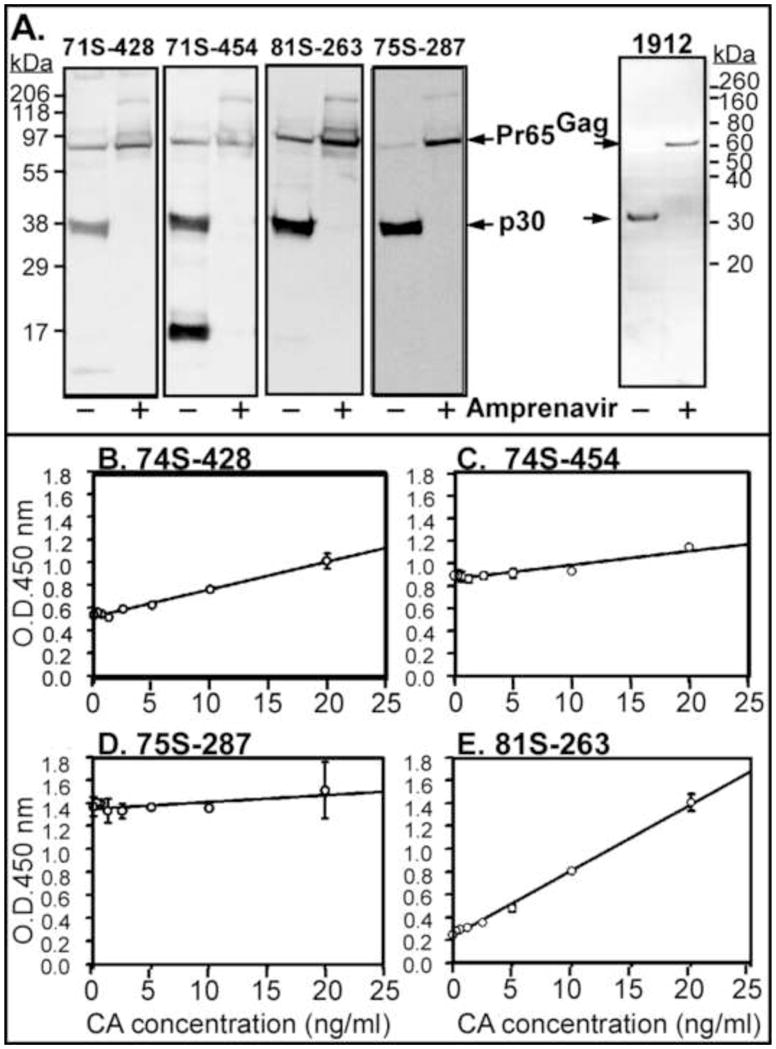
Individual serum numbers are as indicated. (A) Western blot analysis. Virus particle was harvested from D17 virus producing cell treated with or without a protease inhibitor Amprenavir (10mM). The positions of molecular weight standard proteins are shown on the left. (B–E). Comparison of HRP signal (OD450) versus p30 concentration using four goat anti-p30 sera (1:3000). ELISA plates were coated with CRL-1912 monoclonal antibody at 100μg/mL. HRP-conjugated bovine anti-goat antibody was used at 1;5000 dilution. All assays were performed in triplicate. Statistical analysis 74S-454, y=0.0124x + 0.863 (R2=0.891); 74S-428, y=0.024x + 0.501 (R2=0.986); 75S-287, y=0.006x + 1.35 (R2=0.514); 81S-263, y=0.058x+0.233 (R2=0.996).
Serum 81S-263 was selected for further characterization in the ELISA, due to its specificity in the immunoblot, recognition of both the precursor and processed Gag proteins and its dynamic response to a range of CA protein (Fig. 3). The dilutions of 81S-263 serum were examined in the range of 1:1000 to 1:5000 in this ELISA protocol using concentrations of p30 from 0.3 to 20 ng/ml. At a 1:3000 dilution, the maximal slope with the lowest background was observed over a wide range of CA concentrations. With this dilution, the MuLV p30 core antigen protein could be successfully quantified to the ng level.
Figure 3. Titration of the goat anti-p30 81S-253 antibody in response to CA.
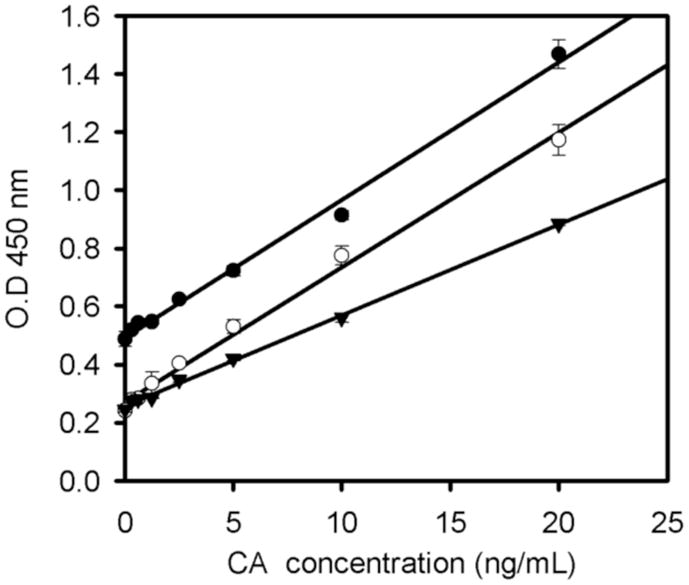
The ELISA trays were coating CRL-1912 monoclonal antibody at 100 μg/ml in the presence of varying levels of CA protein and HRP conjugating bovine anti-goat 2nd antibody (1:5000 dilution). 81S-253 goat anti-p30 serum was diluted to 1:1000 (filled circles), 1:3000 (open circles), and 1:5000 (closed triangles). All assays were performed in triplicate. Statistical analysis: 1:1000 dilution, y=0.047x + 0.49, R2=0.994; 1:3000 dilution, y=0.046x + −.2718, R2=0.994; 1:5000 dilution, y=0.31x + 0.257, R2=0.998. For the maximum sensitivity and p30 concentrations in a defined range, a 1:3000 dilution was optimal.
There are several methods that can be used to quantify retroviral infectious particles, each measuring distinct features. These include determining viral titers by transfer of reporter genes including lacZ or GFP (Mao et al., 2000; Soneoka et al., 1995), normalizing based on the reverse transcriptase activity within viral particle (Goff et al., 1981), and measuring the level of vRNA by qPCR (Carmo et al., 2004). Each assay normalizes based on different properties of the viral particles, namely, the level of Gag, the activity of a pol gene product, the packaging of viral RNA, and the successful gene transfer into a recipient cell. The assembly of viral particles is complex and interrelated and therefore assays that measure distinct features of the viral particles are necessary. Although standardization using reverse transcriptase activity was one of the first methods described to rapidly monitor viral infection, it is not the logical choice to use for studies of the pol gene products, which can have pleiotropic effects (Lu et al., 2004). Alternatively, mutations that can affect viral RNA dimerization and packaging would need to be normalized by alternative assays beyond qPCR. In order to verify that the ELISA protocol is comparable to other protocols, the level of viral particles was determined using these four different quantitative methods. For these experiments, the wildtype virus was compared to a producer cell expressing the integrase (IN) mutant K376A, blocked at viral integration (Schneider et al., 2012). Virus bearing IN K376A was expressed in the vector pNCA-C neo backbone (Schneider et al., 2012) pseudotyped with the VSV-G Env to allow infection into human cells. Virus samples were collected from canine D17 producing cell lines containing the WT or IN K376A viruses and packaging the reporter gene gfp (pGIP) (Mao et al., 2000; Schneider et al., 2012). The two viral preparations had similar levels of RT activity (Fig. 4) and CA levels (by ELISA, WT 2.69 μg/ml vs 2.54 μg/ml for IN K376A), as well as packaged similar levels of vRNA (WT, 7.9 × 109 copy vs IN K376A 7.34 × 109 copies) (Table 1). However, the viral titration, measuring GFP expression in the recipient target cells resulted in an observable difference between WT and IN K376A mutation (Table 1), because the mutation of IN K376A decreases integration (Schneider et al., 2012). As predicted, the IN K376A mutation had no effect on virus assembly, release and maturation but only blocked the integration of the viral genome. With this study we confirmed that the p30 ELISA provided quantification of viral particles in the dynamic range comparable with other quantitative methods. The linear correlation of the viral RNA copy number, RT activity and CA protein shows that this assay can be used quantitatively and performed to estimate infectious particles in concert with other quantitative assays. A ratio between infectious particles with viral RNA, CA, and RT can be applied to the quantitation. Thus, depending on the aspect of viral replication being studied, suitable targets for quantitation are now available.
Figure 4. Comparison of RT activity of virus bearing WT and IN K376A. Autoradiogram of aliquots of viral supernatants assayed for the presence of RT in the cell media.
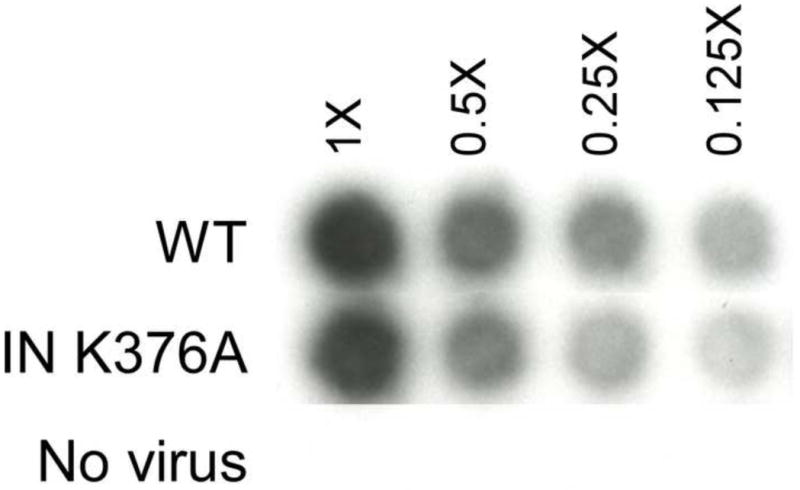
[32P]TMP incorporation catalyzed by RT was measured by exposing the radioactive material bound to DE52 paper (Goff et al., 1981). Viral dilutions are as indicated.
Table 1.
Quantitation of MuLV particles
| MuLV | CA conc. (μg/ml)1 | vRNA copy number2 | Titer (IU/ml)3 |
|---|---|---|---|
| Wildtype | 2.69±0.004 | 7.86±2.55 x109 | 1.15 × 106 |
| IN K376A | 2.54±0.028 | 7.34±1.17 x109 | <100 |
CA concentration determined by ELISA.
Viral RNA was purified by QIAamp Viral RNA mini kit (Qiagen, Valencia CA USA) and cDNA synthesis using Superscript III Reverse Transcriptase primed with Random Hexamers (Invitrogen, Carlsbad, CA USA). vRNA was quantified as previously described using the primers set for minus strand strong stop (MSSS) intermediates (Schneider et al., 2012).
Viral titer was determined using flow cytometric analysis of GFP fluorescence transduction. The producer cell lines expressing the pNCA-C-neo (Schneider et al., 2012) constructs plus pGIP (Chen et al., 2001) were transfected with 2 μg of pHIT-G plasmid DNA (VSVG) using the Fugene 6 (Roche) as described previously described (Schneider et al., 2012). Virus was serial diluted and used to infect TE671 cells. GFP fluorescence as determined by flow cytometry was used to calculate the percentage of GFP-positive cells. The titer (infectious units (IU) per ml) was calculated as: (%) GFP-positive cells x cell number at initial viral exposure)/viral volume (ml) applied when transduction was not saturated (Mao et al., 2000).
Table 2 examines the effect of protease cleavage of the Pr65Gag precursor protein using the p30 ELISA protocol. The monoclonal 1912 and the 81S-263 antibodies both recognize the Gag precursor and cleavage CA products (Fig. 2A). Treatment of cells with amprenavir yielded a slight decrease (25%) in the level of CA detected in viral supernatants. Quantitation of vRNA indicated an increase of vRNA in the presence of amprenavir, which may correspond with the increased affinity of the Pr65Gag for RNA compared with the cleaved NC protein (Karpel, Henderson, and Oroszlan, 1987). Thus the p30 ELISA assay would quantify both cleaved and uncleaved Gag products.
Table 2.
Quantitation of Pr65Gag versus P30 CA
| MuLV | CA conc. (μg/ml)1 | vRNA copy number2 |
|---|---|---|
| Wildtype | 1.75±0.171 | 1.88±0.16 ×109 |
| WT+Amprenavir | 1.31±0.197 | 2.77±0.07 ×109 |
CA concentration determined by ELISA.
Vial RNA was purified by QIAamp Viral RNA mini kit (Qiagen, Valencia CA USA) and cDNA synthesis using Superscript III Reverse Transcriptase primed with Random Hexamers (Invitrogen, Carlsbad, CA USA). vRNA was quantified as previously described using the primers set for minus strand strong stop (MSSS) intermediates (Schneider et al., 2012).
Empirically, it takes 3–5 days to quantify infectious retroviral particles with a titration assay, whereas the method presented here has the ability to quantify vectors in a few hours. Viral titer is the most stringent criteria, as it measures the number of units that successfully complete the infectious life-cycle through viral integration and expression. This incorporates the problems associated with the production of defective particles, including those lacking viral envelopes, viral RNA, or blocked during the replication cycle. The use of IN K376A in this study verified that the particles were identical with respect to CA, RT, and vRNA, but were blocked at viral integration, yielding a diminished viral titer.
The combined data demonstrate that p30 ELISA is a useful method for quantifying viral particles for both basic retrovirologic research as well as applied fields including routine quality control of MuLV-based gene delivery vectors. The ability to rapidly quantify viral particles allows the use of freshly prepared samples, eliminating the effects of freeze-thawing on viral titer. The assay is as sensitive as the commercial kit (VPK-156, Cell Biolabs, San Diego CA USA), is accurate and linear within defined range of p30 concentration, and is less costly. The sensitivity of the p30 ELISA is high enough to perform the characterization of most culture supernatants. In the present study the MuLV vector particles were prepared by methods categorically similar to those used for gene delivery. This may be advantageous for establishing the use of ELISA as a routine assay for gene therapy applications. However, it should be noted that the assay scores positive for both the precursor Pr65Gag as well as the processed CA product. Therefore, mutations that alter the processing of the immature to the mature particle would not be distinguished with this assay.
Supplementary Material
Acknowledgments
This work was support by NIH grant 5RO149932 to M.J.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Dai-Tze Wu, Email: wuta@umdnj.edu.
Sriram Aiyer, Email: aiyerss@umdnj.edu.
Rodrigo A. Villanueva, Email: rodrigo.villanueva@unab.cl.
Monica J. Roth, Email: roth@umdnj.edu.
References
- Åkerström B, Björck L. A physicochemical study of protein G, a molecule with unique immunoglobulin G-binding properties. J Biol Chem. 1986;261:10240–10247. [PubMed] [Google Scholar]
- Carmo M, Peixoto C, Coroadinha AS, Alves PM, Cruz PE, Carrondo MJT. Quantitation of MLV-based retroviral vectors using real-time RT-PCR. Journal of Virological Methods. 2004;119:115–119. doi: 10.1016/j.jviromet.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Chen CC, Rivera A, Ron N, Dougherty JP, Ron Y. A gene therapy approach for treating T-cell-mediated autoimmune diseases. Blood. 2001;97:886–894. doi: 10.1182/blood.v97.4.886. [DOI] [PubMed] [Google Scholar]
- Chesebro B, WWB, Evans L, Wehrly K, Nishio J, Cloyd M. Characterization of monoclonal antibodies reactive with murine leukemia viruses: Use in analysis of strains of Friend MCF and Friend ecotropic murine leukemia virus. Virology. 1983;127:134–148. doi: 10.1016/0042-6822(83)90378-1. [DOI] [PubMed] [Google Scholar]
- Felkner RH, Roth MJ. Mutational analysis of the N-Linked glycosylation sites of the SU envelope protein of Moloney murine leukemia virus. J Virol. 1992;66:4258–4264. doi: 10.1128/jvi.66.7.4258-4264.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff S, Traktman P, Baltimore D. Isolation and properties of Moloney murine leukemia virus mutants: use of a rapid assay for release of virion reverse transcriptase. Journal of Virology. 1981;38:239–248. doi: 10.1128/jvi.38.1.239-248.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpel RL, Henderson LE, Oroszlan S. Interactions of retroviral structural proteins with single-stranded nucleic acids. Journal of Biological Chemistry. 1987;262:4961–7. [PubMed] [Google Scholar]
- Lu R, Limon A, Devroe E, Silver PA, Cherepanov P, Engelman A. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a postnuclear entry step of human immunodeficiency virus type 1 replication. J Virol. 2004;78:12735–12746. doi: 10.1128/JVI.78.23.12735-12746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao AJ, Bechberger J, Lidington D, Galipeau J, Laird DW, Naus CC. Neuronal differentiation and growth control of neuro-2a cells after retroviral gene delivery of connexin43. J Biol Chem. 2000;275:34407–14. doi: 10.1074/jbc.M003917200. [DOI] [PubMed] [Google Scholar]
- Schneider WM, Wu DT, Amin V, Aiyer S, Roth MJ. MuLV IN mutants responsive to HDAC inhibitors enhance transcription from unintegrated retroviral DNA. Virology. 2012;426:188–96. doi: 10.1016/j.virol.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM, Kingsman AJ. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucl Acids Res. 1995;23:629–633. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.