

Abstract
Of the many optical bioassays available, sensing by fluorescence anisotropy have great advantages as it provides a sensitive, instrumentally simple, ratiometric method of detection. However, it is hampered by a severe limitation as the emission lifetime of the label needs to be comparable to the correlation lifetime (tumbling time) of the biomolecule which is labelled. For proteins of moderate size this is in the order of 20–200 ns, which due to practical issues currently limits the choice of labels to the dansyl-type dyes and certain aromatics dyes. These have the significant drawback of UV/blue absorption and emission as well as an often significant solvent sensitivity. Here, we report the synthesis and characterization of a new fluorescent label for high molecular weight biomolecules assay based on the azadioxatriangulenium motif. The NHS ester of the long fluorescence lifetime, red emitting fluorophore: azadioxatriangulenium (ADOTA-NHS) was conjugated to anti-rabbit Immunoglobulin G (antiIgG). The long fluorescence lifetime was exploited to determine the correlation time of the high molecular weight antibody and its complex with rabbit Immuniglobulin G (IgG) with steady-state fluorescence anisotropy and time-resolved methods: solution phase immuno-assay was performed following either steady-state or time-resolved fluorescence anisotropy. By performing a variable temperature experiment it was determined that the binding of the ligand resulted in an increase in correlation time by more than 75 %, and a change in the steady-state anisotropy increase of 18%. The results show that the triangulenium class of dyes can be used in anisotropy assay for detecting binding events involving biomolecules of far larger size than what is possible with the other red emitting organic dyes.
Introduction
Fluorescence polarization assay is one of the most common methods in drug development and other research areas employing high-throughput screening.1–4 The sizes of molecules that can be detected in these assays are limited as the emission lifetime of the fluorescent label has to be well-matched with the rotational correlation time of the molecules and complexes under investigation. Most efficient organic fluorophores have short lifetimes, as a consequence polarization assay involving organic dyes are limited to the case of small labelled proteins and peptides binding to much larger biomolecules.1 Currently, the fluorescent labels capable of detecting the slow rotational motion of large biomolecules are small aromatic molecules such as dansyl and pyrene derivatives. These have lifetimes in the 10–30 ns range, but have the considerable drawback of absorption and/or emission in the UV as well as significantly solvent dependent emission properties.5–8 Furthermore, the highly energetic UV excitation wavelengths are not suited for cellular applications as they result in enhanced photodegradation, significant auto-fluorescence and interference from scattering phenomena.1, 9, 10 Following excitation at wavelengths out of the blue region of the spectrum emissive transition metal complexes have been used to detect large biomolecules such as Immunoglobulin G.2, 11
However, the initial anisotropy for such complexes is intrinsically low, decreasing the dynamic range of the measurement. Furthermore, the emission lifetimes of these complexes are in the micro- to milli-second range. These two facts imply that the metal complexes are not ideally suited for detection of medium sized biomolecules. The azaoxa-triangulenium class of dyes,12–14 in the following represented by azadioxatriangulenium (ADOTA+, Figure 1), are bright organic dyes with a long fluorescence lifetime (≈25 ns) and emission in the red.15, 16 The fluorescence lifetimes are comparable to those of dansyl and pyrene. However, the emission at 550–600 nm results in much reduced interference from protein auto-fluorescence and Rayleigh scattering.2, 17 Here, we report the synthesis and first assay-type study using this new long lifetime fluorescence polarization probe.
Figure 1.

Structure of ADOTA-NHS
Results and Discussion
The synthesis of the amine reactive ADOTA+ dye: ADOTA-NHS; is shown in Scheme 1. The procedure follows classical triangulenium dye synthesis.12, 18, 19 The carboxylic acid group was introduced in the triangulenium skeleton using methyl 4-amino-butanoate as nucleophile in a substitution reaction on the readily available tris(dimethoxyphenyl)methylium ion (1).12, 20 The resulting acridinium salt (2) was isolated in 73–91 % yield. 2 was subjugated to pyridine hydrochloride mediated ring-closure to form the azadioxatriangulenium dye (3); this proceed in an isolated yield of 94 %. From 3, the activated ester (ADOTA-NHS, 4) was obtained in 40 % by reaction with TSTU. Detailed synthetic procedures and full spectroscopic characterization of the new compounds 2, 3 and 4 are included as Supporting Information.
Scheme 1.

Synthesis of ADOTA-NHS
To demonstrate the properties of ADOTA-NHS as a biolabel, the dye was conjugated to anti-rabbit IgG (antiIgG in the following) with a standard labelling protocol (see Supporting Information for details). Using absorption spectroscopy we determined that the labelling procedure resulted in an average of 3.3 labels per antiIgG. The free ADOTA+ dye is solvent insensitive; in water its emission quantum yield is determined to 41% and its fluorescence decay is monoexponential with a fluorescence lifetime of τfl = 20.1 ns (see Supporting Information Figure S1 for details). These values compare well with the reported quantum yield of 49% and lifetime of 23.2 ns in acetonitrile16, 21—where the ion is known to be well solvated—and indicate that despite being an organic molecule of moderate size, the dye is well behaved in water. After conjugation to antiIgG, the fluorescence intensity decay can be fitted to a biexponential decay model and the fluorescence lifetime has been reduced to an amplitude averaged lifetime of <τ>av = 10.1 ns. The first of the components account for 61 % of the amplitude and have a fluorescence lifetime of τ1 = 14.5 ns, where the second component is much shorter lived with a fluorescence lifetime of only τ2 = 3 ns. The biexponential decay characteristics of the dye-conjugate are a consequence of labels situated in several different microenvironments. The amplitudes of the two components in the fit of the time-resolved emission profile show that 39% of the emitting labels are situated close to a fluorescence quencher on the protein surface (τ2 = 3 ns vs. τfl = 20.1 ns), where 61% of the population of emitting labels is only moderately quenched by the surrounding protein surface (τ1 = 14.5 ns vs. τfl = 20.1 ns). As ADOTA+ has been shown to be quenched by amines,16 the quenching is expected to be found in a fraction of the labels in close proximity to the amino groups of e.g. lysine.22 In the antiIgG-IgG complex the quenched fraction rises slightly from 39 % to 41%, this is in agreement with the assumption of quenching by surface amino groups, as the total number of these will increase in the event of binding. The photostability of ADOTA+ is similar to that of rhodamine 6G, see supporting information for details.
The fluorescence anisotropy of ADOTA+ is r0 = 0.38 in rigid media21, very close to the ideal value of r0 = 0.4.22 As the fundamental anisotropy and the fluorescence lifetime are known, we can investigate the motion of the labelled biomolecules by following anisotropy of the emission (r) and using the Perrin equation (Equation 1).
| Eq 1 |
In eq. 1 θ is the rotational correlation time of the biomolecule, and D is the rotational diffusion coefficient. Even if the biomolecules are immobilized the measured anisotropy for the dye label will be considerably less than that of the immobilized dye. This is due to the local motion of the biomolecule around the label-biomolecule linking site and the motion of the label itself; the dye will never be completely constrained. Here, flexibility must be expected from the propyl linker chain, even though its rotation has some constrains,23 as well as from the methylene groups in lysine.
The measured anisotropy is as expected lower than r0, even though the overall motion of the antibody is limited. Figure 2 shows the fluorescence emission and excitation anisotropy of ADOTA-antiIgG and the ADOTA-antiIgG-IgG bioconjugates superimposed on the ADOTA-NHS spectra. At room temperature the maximum anisotropy obtained for ADOTA-antiIgG-IgG is r = 0.13 while that of ADOTA-antiIgG is r = 0.11. These values are roughly a factor of three lower than the immobilized value. The contribution of local motion of the label to the measured anisotropy can be estimated by extrapolating a fit of 1/r against temperature to T = 0.22 The intercept of this fit and the y-axis yields the apparent maximum anisotropy r0app, which corresponds to the anisotropy in the absence of the slow overall motion of the biomolecule. By using the apparent anisotropy r0app in the Perrin equation a more accurate determination of the hydrodynamic volume of the protein can be performed.
Figure 2.

Fluorescence emission and excitation anisotropy of ADOTA-antiIgG (dash) and ADOTA-antiIgG-IgG (dot) superimposed on the fluorescence emission and excitation spectrum of ADOTA-antiIgG.
The binding of ADOTA-antiIgG to the complimentary antibody is accompanied by a numerically large increase in molar weight, but the relatively increase is only a factor of 2. When using the Perrin equation the binding is measured as a change of Δr = 0.02. This is a relatively small change compared to experiments where a small ligand bind to a larger protein, where the value goes from r ≈ 0.00 up to r = 0.3–0.4.2 It is however clearly measurable, even with the considerable quenching of the label by quenchers in the biomolecules. After determining the fluorescence anisotropy at several temperatures the molecular weight (M) can be determined for ADOTA-antiIgG and the ADOTA-antiIgG-IgG complex by rewriting and reorganizing the Perrin equation in Equation 1 to:22
| Eq 2 |
| Eq 3 |
In eq. 2 and eq. 3 the factors η, V, R, and T are the solvent viscosity, biomolecules volume, gas constant, and temperature respectively. The factors v and h in eq. 3 is the density of the biomolecules and its hydration respectively. From the slope of a plot of 1/r against T the volume of the biomolecules can be calculated; a set of values are compiled in Table 1. Note that the Perrin equation inherently assumes that the rotor is spherical, and the results will reflect this assumption, see the discussion below.
Table 1.
Summary of results from a variable temperature study of the time-resolved and steady-state fluorescence anisotropy of anti-rabbit IgG and an anti-rabbit IgG - rabbit IgG complex.
| antiIgG | antiIgG-IgG | Δc | |
|---|---|---|---|
| r0 | 0.36 ± 0.001 | ||
| r0app | 0.18 ± 0.002 | 0.19 ± 0.004 | 0.01 |
| r500 nm (20°C) | 0.11 ± 0.004 | 0.13 ± 0.005 | 0.02 (+18 %) |
| θ1 (20°C) | 52 ± 15 ns | 91 ± 30 ns | 39 ns (+75 %) |
| Ma | ~150,000 Da | ~300,000 Da | |
| MPerrinb | 187,900 ± 18% Da | 280,900 ± 18% Da | 107,000 Da |
| Mθb | 150,500 ± 1% Da | 258,700 ± 1% Da | 108,200 Da |
Masses taken from ref 26.
Masses calculated assuming antiIgG and antiIgG-IgG are spherical rotors with zero net hydration, see text for detail.
difference between the free antiIgG and the complex.
A set of assumptions are made when calculating the molecular weight of biomolecule rotors from the correlation time: i) the rotor is assumed to be either oblate, prolate or spherical, ii) a hydration number describing the co-rotating body of water is assumed and iii) the hydration is assumed to uniformly distributed over the biomolecule. For IgG and IgG-IgG complexes none of these assumptions are valid. IgG is a T-shaped rotor, the shape of the complex is even more complicated; and the individual domains of IgG have independent degrees of freedom that allows movement of each segment.24, 25 The hydration of a complex shape will be higher than a similar hydration of a sphere: standard hydration of small proteins = 0.23 ml/g22, typical hydration of IgG = 0.59 ml/g.24 Finally, the assumption of a uniform hydration will never be true, when monitoring a binding event, as the binding will release significant numbers of surface bound water molecules in creating the protein-protein interface in the complex. All these issues can be addressed in an extensive study using ADOTA+ and selectively labeling the different domains of IgG, with this approach the rotational correlation of each part of IgG can be determined individually.
The values in Table 1 are calculated using the crude assumption that the hydration is 0 ml/g and that antiIgG and antiIgG-IgG are spherical rotors. If a density of the biomolecule of 0.75 ml/g and the hydration of 0.23 ml/g22 are used the molecular weight of the biomolecules become: MantiIgG = 144 kDa, Mcomplex = 211 kDa and MIgG = 71 kDa. The apparent weight of antiIgG itself is close to the expected 150 kDa, but the weight of the complex and IgG is underestimated due to the factors described above.
The difference in fluorescence anisotropy of the binding of antiIgG to an IgG of similar size is detectable, and this difference can be correlated to the change in size of the complex. While a large change in anisotropy upon binding of IgG to antiIgG would be preferable, the change here is clear and easily detectable on standard equipment. Developing red emitting long-lifetime probes that are less quenched on a protein surface, and thus have longer lifetimes that are even better matched to biomolecules of this size is a current focus of our research.
More detailed information on the motion of the dye-biomolecule conjugate can be extracted from time-resolved anisotropy measurements. Here, we are interested in separating the rotational diffusion of ADOTA-antiIgG from the local motion of the dye. This allows for a direct comparison of the rotational diffusion of ADOTA-antiIgG to that of the ADOTA-antiIgG-IgG complex, without having the signal polluted by the anisotropy loss arising from local motion. Figure 3 shows the correlation time of the antibody and the antiIgG-IgG complex as a function of temperature next to the same experiment using steady-state fluorescence anisotropy. The results of the two experiments are summarized in Table 1. For ADOTA-antiIgG at 20° C the anisotropy decay is bi-exponential with correlation times of θ1 = 52 ns and θ2 = 1.1 ns (see Supporting Information Figure S2 for details). After complexation with the IgG, the longer component of the correlation time increases to θ1 = 91 ns, while the shorter component increases to θ2 = 1.4 ns. θ2 is associated to the local movement of the label and is responsible for the majority of the fluorescence anisotropy loss in the labelled antiIgG. θ1 is attributed to the overall rotational motion of the antibody. That the anisotropy decay only contains a single long correlation time for both ADOTA-antiIgG and the ADOTA-antiIgG-IgG complex indicates that the overall motion of both biomolecules can be approximated to that of a sphere in solution. This simplifies the steady-state analysis and justifies the use of the Perrin equation to directly calculate the molecular weight through equation 3.22 The observation indicating a spherical rotor can be due to the random distribution of the 3–4 labels on each IgG molecule, further study will show if this is the case.
Figure 3.
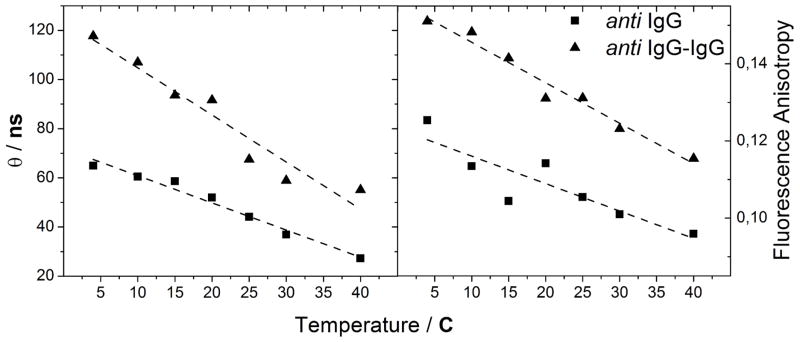
Rotational correlation time (left) from TCSPC traces, and steady-state fluorescence anisotropy (right) of ADOTA-antiIgG (squares) and ADOTA-antiIgG-IgG (triangles) as a function of temperature. Linear fits to the data are inserted to guide the eye.
The advantage of time-resolved measurements can be seen when observing the large increase in correlation time on ligand binding relative to the increase in steady-state anisotropy. The source of the relatively small change measured in steady-state anisotropy is the large anisotropy-loss due to rapid local motion of the dye. Time-resolved measurements bypass this problem by separating the contributions in the steady-state anisotropies into components. Here, the local and overall motions are on different time scales and separation of these components is easy. The advantages of the time-resolved methods are further demonstrated as the error in the measurement is an order of magnitude smaller than for the steady-state measurement after averaging over all temperatures. The absolute values of the molecular weight from these time-resolved measurements are shown in Table 1 with no hydration. If a density of the biomolecule of 0.75 ml/g and a hydration of 0.23 ml/g are used the numbers become: MantiIgG = 129 kDa and Mcomplex = 232 kDa for ADOTA-antiIgG and the ADOTA-antiIgG-IgG complex respectively. These number are significantly lower than the accepted values.26, highlighting the errors of the simplified treatment of the rotational motion and the hydration.
The time-resolved and the steady-state measurements do not agree on a molecular weight of the ligand MIgG =103 kDa and MIgG =71, demonstrating the inherent difficulty in using these methods to obtain the molecular weight of large molecules; a difficulty possibly originating in the hydration of the biomolecules. Both methods can be used for detection of binding events in molecular sensing and binding assays, where the focus is on a relative changes in a parameter rendering the actual mass change irrelevant.
Conclusion
We have synthesized the red-emitting, long lifetime fluorescent label ADOTA-NHS (4). The dye is readily conjugated to biomolecules with standard NHS-ester coupling protocols, and we have shown that the it can be used in anisotropy based binding assay; even in the very difficult case of a large biomolecules binding to a similar size ligand. We believe that this class of long-lifetime, high emission yield red dyes have great potential as biolabels both in ordinary anisotropy assay and in complex biological systems where UV/blue background emission is a major concern, but long lifetimes are desirable. We have demonstrated that detection can be achieved both by steady-state and time-resolved fluorescence anisotropy methods. The time-resolved methods give more accurate absolute values, smaller error, and a larger dynamic range. ADOTA+ is uniquely suited to this purpose with a long fluorescence lifetime (τfl = 20.1 ns) and emission in the red (λfl = 560 nm).
Methods and Materials
All chemicals were used as received. For the general synthesis of triangulenium dyes see e.g. reference 12, 13 and 14. For label synthesis and labeling procedure see reference 27 and 28.
Optical measurements
Absorption spectra were measured on a Cary 50 UV/Vis spectrograph. Steady state fluorescence spectra were measured in a Cary Eclipse Fluorescence spectrometer equipped with Cary polarizers. All measurements were performed at an angle of 90 degrees between excitation and emission, and all samples had absorbances below 0.05 at the excitation wavelength. Time resolved fluorescence measurements were done on a PicoQuant FT200 TCSPC instrument. The excitation beam was the polarized output of a 470 nm diode laser, and emission side was equipped with a rotatable Glan-Thompson prism. The TCSPC traces were analysed using the PicoQuant Fluofit package version 4.2.1. Emission quantum yields were measured using the standard procedure22 with ADOTA+ in acetonitrile as a reference. Three solutions of absorbance below 0.05 at the excitation wavelength were used for both sample and reference. Fluorescence lifetimes were measured in a PicoQuant FT200 instrument at magic angle polarization between excitation and emission using a 470 nm laser diode. Sample absorbance was kept below 0.05 at the excitation wavelength.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant: R01EB12003 and the Danish Council for Independent Research, Technology and Production Sciences (grant 10-093546).
Footnotes
Supporting Information Available: Absorption and emission spectra and temporal emission profiles are included as supporting information.
References
- 1.Owicki JC. Fluorescence polarization and anisotropy in high throughput screening: Perspectives and primer. J Biomol Screen. 2000;5:297–306. doi: 10.1177/108705710000500501. [DOI] [PubMed] [Google Scholar]
- 2.Pope AJ, Haupts UM, Moore KJ. Homogeneous fluorescence readouts for miniaturized high-throughput screening: theory and practice. Drug Discov Today. 1999;4:350–362. doi: 10.1016/s1359-6446(99)01340-9. [DOI] [PubMed] [Google Scholar]
- 3.Inglese J, Johnson RL, Simeonov A, Xia MH, Zheng W, Austin CP, Auld DS. High-throughput screening assays for the identification of chemical probes. Nature Chem Bio. 2007;3:466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 4.Giepmans BNG, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 5.Grabowski JJ, Bertozzi CR, Jacobsen JR, Jain A, Marzluff EM, Suh AY. Fluorescence Probes in Biochemistry - an Examination of the Nonfluorescent Behavior of Dansylamide by Photoacoustic Calorimetry. Anal Biochem. 1992;207:214–226. doi: 10.1016/0003-2697(92)90003-p. [DOI] [PubMed] [Google Scholar]
- 6.Grieser F, Thistlethwaite P, Urquhart R, Patterson LK. Photophysical Behavior in Spread Monolayers - Dansyl Fluorescence as a Probe for Polarity at the Air-Water-Interface. J Phys Chem. 1987;91:5286–5291. [Google Scholar]
- 7.Winnik FM. Photophysics of Preassociated Pyrenes in Aqueous Polymer-Solutions and in Other Organized Media. Chem Rev. 1993;93:587–614. [Google Scholar]
- 8.Dong DC, Winnik MA. The Py Scale of Solvent Polarities. Can J Chem. 1984;62:2560–2565. [Google Scholar]
- 9.Aubin JE. Autofluorescence of Viable Cultured Mammalian-Cells. J Histochem Cytochem. 1979;27:36–43. doi: 10.1177/27.1.220325. [DOI] [PubMed] [Google Scholar]
- 10.Benson RC, Meyer RA, Zaruba ME, Mckhann GM. Cellular Autofluorescence - Is It Due to Flavins. J Histochem Cytochem. 1979;27:44–48. doi: 10.1177/27.1.438504. [DOI] [PubMed] [Google Scholar]
- 11.Sakamoto T, Mahara A, Munaka T, Yamagata K, Iwase R, Yamaoka T, Murakami A. Time-resolved luminescence anisotropy-based detection of immunoglobulin G using long-lifetime Ru(II) complex-labeled protein A. Anal Biochem. 2004;329:142–144. doi: 10.1016/j.ab.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Laursen BW, Krebs FC. Synthesis, structure, and properties of azatriangulenium salts. Chem Eur J. 2001;7:1773–1783. doi: 10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 13.Hammershoj P, Sørensen TJ, Han BH, Laursen BW. Base-Assisted One-Pot Synthesis of N,N′,N″-Triaryltriazatriangulenium Dyes: Enhanced Fluorescence Efficiency by Steric Constraints. J Org Chem. 2012;77:5606–5612. doi: 10.1021/jo3007732. [DOI] [PubMed] [Google Scholar]
- 14.Laursen BW, Krebs FC. Synthesis of a triazatriangulenium salt. Angew Chem Int Ed. 2000;39:3432–3434. doi: 10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 15.Sørensen TJ, Laursen BW, Luchowski R, Shtoyko T, Akopova I, Gryczynski Z, Gryczynski I. Enhanced fluorescence emission of Me-ADOTA(+) by self-assembled silver nanoparticles on a gold film. Chem Phys Lett. 2009;476:46–50. doi: 10.1016/j.cplett.2009.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dileesh S, Gopidas KR. Photoinduced electron transfer in azatriangulenium salts. J Photochem Photophys A-Chem. 2004;162:115–120. [Google Scholar]
- 17.Doody MA, Baker GA, Pandey S, Bright FV. Affinity and Mobility of Polyclonal Anti-Dansyl Antibodies Sequestered within Sol-Gel-Derived Biogels. Chem Mater. 2000;12:1142–1147. [Google Scholar]
- 18.Laursen BW, Krebs FC, Nielsen MF, Bechgaard K, Christensen JB, Harrit N. 2,6,10-Tris(dialkylamino)trioxatriangulenium ions. Synthesis, structure, and properties of exceptionally stable carbenium ions. J Am Chem Soc. 1998;120:12255–12263. [Google Scholar]
- 19.Sørensen TJ, Laursen BW. Synthesis and Optical Properties of Trioxatriangulenium Dyes with One and Two Peripheral Amino Substituents. J Org Chem. 2010;75:6182–6190. doi: 10.1021/jo1009917. [DOI] [PubMed] [Google Scholar]
- 20.Martin JC, Smith RG. Factors Influencing Basicities of Triarylcarbinols Synthesis of Sesquixantheniol. J Am Chem Soc. 1964;86:2252–2256. [Google Scholar]
- 21.Thyrhaug E, Sørensen TJ, Gryczynski I, Gryczynski Z, Laursen BW. Electronic Transitions in Triangulenium Dyes. J Phys Chem A. 2013 doi: 10.1021/jp312376k. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer-Verlag New York Inc; New York: 2006. [Google Scholar]
- 23.Sørensen TJ, Hildebrandt CB, Elm J, Andreasen JW, Madsen AO, Westerlund F, Laursen BW. Large area, soft crystalline thin films of N,N′, N″-trialkyltriazatriangulenium salts with homeotropic alignment of the discotic cores in a lamellar lattice. J Mater Chem. 2012;22:4797–4805. [Google Scholar]
- 24.Carrasco B, Garcia de la Torre J, Davis KG, Jones S, Athwal D, Walters C, Burton DR, Harding SE. Crystallohydrodynamics for solving the hydration problem for multi-domain proteins: open physiological conformations for human IgG. Biophys Chem. 2001;93:181–196. doi: 10.1016/s0301-4622(01)00220-4. [DOI] [PubMed] [Google Scholar]
- 25.Hall CG, Abraham GN. Size, shape, and hydration of a self-associating human IgG myeloma protein: Axial asymmetry as a contributing factor in serum hyperviscosity. Arch Biochem Biophys. 1984;233:330–337. doi: 10.1016/0003-9861(84)90453-3. [DOI] [PubMed] [Google Scholar]
- 26.Painter RH. In: Encyclopedia of Immunology. Peter JD, editor. Elsevier; Oxford: 1998. pp. 1208–1211. [Google Scholar]
- 27.Rich RM, Stankowska DL, Maliwa lBP, Sørensen TJ, Laursen BW, Krishnamoorthy RR, Gryczynski Z, Borejdo J, Gryczynski I, Fudala R. Elimination of autofluorescence background from fluorescence tissue images using time-gated detection and the AzaDiOxaTriAngulenium (ADOTA) fluorophore. Analytical and Bioanalytical Chemistry. 2013;405:2065–2075. doi: 10.1007/s00216-012-6623-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rich RM, Mummert M, Gryczynski Z, Borejdo J, Sørensen TJ, Laursen BW, Foldes-Papp Z, 1, Gryczynsi I, Fudala R. Elimination of autofluorescence in Fluorescence Correlation Spectroscopy by using the AzaDiOxaTriAngulenium (ADOTA) fluorophore in combination with time correlated single photon counting (TCSPC) doi: 10.1007/s00216-013-6879-0. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.