

Selenium is an essential micronutrient for animals.[1] Humans contain 25 presumably essential selenoproteins[2] in which selenium is found in the form of Sec.[3] In this 21st genetically encoded amino acid[4] the thiol moiety of Cys is replaced by a selenol group. In all Sec-decoding organisms, Sec biosynthesis (Scheme 1B) starts with the acylation of tRNASec by seryl-tRNA synthetase (SerRS) to form Ser-tRNASec (reviewed in[5]). In bacteria, conversion of Ser-tRNASec to Sec-tRNASec is achieved by SelA (reviewed in[4]). In contrast, archaea and eukaryotes employ an additional phosphorylation step. O-phosphoseryl-tRNASec kinase (PSTK) phosphorylates the tRNA-bound Ser moiety of Ser-tRNASec to form O-phosphoseryl-tRNASec (Sep-tRNASec),[6] the substrate for SepSecS that catalyzes the tRNA-dependent Sep to Sec conversion.[7] The selenium donor for both SelA and SepSecS is selenophosphate (reviewed in[4, 7b]).
Scheme 1.

Aminoacyl-tRNA formation and first steps of protein synthesis. (A) Canonical amino acids: aa-tRNA gets delivered by EF-Tu to the ribosome. (B) Selenocysteine gets formed while bound to tRNA; Sec-tRNA transfer to the ribosome and accurate codon recognition are achieved by SelB (Sec-specific elongation factor) and the SECIS element (RNA structure within the open reading frame of bacterial selenoprotein mRNAs).
During selenoprotein synthesis Sec is co-translationally incorporated by a re-programmed UGA stop codon. A specialized elongation factor (SelB in bacteria) and an RNA structural signal (SECIS element) located within the bacterial ORF sequence are required for unambiguous Stop to Sec recoding.[4] EF-Tu does not recognize Sec-tRNASec and also discriminates against Ser-tRNASec.[4]
Selenium and sulfur are in the same group of elements in the periodic table and share certain properties (e.g., size, electronegativity, major oxidation states); yet, Cys and Sec are distinguished by different electrode potentials,[8] nucleophilicity (Cys < Sec),[9] and side-chain pKa (8.3 for Cys vs 5.2 for Sec).[10] Thus, selenoproteins have unique properties[11]. Sec is frequently found as an enzyme active site residue endowing these proteins (e.g., redox enzymes) with superior catalytic activities. Sec to Cys replacements in selenoenzymes may lead to 10 to 1,000-fold activity loss (reviewed in[11b]). While disulfides occur frequently in proteins to increase stability or provide redox functions, diselenides are much less frequent.[12] The occurrence of diselenides in proteins has exciting biological and biomedical significance, as they are more stable than disulfides[13] and sometimes even resistant to reduction by DTT.[12]
These Sec-dependent properties advocate that inclusion of Sec in proteins should be a promising tool in designing unique proteins for various applications (e.g., X-ray crystallography, PET studies, protein folding, NMR, electron paramagnetic resonance spectroscopy).[14] There are several current strategies to produce selenoproteins. First, the E. coli Sec insertion machinery can be exploited for heterologous overexpression of selenoproteins,[15] but its use is severely limited by sequence constraints of the SECIS sequence which inhibits facile site-directed replacement of any amino acid with Sec. Mammalian Sec insertion appears to be somewhat easier.[16] Second, solid-phase chemical synthesis of Sec-containing peptides[17] as well as site-specific chemical modification of existing proteins (e.g.,[18]) are limited by constraints on the sequence context. Finally, use of a Cys-auxotrophic E. coli strain randomly replaces up to 80% of all Cys residues in a protein with Sec.[19] Consequently, the need arises to develop a more versatile site-directed Sec insertion system.
Here we describe a system for general selenoprotein synthesis that allows site-specific insertion of Sec into any desired position of a protein. The system is based on a synthetic tRNAUTu (namely a tRNA for Sec (U) and recognized by EF-Tu) which must be a substrate for SerRS and for SelA to be converted to Sec-tRNAUTu. To obviate the need for SelB and the SECIS element, this Sec-tRNA was also designed to be carried by EF-Tu to the ribosome, where the tRNA anticodon decodes the desired mRNA codon. Thus, tRNAUTu should participate in protein synthesis like any canonical tRNA species (Scheme 1A).
We expected that our tRNAUTu design would result in some chimera of E. coli tRNASer and tRNASec (Fig. 1). Making mutations throughout the tRNA revealed that only tRNA variants with changes in the acceptor helix or the tRNA core were active SelA substrates. The tRNASec acceptor helix has an additional 8th base pair (bp); this longer acceptor helix is important for tRNASec recognition by E. coli SelA, and it precludes tRNASec from being recognized by EF-Tu.[20] However, tRNASer is a regular substrate for EF-Tu but not for SelA.[21] Thus, some elements of EF-Tu and SelA recognition could be mutually exclusive. tRNASer and other canonical tRNAs contain three bp (49–65, 50–64, 51–63; highlighted in red in Fig. 1) that provide the thermodynamic binding specificity for EF-Tu.[22] Different bases are found in those locations in tRNASec which appear to contribute to the incompatibility between tRNASec and EF-Tu.[23] Our final tRNAUTu design (Fig. 1) included anticodons that recognize the stop codons opal UGA (tRNAUTuop) and amber UAG (tRNAUTuam).
Fig. 1.

Secondary structure of E. coli tRNASer and tRNASec, and tRNAUTu. E. coli tRNASer is the major scaffold of tRNAUTu, the acceptor stem originates from tRNASec (blue), and recognition elements for EF-Tu were retained from tRNASer (red). The amber anticodon CUA (orange) is tRNAUTuam, while the opal anticodon UCA (green) defines tRNAUTuop.
Given the major challenge of designing a tRNA required to be an acceptable substrate for three major E. coli proteins, we first used in vivo experiments as definitive tests of our designs. We tested tRNAUTu variants in vivo for selenoprotein synthesis in three experiments. When grown anaerobically, E. coli produces formate dehydrogenase H (FDHH) containing an essential Sec at position 140; its activity in the presence of formate enables the cells to reduce benzyl viologen (BV) to generate a purple color.[24] An E. coli ΔselA, ΔselB, ΔfdhF strain was transformed with the appropriate combinations of plasmid-encoded selA, selB, and either an amber (fdhFUAG140, tRNAUTuam) or opal (fdhFUGA140, tRNAUTuop) reporter system for Sec insertion (Fig. 2A). The data show that tRNAUTuam when co-expressed with SelA successfully read the amber codon of fdhFam (Fig. 2A4). The BV color was almost as intense as in the positive control (Fig. 2A2), in which E. coli tRNASec in combination with the homologous Sec insertion machinery (SelA, SelB, and SECIS) translates the opal codon in fdhFop to produce active FDHH. Furthermore, no unspecific translation of fdhFam by E. coli tRNASec is observed (Fig. 2A3). Translation of fdhFop by tRNAUTuop seems to be less efficient than of fdhFam by tRNAUTuam judged by the much slower appearance of colored cells (Fig. 2A1).
Fig. 2.

(A) tRNAUTu mediates functional Sec incorporation in FDHH. An E. coli ΔselAΔselBΔfdhf deletion strain was complemented with E. coli SelA, M. jannaschii PSTK, and (A1) tRNAUTuop and FDHH op; (A4) tRNAUTuam and FDHH am. Controls lacked (A2) tRNAUTuop or (A3) tRNAUTuam, and tested FDHH op and FDHH am with E. coli tRNASec and recombinant selB. FDHH activity visualized by the purple color in the benzyl viologen assay. (B) 75Se incorporation into E. coli FDHH. The E. coli culture described in Fig. 2A4 was grown in the presence of [75Se]selenite. 6xHis-tagged FDHH protein was purified and analyzed by SDS-Page (lane 1) followed by autoradiography (lane 2); FDHH corresponds to the protein band of ~80 kDa. (C) ThyA C146U restores thymine prototrophy. An E. coli ΔselAΔselBΔthyA deletion strain was complemented with wild type thyA+, thyA146Ser or thyA146am alongside with tRNAUTuam and SelA. All clones showed growth on M9 minimal medium supplemented with thymine (C1) while only thyA146am (expressing C146U ThyA) was able to reconstitute the wild type phenotype (ThyA+) on M9 minimal medium in the absence of thymine (C2).
The second experiment demonstrated direct Sec insertion into FDHH. We incubated the culture described in Fig. 2A1 with [75Se]selenite and ran the extracted protein on SDS-PAGE (Fig. 2B). The appearance of a radioactive 80kDa band corresponds to 75Se-labeled FDHH. The second radioactive band (~40 kDA) is an FDHH degradation product as described earlier.[7a]
Thirdly, we functionally replaced Cys in an enzyme active site with Sec. Thymidylate synthase (ThyA) catalyzes the conversion of deoxyuridylate to thymidylate, and its thyA gene in contrast to fdhF is devoid of a SECIS element. An E. coli ΔthyA strain that carries a deletion of the endogenous thyA gene is thymine auxotrophic. The enzyme’s active site residue at position 146 is Cys. Replacement by Ser reduces activity 5,000-fold.[25] To check whether Cys146 can be replaced by Sec we transformed an E. coli ΔthyA, ΔselA, ΔselB strain with the requisite plasmids and checked for growth on minimal medium in presence and absence of thymine (Fig. 2C). As expected complementation with thyA+ and with thyA146am/tRNAUTuam restored prototrophic growth, while thyA146Ser or empty vector did not. Thus, tRNAUTuam permitted insertion of Sec which takes on the function of the active site Cys. Taken together, the three experiments above established the suitability of tRNAUTu for UAG-directed Sec insertion.
We endeavored to determine the efficiency and accuracy of the current system and to discover which of the components (SerRS, SelA, EF-Tu, or tRNAUTu) might be limiting the reaction. Serylation kinetics with pure E. coli SerRS (Table S1) revealed that tRNAUTuam is as good a substrate as tRNASec and tRNASerUCA; thus serylation of tRNAUTu is no impediment. Given that Sec-tRNAUTu is formed from a Ser-tRNAUTu intermediate, it is possible that some Ser misincorporation may occur. This was no concern in the cases of ThyA and FDHH which are inactive when Ser is present in place of Sec.
As a SelA in vitro experiment showed that the designed Ser-tRNAUTuam is less efficiently converted to Sec-tRNA than the natural substrate for SelA Ser-tRNASer (Fig. S1), we included PSTK (the eukaryotic kinase that converts Ser-tRNASec to Sep-tRNASec) to our reaction mixtures; in this way we expected PSTK to reduce the amount of Ser-tRNAUTu by converting it to Sep-tRNAUTu (Fig. S2). This aminoacyl-tRNA that will not bind to EF-Tu[26] and thus not be transported to the ribosome, while it is a likely substrate for SelA-dependent conversion to Sec-tRNAUTu (Fig. S2B, lanes 2,3).[7c] Ser incorporation may also be due to preferential binding of EF-Tu to Ser-tRNAUTu compared to Sec-tRNAUTu. However, Thiol-Sepharose might afford a separation of a Sec-containing protein from a mixture with its Ser homolog; therefore we proceeded with tRNAUTu-mediated Sec incorporation into a bacterial and a human test protein.
Glutaredoxins are glutathione-dependent reductases that regulate the cellular redox state.[27] E. coli glutaredoxin (Grx1) is a small (85 amino acids) redox-active disulfide (C11/C14)-containing monomeric protein with glutathione-disulfide oxidoreductase activity.[28] Grx1 can be easily purified by Thiol-Sepharose chromatography[29] and has been extensively studied.[27] Partial chemical synthesis afforded the glutathione peroxidase mimic selenoglutaredoxin, a Sec-containing Grx1 variant (C11U/C14S), that was analyzed for its ability to catalyze thiol-disulfide exchange reactions, as well as for its peroxidase activity.[30] This selenoprotein showed superior catalytic properties compared to its Cys homolog (C11/C14S).[30] Thus, we decided to biosynthesize selenoglutaredoxin by tRNAUTu-mediated overexpression in E. coli followed by purification on activated Thiol-Sepharose,[29] in order to compare its properties to the one described earlier.[30] In this way we obtained the Grx1 variants C11U/C14S and C11S/C14S in pure form in about 50% yield (Fig. S3). Incorporation of Sec at position 11 was confirmed by mass spectroscopy (Fig. 3, Fig. S4). The analysis also showed the selenoglutaredoxin to be free of the C11S/C14S Grx1 variant.
Fig. 3.

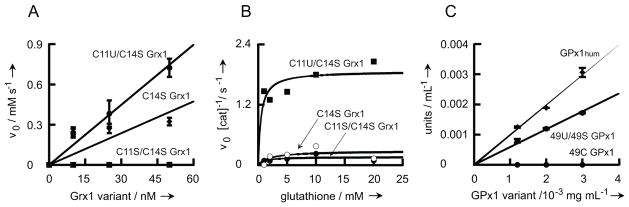
Characterization of Grx1 and GPx1 mutants. (A) Glutathione disulfide oxidoreductase activity of Grx1 variants. Pure C11U/C14S Grx1, C11S/C14S Grx1 and C11/C14S Grx1 were tested for disulfide oxidoreductase activity. NADPH consumption was followed at 340 nm as a function of Grx1 concentration. (B) Peroxidase activity of Grx1 variants. Pure C11U/C14S Grx1, C11S/C14S Grx1 and C11/C14S Grx1 were tested for peroxidase activity. NADPH consumption was monitored at 340 nm as a function of reduced glutathione concentration. (C) GPx1 peroxidase activity. Peroxidase activity of recombinant Sec containing 49U GPx1 and Cys containing 49C GPx1 was compared to GPx1hum from human erythrocytes. GPx1 activity was determined with the Sigma cellular activity assay kit. Experiments shown in figures A & C were performed in triplicate, and bars indicate the standard error of the mean.
Varying the conditions of protein expression, such as gene dosage or time of selenoprotein induction (see Supplementary Text and Fig. S5), increased selenoprotein yield to about 65%, while the remainder of the protein mixture was the Ser homolog. In certain cases (e.g., Grx1) Thiol-Sepharose chromatography will effectively separate the mixture.
Comparison of our selenoglutaredoxin with its Cys (C11/C14S) and Ser (C11S/C14S) homologs was undertaken using two standard assays. Like wild-type Grx1, C14S Grx1 efficiently reduces glutathionyl mixed disulfides;[31] this is measured by the thiol-disulfide interchange between β-hydroxyethyldisulfide and glutathione.[28] Our selenoglutaredoxin exhibited improved activity over that of the Cys homolog C14S Grx1 (90 vs 47 units/mg) (Fig. 4A). The values agree well with previously published data.[30] No activity above background could be detected for the Ser homolog C11S/C14S Grx1.
Fig. 4.
Mass spectroscopic confirmation of Sec incorporation. The presence of selenocysteine at amino acid position 11 in pure C11U/C14S Grx1 was confirmed by mass spectroscopy. Shown is the MS/MS spectrum of the trypsin-digested Sec-containing fragment S9G10U11P12Y13S14V15R16. Fragments observed in the second mass spectrometric analysis of this peptide are labeled b3, y2, y3, y4 and y5. The unit m/z describes the mass-to-charge ratio.
Selenoglutaredoxin functions inefficiently as a glutathione peroxidase (GPx),[30] catalyzing the reduction of peroxides to the corresponding alcohols by glutathione.[32] Consequently, we also examined the GPx activity of our selenoglutaredoxin and its Cys and Ser homologs. C11U/C14S Grx1 showed modest peroxidase activity (~2×10−3 s−1) in accordance with published data.[30] Both, C11S/C14S Grx1 and C14S Grx1 were significantly less active (Fig. 4B).
Human glutathione peroxidase (GPx1), a key component of the mammalian antioxidant defense, is a 217 amino acid selenoprotein with an essential active site Sec residue (position 49); replacement by Cys leads to 1000-fold activity loss, while Ser substitution renders the enzyme inactive.[33] Synthesis of eukaryotic selenoproteins (e.g., GPx1) relies on a SECIS element in the 3′ untranslated mRNA.[16a] Not surprisingly, human Sec-containing GPx1 has not been expressed in E. coli. Therefore we attempted to produce human GPx1 by tRNAUTu-mediated expression in E. coli. This yielded a mixture of two GPx1 variants, U49 GPx1 and U49S GPx1 in approximately equal amounts as deduced from mass spec via peak integration (Fig. S6). Glutathione peroxidase activity of the protein mixture was 6 units/mg, which is similar to 10 units/mg determined for a commercially available pure human GPx1 preparation. The Cys homolog U49C GPx1 was not active under these conditions (Fig. 4C).
The data presented here reveal the success of tRNAUTu mediated site-specific Sec incorporation as exemplified by four selenoproteins, three of bacterial and one of human origin. The remaining challenge is to achieve complete conversion of tRNAUTu-bound Ser to Sec. Structural knowledge of tRNA complexes with SelA and with EF-Tu will inspire further rewiring of translation to facilitate optimal EF-Tu-dependent Sec incorporation. We successfully uncoupled selenoprotein synthesis from the SECIS/SelB-dependent codon reading; our tRNA design efforts establish that Sec can be accommodated to ‘normal’ mRNA translation as if it were a canonical amino acid. Our system has general utility in protein engineering, molecular biology, and disease research.
Supplementary Material
Footnotes
We thank David Lewin, Lynn Sherrer, Patrick O’Donoghue for enthusiastic discussions, and Dan Su for experimental advice. We are grateful to TuKiet Lam and Edward Voss (W.M. Keck Foundation Biotechnology Resource Laboratory, Yale University) for their help with the FT-ICR mass spectral analyses. M.J.H. and M.J.B. acknowledge Feodor Lynen Postdoctoral Fellowships from the Alexander von Humboldt Foundation (Bonn, Germany). C.A. was a Postdoctoral Fellow of the Swiss National Science Foundation. This work was supported by grants from the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, DOE (DE-FG02-98ER20311), from the National Institute of General Medical Sciences (GM 22854), and by DARPA contract N66001-12-C-4020.
References
- 1.Rayman MP. Lancet. 2000;356:233–241. doi: 10.1016/S0140-6736(00)02490-9. [DOI] [PubMed] [Google Scholar]
- 2.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 3.Cone JE, Del Rio RM, Davis JN, Stadtman TC. Proc Natl Acad Sci U S A. 1976;73:2659–2663. doi: 10.1073/pnas.73.8.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Böck A, Thanbichler M, Rother M, Resch A. In: Aminoacyl-tRNA synthetases. Ibba M, Franklyn CS, Cusack S, editors. Landes Bioscience; Georgetown, TX: 2005. pp. 320–327. [Google Scholar]
- 5.Ambrogelly A, Palioura S, Söll D. Nat Chem Biol. 2007;3:29–35. doi: 10.1038/nchembio847. [DOI] [PubMed] [Google Scholar]
- 6.a Chiba S, Itoh Y, Sekine S, Yokoyama S. Mol Cell. 2010;39:410–420. doi: 10.1016/j.molcel.2010.07.018. [DOI] [PubMed] [Google Scholar]; b Sherrer RL, O’Donoghue P, Söll D. Nucleic Acids Res. 2008;36:1247–1259. doi: 10.1093/nar/gkm1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Yuan J, Palioura S, Salazar JC, Su D, O’Donoghue P, Hohn MJ, Cardoso AM, Whitman WB, Söll D. Proc Natl Acad Sci U S A. 2006;103:18923–18927. doi: 10.1073/pnas.0609703104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Palioura S, Sherrer RL, Steitz TA, Söll D, Simonovic M. Science. 2009;325:321–325. doi: 10.1126/science.1173755. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Xu XM, Carlson BA, Mix H, Zhang Y, Saira K, Glass RS, Berry MJ, Gladyshev VN, Hatfield DL. PLoS Biol. 2007;5:4. doi: 10.1371/journal.pbio.0050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nauser T, Dockheer S, Kissner R, Koppenol WH. Biochemistry. 2006;45:6038–6043. doi: 10.1021/bi0602260. [DOI] [PubMed] [Google Scholar]
- 9.Singh R, Whitesides GM. J Org Chem. 1991;56:6931–6933. [Google Scholar]
- 10.Huber RE, Criddle RS. Arch Biochem Biophys. 1967;122:164–173. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
- 11.a Arner ES. Exp Cell Res. 2010;316:1296–1303. doi: 10.1016/j.yexcr.2010.02.032. [DOI] [PubMed] [Google Scholar]; b Beld J, Woycechowsky KJ, Hilvert D. In: Oxidative Folding of Peptides and Proteins. Buchner J, Moroder L, editors. The Royal Society of Chemistry; 2009. pp. 253–273. [Google Scholar]
- 12.Shchedrina VA, Novoselov SV, Malinouski MY, Gladyshev VN. Proc Natl Acad Sci U S A. 2007;104:13919–13924. doi: 10.1073/pnas.0703448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a Beld J, Woycechowsky KJ, Hilvert D. Biochemistry. 2007;46:5382–5390. doi: 10.1021/bi700124p. [DOI] [PubMed] [Google Scholar]; b Metanis N, Hilvert D. Angew Chem Int Ed Engl. 2012;51:5585–5588. doi: 10.1002/anie.201109129. [DOI] [PubMed] [Google Scholar]
- 14.Johansson L, Gafvelin G, Arner ES. Biochim Biophys Acta. 2005;1726:1–13. doi: 10.1016/j.bbagen.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 15.a Aldag C, Gromov IA, Garcia-Rubio I, von Koenig K, Schlichting I, Jaun B, Hilvert D. Proc Natl Acad Sci U S A. 2009;106:5481–5486. doi: 10.1073/pnas.0810503106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Arner ES. Meth Enzymol. 2002;347:226–235. doi: 10.1016/s0076-6879(02)47022-x. [DOI] [PubMed] [Google Scholar]
- 16.a Novoselov SV, Lobanov AV, Hua D, Kasaikina MV, Hatfield DL, Gladyshev VN. Proc Natl Acad Sci U S A. 2007;104:7857–7862. doi: 10.1073/pnas.0610683104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu H, Yin L, Board PG, Han X, Fan Z, Fang J, Lu Z, Zhang Y, Wei J. Protein Expr Purif. 2012;84:59–63. doi: 10.1016/j.pep.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 17.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 18.Wu ZP, Hilvert D. J Am Chem Soc. 1989;111:4513–4514. [Google Scholar]
- 19.Johansson L, Chen C, Thorell JO, Fredriksson A, Stone-Elander S, Gafvelin G, Arner ES. Nat Methods. 2004;1:61–66. doi: 10.1038/nmeth707. [DOI] [PubMed] [Google Scholar]
- 20.Baron C, Böck A. J Biol Chem. 1991;266:20375–20379. [PubMed] [Google Scholar]
- 21.Forchhammer K, Böck A. J Biol Chem. 1991;266:6324–6328. [PubMed] [Google Scholar]
- 22.Schrader JM, Chapman SJ, Uhlenbeck OC. J Mol Biol. 2009;386:1255–1264. doi: 10.1016/j.jmb.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudinger J, Hillenbrandt R, Sprinzl M, Giege R. EMBO J. 1996;15:650–657. [PMC free article] [PubMed] [Google Scholar]
- 24.Lacourciere GM, Levine RL, Stadtman TC. Proc Natl Acad Sci U S A. 2002;99:9150–9153. doi: 10.1073/pnas.142291199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dev IK, Yates BB, Leong J, Dallas WS. Proc Natl Acad Sci U S A. 1988;85:1472–1476. doi: 10.1073/pnas.85.5.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Soll D. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lillig CH, Berndt C, Holmgren A. Biochim Biophys Acta. 2008;1780:1304–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Holmgren A, Åslund F. Methods Enzymol. 1995;252:283–292. doi: 10.1016/0076-6879(95)52031-7. [DOI] [PubMed] [Google Scholar]
- 29.Åslund F, Ehn B, Miranda-Vizuete A, Pueyo C, Holmgren A. Proc Natl Acad Sci U S A. 1994;91:9813–9817. doi: 10.1073/pnas.91.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casi G, Roelfes G, Hilvert D. Chembiochem. 2008;9:1623–1631. doi: 10.1002/cbic.200700745. [DOI] [PubMed] [Google Scholar]
- 31.Bushweller JH, Aslund F, Wüthrich K, Holmgren A. Biochemistry. 1992;31:9288–9293. doi: 10.1021/bi00153a023. [DOI] [PubMed] [Google Scholar]
- 32.Ursini F, Maiorino M, Brigeliusflohe R, Aumann KD, Roveri A, Schomburg D, Flohe L. Methods Enzymol. 1995;252:38–53. doi: 10.1016/0076-6879(95)52007-4. [DOI] [PubMed] [Google Scholar]
- 33.Rocher C, Lalanne JL, Chaudiere J. Eur J Biochem. 1992;205:955–960. doi: 10.1111/j.1432-1033.1992.tb16862.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.