

Abstract
Endocannabinoids play essential roles in synaptic plasticity; thus, their dysfunction often causes impairments in memory or cognition. However, it is not well understood whether deficits in the endocannabinoid system account for the cognitive symptoms of schizophrenia. Here, we show that endocannabinoid-mediated synaptic regulation is impaired by the prolonged elevation of neuregulin-1, the abnormality of which is a hallmark in many patients with schizophrenia. When rat hippocampal slices were chronically treated with neuregulin-1, the degradation of 2-arachidonoylglycerol (2-AG), one of the major endocannabinoids, was enhanced due to the increased expression of its degradative enzyme, monoacylglycerol lipase. As a result, the time course of depolarization-induced 2-AG signaling was shortened, and the magnitude of 2-AG-dependent long-term depression of inhibitory synapses was reduced. Our study reveals that an alteration in the signaling of 2-AG contributes to hippocampal synaptic dysfunction in a hyper-neuregulin-1 condition and thus provides novel insights into potential schizophrenic therapeutics that target the endocannabinoid system.
Introduction
Endocannabinoids (eCBs) are involved in cognitive and emotional behaviors via the regulation of synaptic plasticity (Zanettini et al., 2011; Castillo et al., 2012). Therefore, the dysfunction of the eCB system is implicated in many psychiatric disorders; however, the role of eCBs in schizophrenia is unclear (Marco et al., 2011). eCBs are released from neurons upon an increase in intracellular calcium and/or the activation of G-proteins. Then, eCBs stimulate presynaptic type 1 cannabinoid receptors (CB1Rs) to block neurotransmission, before being taken up into cells for enzymatic degradation. CB1Rs are widely expressed in the brain, including areas involved in schizophrenia, such as the prefrontal cortex and hippocampus. Alterations in the eCB system are found in some schizophrenic patients and animal models, implying the involvement of eCBs in this disease (Giuffrida et al., 2004; D'Souza et al., 2005; Boucher et al., 2007b; Vigano et al., 2009); however, the pathological mechanisms of eCBs are uncertain.
The expression and function of neuregulin-1 (NRG1), a growth factor, and its receptor, ErbB receptor tyrosine kinase, are often altered (either increased or decreased) in many patients with schizophrenia (Mei and Xiong, 2008; Banerjee et al., 2010; Buonanno, 2010; Rico and Marin, 2011). These observations suggest that a normal range of NRG1-ErbB signaling is essential for cognitive integrity. NRG1 is expressed primarily in glutamatergic neurons and also in interneurons and astrocytes (Bernstein et al., 2006; Liu et al., 2011). Among four types of ErbB (ErbB1-4), only ErbB4 both binds to NRG1 and possesses an active tyrosine kinase domain; additionally, ErbB4 is the major ErbB that has been implicated in schizophrenia (Mei and Xiong, 2008; Banerjee et al., 2010; Buonanno, 2010). ErbB4 in the brain is expressed largely in various types of γ-amino acid butyric acid (GABA)-ergic interneurons (Yau et al., 2003; Vullhorst et al., 2009; Neddens and Buonanno, 2010). In the stratum radiatum of the hippocampal CA1 area, 20% and 6% of ErbB4-expressing interneurons coexpress cholecystokinin (CCK) and parvalbumin (PV), respectively (Neddens and Buonanno, 2010). In contrast, PV interneurons compose 50–60% of ErbB4-expressing cells in the hippocampal dentate hilus (Neddens and Buonanno, 2010) and neocortex (Abe et al., 2011).
Despite the diversity of interneurons, studies on the NRG1-ErbB4 function in interneurons have been limited to PV interneurons (Chen et al., 2010; Wen et al., 2010; Li et al., 2012; Shamir et al., 2012; Tan et al., 2012), in part because of the high incidence of PV/ErbB4 coexpression in some brain areas. Along with PV cells, CCK-expressing interneurons form a major basket cell population. Among many differences between PV and CCK interneurons, the presence of CB1Rs in CCK, but not PV, interneurons (Katona et al., 1999) is one of the sharpest contrasts. Because NRG1-ErbB4 signaling in CCK cells is not well known, the role of eCBs in NRG1-mediated pathology remains elusive.
Here, we report that the chronic elevation of NRG1 in hippocampal slice cultures curtails the action of 2-arachidonolyglycerol (2-AG), one of the major eCBs, by increasing the expression of a 2-AG degradative enzyme, monoacylglycerol lipase (MGL).
Materials and Methods
Hippocampal slice culture.
Organotypic slice cultures were prepared from isolated hippocampi of 14- to 15-day-old male Sprague Dawley rats (Harlan Laboratories) that were isoflurane-anesthetized and decapitated. Slices (350 μm thick) were made with a vibrating slicer (Leica VT1200S) in ice-cold saline, which consisted of 108 mm NaCl, 2.5 mm KCl, 45 mm NaHCO3, 1 mm NaH2PO4, 0.5 mm CaCl2, 5 mm MgSO4, 12 mm glucose, and 0.5 mm ascorbic acid (300–305 mOsm), and were bubbled with carbogen (95% O2 and 5% CO2). After washing the slices with 37°C culture medium, we placed them on culture membranes (Millipore) at the interface of the culture medium and air at 37°C. The medium was exchanged every 2–3 d and was composed of 50% Basal Medium Eagle, 25% Earle's salt solution, 25% horse serum, 2 mm l-glutamine, 10 mm HEPES, and additional 5 mm glucose. The protocol was approved by the Institutional Animal Care and Use Committee of Georgia Regents University.
Electrophysiology.
Recordings were made from hippocampal slice cultures at 19–25 DIV. The treatment of slice cultures with NRG1 (5 nm; β-type epidermal growth factor domain) started as early as 11 DIV and lasted 8–11 d, except for the experiment illustrated in Figure 2C, D. In all electrophysiological experiments, we blocked the evoked GABA release from CB1R-negative synapses by preincubating slices for 15–30 min with 300 nm ω-agatoxin IVA (Wilson et al., 2001), an irreversible inhibitor of P/Q-type calcium channels. Whole-cell voltage-clamp recordings from CA1 pyramidal neurons were made at a holding potential of −65 mV at 32 ± 0.5°C while the recording chamber was perfused with bath solution at 1.6–1.8 ml/min. The bath solution contained 128 mm NaCl, 2.5 mm KCl, 26 mm NaHCO3, 1 mm NaH2PO4, 2 mm CaCl2, 2 mm MgSO4, and 12 mm glucose (300–305 mOsm) and was equilibrated with carbogen. The electrode resistance in the bath solution was 3–5 MΩ, and the series resistance (<20 MΩ) was stable within 15%. Signals were amplified by a Multiclamp 700B amplifier (Molecular Devices), filtered at 2 kHz, and digitized at 50 kHz by a Digidata 1440A and the Clampex 10 program (Molecular Devices). The pipette solution contained 129 mm K-gluconate, 2 mm NaCl, 1 mm QX314-Cl, 4 mm MgSO4, 0.2 mm CaCl2, 2 mm EGTA, 3 mm ATP-Na, 0.3 mm GTP-Na, 10 mm HEPES, and 10 mm phosphocreatine di(tris) (pH 7.2 with ∼15 mm KOH, 290–295 mOsm). In the experiments of the long-term depression of inhibitory synapses (iLTD), CaCl2 and EGTA in the pipette solution were replaced with 20 mm BAPTA, and K-gluconate was reduced to 86 mm. We used 20 mm BAPTA in the iLTD pipette solution because a high concentration of calcium buffer does not affect iLTD (Chevaleyre and Castillo, 2003) but blocks the metabotropic glutamate receptor (mGluR)-dependent GABAergic long-term potentiation (LTP) (Patenaude et al., 2003), which might obscure the expression of iLTD, as was occasionally observed with 2 mm EGTA in pipettes in our pilot experiment (data not shown).
Figure 2.

The chronic blockade of ErbB or short-term treatment with NRG1 has no effect on DSI. A, eIPSCs were recorded from cells that were treated with either vehicle or PD158780 (10 μm) for 8–11 d. The time after depolarization (0 mV, 5 s) is indicated above the traces. B, The group data of DSI from vehicle- or PD158780-treated cells. Normalized eIPSC amplitudes of the two groups were not different from each other. C, The short-term (1–3 d) treatment of slice cultures with 5 nm NRG1 did not alter DSI. D, The normalized eIPSC amplitude in NRG1-treated cells was similar to that in control cells during the recovery from DSI. E, The time constant of DSI decay in PD158780-treated cells was not different from that in sister control cells. After 1–3 d of treatment with NRG1, the time constant was also similar to that of sister control cells. Error bars indicate SEM.
The evoked IPSC (eIPSC) was recorded in the presence of NBQX (5 μm) and CGP37849 (5 μm) and was elicited by a stimulus via a θ glass electrode (10–20 μm tip diameter) that was filled with the bath solution and was within the CA1 stratum pyramidale. The presynaptic stimulus was delivered every 6 s in the depolarization-induced suppression of inhibition (DSI) experiments and every 12 s in all other eIPSC recordings. The vehicle for most cannabinoid drugs was ethanol, which has little effect on eIPSC amplitudes (Kim and Alger, 2010): 0.01–0.036% (v/v) for methanandamide (mAEA), 0.01% for SR141716, 0.01–0.02% for AM404, and 0.04% for PD158780. We prepared the JZL184 stock solution by dissolving JZL184 in DMSO to 100 mm and then diluting to 10 mm with ethanol. When slices were treated with both PD158780 and NRG1, PD158780 preceded NRG1 by 0.5–1 h. NBQX, CGP37849, gabazine, and mAEA were purchased from Abcam; NRG1, AM404, DHPG, and PD158780 were from R&D Systems; URB597 was from Cayman Chemical; and JZL184 was from R&D Systems or Cayman Chemical. ω-Agatoxin IVA was from Peptide International. Basal Medium Eagle and horse serum were from Invitrogen. Other chemicals were obtained from Sigma-Aldrich. SR141716 was a gift from the National Institute on Drug Abuse.
Western blot.
Proteins for one set of Western blot experiments were extracted from six cultured slices. After being washed with carbogenated bath solution, slices were transferred to RIPA buffer, which was composed of 150 mm NaCl, 1% NP-40, 0.25% deoxycholate, 0.1% SDS, 50 mm Tris, and 1 mm EDTA (pH 8.0; supplemented with protease inhibitor mixture), and were homogenized by sonication for 30 s at 2–5°C. The homogenates were centrifuged for 10 min at 4°C. The protein samples were denatured at 70°C for 5 min, run on a SDS-PAGE gel, and then transferred onto nitrocellulose membrane (Bio-Rad). The membranes were washed with PBS containing Tween 20, blocked with 5% BSA in Tween PBS, and probed with one of the primary antibodies: rabbit anti-MGL (1:500) or rabbit anti-CB1R (1:500) antibody (Cayman Chemical). After overnight probing at 4°C, the membranes were washed with Tween PBS for 5 × 5 min and were incubated with HRP-conjugated anti-rabbit IgG (1:3000; Bio-Rad) for 1 h at 22–24°C. Bands were visualized with a chemiluminescent detection system (Pierce). Then, the membranes were reused for two cycles of stripping/reprobing with an unused primary antibody (MGL or CB1R) or rabbit anti-β-actin antibody (1:100; Santa Cruz Biotechnology). Western blot films were scanned on a film scanner (Epson Perfection V700) at 1200 DPI, and the grayscale of bands were measured with ImageJ software (National Institutes of Health). Band intensities, after being subtracted by band-free background intensity, were normalized to the β-actin signal from the same lanes. In each experiment, the signals of NRG1-treated slices were normalized to that of sister control slices.
Data analysis.
All DSI experiments were performed >15 min after the establishment of a whole-cell recording for the stabilization of the intracellular condition. Postsynaptic cells were depolarized every 3 min; and for a given cell, 2–5 DSI trials were averaged, except for the DSIs in the presence of JZL184 and AM404; we applied JZL184 or AM404 for 20–30 min without depolarization to avoid any potential accumulation of eCBs in the extracellular space and then recorded 1 or 2 DSI episodes. The first IPSC after each depolarization was evoked 2 s after the termination of depolarization in all DSI experiments. A recovery of the eIPSC amplitudes from DSI was fitted with an exponential function, f(t) = A e−t/τ + C, where t is the time of eIPSC relative to the first postdepolarization eIPSC, τ is a time constant, and A and C are adjustment variables. The fitting was performed in a 90 s range that started from the first postdepolarization eIPSC, except for the DSIs in JZL184. In the presence of JZL184, because the initiation of recovery from DSI was delayed by ∼5 s, the fitting was conducted for a 100 s range that started from the second eIPSC. The peak amplitudes of eIPSCs were measured over a 0.4 ms window. Drug-induced changes in eIPSC amplitudes were measured when the changes reached a steady state: 10–13 min after the onset of SR141716, 15–20 min after AM404, 12–15 min after 0.4 μm DHPG, 47–52 min after 50 μm DHPG for iLTD, and at variable time points determined blindly for mAEA. For short-term plasticity, the mean amplitude of the second or third eIPSC in a 20 Hz train was normalized to the mean of the first amplitude; the mean amplitudes were obtained from 15–70 eIPSCs. Stimulus artifacts of eIPSCs were graphically truncated in the figures for clarity.
The data from NRG1-treated slices were compared only with those from sister control slices, except for the analysis in Figure 6D (i.e., comparisons with non-JZL184 control cells). Comparisons between two groups were made using t tests with a two-tailed confidence level of p < 0.05. Multiple comparisons were performed with one- or two-way ANOVA. When ANOVA revealed a significant difference (p < 0.05), Bonferroni t tests were used for pairwise comparisons.
Figure 6.
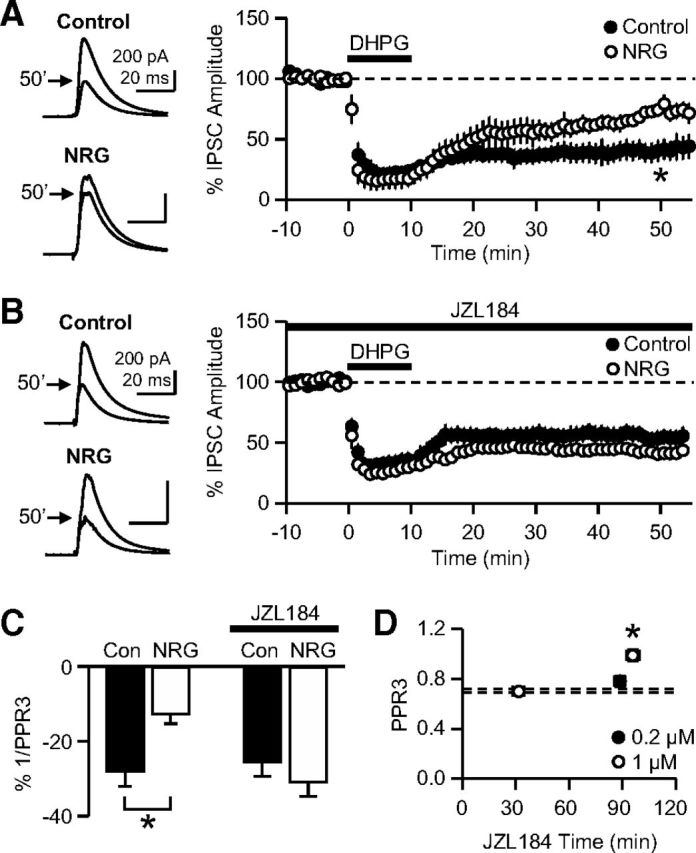
The chronic treatment with NRG1 impairs iLTD. A, iLTD was induced by a 10 min application of DHPG (50 μm), a group I mGluR agonist. The magnitude of iLTD at 47–52 min in NRG1-treated cells was significantly smaller than that in control cells. *p = 0.008 (t test). B, The iLTD experiments were performed in the presence of JZL184 (0.2 μm) in the extracellular solution. The magnitude of iLTD at 47–52 min in NRG1-treated neurons was not significantly different from the control value. C, An iLTD-induced decrease in 1/PPR3 (PPR3 = third IPSC/first IPSC evoked at 20 Hz), as an indication of relative Pr, was significantly smaller in NRG1-treated cells than in sister control cells. *p = 0.015 (t test). In JZL184, there was no difference in the iLTD-induced decrease in 1/PPR3 between the two groups. D, In control slices (i.e., not treated with NRG1), PPR3 was measured after various periods of JZL184 application at 0.2 or 1 μm. Dashed lines indicate the PPR3 (mean ± SEM) of control slices without JZL184 (replotted from Fig. 5C). *p < 0.005 (Bonferroni t tests vs other data points, including the non-JZL184 control, after ANOVA). Error bars indicate SEM.
Results
Chronic treatment with NRG1 shortens the duration of DSI
To chronically increase NRG1 signaling, we treated hippocampal slice cultures for 8–11 d with NRG1 peptide at 5 nm, which is the maximum dose for in vitro effects (Woo et al., 2007). The slice culture method was well suited to our study because (1) NRG1 could be homogeneously applied to all cells; (2) only the effects directly on the hippocampus could be examined; and (3) life-long compensation, which could occur in transgenic mice, was minimized. The function of 2-AG was assayed as DSI, which is 2-AG-mediated retrograde signaling onto CB1R-positive GABAergic terminals (Castillo et al., 2012). Monosynaptic eIPSCs were recorded from CA1 pyramidal neurons after NRG1 was washed from slices for 0.5–1 h (Fig. 1A–E).
Figure 1.
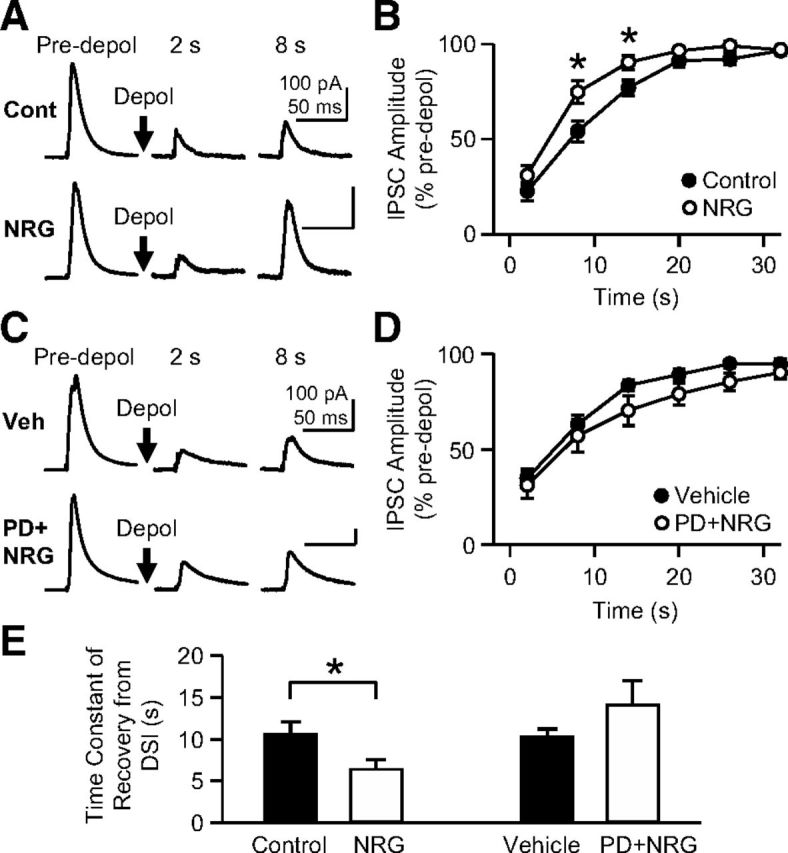
The chronic treatment of hippocampal slice cultures with NRG1 (5 nm, 8–11 d) shortens the duration of DSI. A, eIPSCs were recorded from CA1 pyramidal neurons, and the recovery of the amplitude after depolarization (0 mV, 5 s) was examined in control and NRG1-treated slices. The time after depolarization is indicated above the traces. B, Mean eIPSC amplitudes normalized to the predepolarization baseline were plotted against the time after the end of depolarization. *p < 0.05 (Bonferroni t test after two-way ANOVA for the range of 2–20 s). C, PD158780, an ErbB inhibitor, blocked the effect of NRG1 on the eIPSC recovery from the DSI (0 mV, 5 s). Slices were treated with 10 μm PD158780 and 5 nm NRG1 or only with the vehicle for PD158780 without NRG1 for 8–11 d. D, The group data of DSI from vehicle- or PD158780/NRG1-treated cells. There was no significant difference in eIPSC amplitudes between the two groups in the range of 2–20 s. E, The time constant of DSI decay, when obtained from a single exponential fitting, in NRG1-treated cells was significantly smaller than that in sister control cells. *p = 0.046 (t test). The time constant in PD158780/NRG1-treated cells was not different from that in vehicle-treated cells. Error bars indicate SEM.
Two seconds after the depolarization (to 0 mV for 5 s) of postsynaptic cells, eIPSC amplitudes in NRG1-treated cells were suppressed to 31 ± 5% (n = 10) of the predepolarization baseline, which was similar to that in control cells in sister cultures (22 ± 5%; n = 11) (p = 0.19, Bonferroni t test after two-way ANOVA; Fig. 1A,B). However, 8 s after depolarization, the eIPSCs in NRG1-treated slices recovered to 75 ± 6% of the baseline, whereas the control eIPSCs rose only to 54 ± 6% of the baseline (p = 0.002, Bonferroni t test; Fig. 1A,B). The difference between control and NRG1 groups persisted at 14 s after depolarization (p = 0.039, Bonferroni t test; Fig. 1B). The NRG1-induced faster recovery from DSI was also evident when the time constant of the eIPSC recovery, which was obtained from a single exponential curve fitting, was compared; the time constant was 10.6 ± 1.5 s in control cells and 6.4 ± 1.2 s in NRG1-treated cells (p = 0.046, t test; Fig. 1E). This result indicates that the chronic treatment with NRG1 curtails depolarization-induced 2-AG signaling in hippocampal pyramidal neurons.
We tested whether the effect of NRG1 on DSI was mediated by ErbB receptors. Slice cultures were treated with both NRG1 (5 nm) and PD158780 (10 μm), an inhibitor of ErbB1, ErbB2, and ErbB4, for 8–11 d. In PD158780/NRG1-treated cells (n = 12), NRG1 failed to shorten the duration of DSI compared with that in control cells (n = 10), which were treated with the vehicle for PD158780 (without NRG1) (p = 0.066, two-way ANOVA; Fig. 1C,D). The time constant of recovery from DSI in the PD158780/NRG1 group (14.1 ± 2.9 s) was also similar to that in the vehicle group (10.2 ± 1.0 s) (p = 0.26, t test; Fig. 1E). This result suggests that the NRG1 effect on DSI is mediated by ErbB receptors.
We next examined whether a milder action of NRG1, by either endogenous low levels of NRG1 or a short-term treatment with NRG1, could modulate DSI (Fig. 2A–E). Because both upregulation and downregulation of NRG1 signaling can induce schizophrenia phenotypes in humans (Hashimoto et al., 2004; Petryshen et al., 2005; Hahn et al., 2006; Law et al., 2006, 2007; Bertram et al., 2007; Chong et al., 2008; Barakat et al., 2010; Shibuya et al., 2010) and rodents (Ehrlichman et al., 2009; Chen et al., 2010; Duffy et al., 2010; Kato et al., 2010; Deakin et al., 2012), we hypothesized that the chronic blockade of ErbB, as a model of the hypofunction of endogenous basal NRG1, might also alter the recovery from DSI. When slice cultures were treated with 10 μm PD158780 (without NRG1), the time course of DSI decay (n = 8) was similar to that of sister control cells (n = 8) (p = 0.51, two-way ANOVA; Fig. 2A,B). Additionally, the time constant of DSI decay in PD158780-treated cells (13.3 ± 0.7 s) was similar to that in sister control cells (13.8 ± 1.0 s) (p = 0.70, t test; Fig. 2E). Although bidirectional changes in NRG1 may cause schizophrenia symptoms, the modulation of the eCB system appears to be specific to the hyperfunction of NRG1 and not to the downregulation of basal NRG1 activity in our experimental conditions.
Next, we tested whether the treatment with 5 nm NRG1 for a shorter period, 1–3 d, could also curtail the DSI time course. In this condition, the DSI kinetics in NRG1-treated cells (n = 8) were not significantly different from those in control cells (n = 7) (p = 0.46, two-way ANOVA; Fig. 2C,D). The time constant of DSI decay was also similar in the two groups (12.1 ± 3.5 s for control and 13.7 ± 4.8 s for treated cells; p = 0.80, t test; Fig. 2E). Together, these results indicate that the prolonged and strong stimulation of ErbB receptors was necessary for the NRG1 effect on DSI to occur. In all other subsequent experiments, the treatment with NRG1 lasted 8–11 d. The NRG1-induced change in DSI was an entirely CB1R-dependent phenomenon because the DSI in both groups could be blocked by 1 μm SR141716, a CB1R antagonist. In SR141716, the mean eIPSC amplitudes at 2 s after depolarization (0 mV, 5 s) were 91 ± 3% (n = 6) of the baseline in control cells and 94 ± 2% (n = 8) in cells treated with NRG1 (5 nm, 8–11 d).
NRG1 treatment preserves CB1R expression and function
To test whether a reduction in CB1R function is involved in the faster recovery from DSI, we probed the sensitivity of agatoxin-resistant eIPSCs to mAEA, a specific CB1R agonist. The suppression of eIPSC amplitudes caused by 0.1 μm mAEA in NRG1-treated cells (60 ± 10% of baseline; n = 11) was not significantly different from that in control cells (70 ± 4% of baseline; n = 8) (p = 0.41, t test; Fig. 3A,B). The effect of 5 μm mAEA in NRG1-treated cells (12 ± 5% of baseline; n = 6) was also similar to that in control cells (11 ± 5% of baseline; n = 5) (p = 0.87, t test; Fig. 3A,B). This finding indicates that the chronic treatment with NRG1 did not alter the CB1R function. In addition, the expression of CB1R was not affected by the NRG1 treatment when examined with a Western blot method; the level of CB1R expression in NRG1-treated slices was 100 ± 6% of the control level (n = 6 experiments; p = 0.98, one-sample t test vs 100%; Fig. 3C,D).
Figure 3.

Treatment with NRG1 (5 nm, 8–11 d) affects neither the function nor the expression of CB1Rs. A, The suppression of eIPSC amplitudes caused by mAEA (0.1 or 5 μm), a CB1R agonist, in NRG1-treated slices was not different from that in control slices. The traces are from four different representative cells. B, The group data of the mAEA effect on the eIPSC amplitude. C, Western blot analysis shows that the level of CB1R expression was unaffected by NRG1 treatment. In each experiment, the CB1R signal of NRG1-treated slices was normalized to that of control slices after being normalized to β-actin intensity. D, The group data from the Western blot assay of CB1R expression. Error bars indicate SEM.
Enhanced expression and function of MGL contribute to the faster recovery from DSI
Because changes in 2-AG degradation often affect the DSI time course (Kim and Alger, 2004; Makara et al., 2005; Pan et al., 2009), we hypothesized that the NRG1-induced facilitation of IPSC recovery from DSI was the result of an increase in the function and/or expression of MGL, which is a major degradative enzyme for 2-AG. First, we directly probed changes in the protein levels of MGL. Western blot assays showed that the expression of MGL in NRG1-treated slices was 20 ± 5% higher than that in control slices (n = 5 experiments; p = 0.016, one-sample t test vs 100%; Fig. 4A,B). If the increase in MGL expression contributes to the faster decay of DSI, a blockade of MGL would equalize the difference in the DSI time course between control and NRG1-treated cells (e.g., in the presence of JZL184, a specific MGL inhibitor). We applied JZL184 (1 μm) to the bath solution for 25–30 min and then measured DSI. JZL184 slowed the IPSC recovery from DSI in both control and NRG1-treated cells, as predicted (Pan et al., 2009, 2011), resulting in no difference in the DSI time course between control (n = 8) and NRG1-treated cells (n = 9) (p = 0.42, two-way ANOVA; Fig. 4C,D). The time constant of DSI decay in NRG1-treated cells (25.6 ± 6.2 s) was also similar to the control value (22.8 ± 3.8 s) (p = 0.71, t test; Fig. 4G). This result shows that MGL activity is necessary for NRG1-treated cells to display the shorter DSI time course, and thus implies that MGL is involved in the effect of chronic NRG1 treatment on the DSI time course.
Figure 4.

The NRG1-induced enhancement of MGL expression and/or function contributes to the facilitated recovery from DSI. A, Western blot analysis shows that the level of MGL expression was increased by NRG1 treatment. The blot in Figure 3C was reprobed with an anti-MGL antibody. B, The quantitative group data from Western blotting for MGL. *p = 0.016 (one-sample t test vs 100%). C, eIPSCs were recorded from either a control or NRG1-treated (5 nm, 8–11 d) cell in the presence of JZL184 (1 μm) in the extracellular solution. Depol, postsynaptic depolarization (0 mV, 5 s). D, The group data of the DSI measured in 1 μm JZL184 show no difference between control and NRG1-treated groups. E, In the presence of 1 μm URB597 in the bath solution, the eIPSC recovery from DSI (0 mV, 5 s) in NRG1-treated cells was faster than that in control cells. *p < 0.005 (Bonferroni t test after two-way ANOVA). F, In the presence of 20 μm AM404 in the bath solution, the eIPSC recovery from DSI (0 mV, 5 s) in NRG1-treated cells was not significantly different from that in control cells. G, In the presence of 1 μm JZL184 or 20 μm AM404, the time constant of DSI decay in NRG1-treated cells was not significantly different from that in respective sister control cells. In 1 μm URB597, the time constant in NRG1-treated cells was significantly smaller than that in sister control cells. *p = 0.021 (t test). Error bars indicate SEM.
We investigated whether the NRG1-induced change in DSI was specific to MGL and not to another eCB degradative enzyme, fatty acid amide hydrolase (FAAH), which hydrolyzes another major eCB anandamide. We measured DSI in the presence of URB597, a specific FAAH inhibitor, in the extracellular solution during the eIPSC recording. The eIPSC amplitudes at 8 and 14 s after depolarization in NRG1-treated cells (n = 6) were significantly larger than those in control cells (n = 7) (p < 0.005, Bonferroni t test after two-way ANOVA; Fig. 4E). The time constant of DSI decay in NRG1-treated cells (7.1 ± 0.7 s) was smaller than that in sister control cells (13.7 ± 2.2 s) (p = 0.021, t test; Fig. 4G). This result indicates that the effect of NRG1 on DSI is not dependent on FAAH.
Because 2-AG degradation drives its uptake into cells via eCB transporters, an inhibitor of the eCB transporter would also abolish the difference in the DSI duration between control and NRG1-treated slices, as JZL184 did. When recorded in the presence of AM404 (20 μm), an eCB transporter inhibitor, the eIPSC amplitudes during the recovery from DSI in NRG1-treated cells (n = 15) were not different from those in control cells (n = 14) (p = 0.10, two-way ANOVA; Fig. 4F). The recovery time constant in NRG1-treated cells (17.3 ± 1.7 s) was also similar to the control value from sister cultures (16.7 ± 2.7 s) (p = 0.85, t test; Fig. 4G). Together, these results suggest that the chronic treatment with NRG1 enhanced the MGL expression, and thus facilitated the uptake and degradation of 2-AG, resulting in a faster recovery from DSI.
NRG1 treatment does not alter constitutive eCB signaling
If MGL is knocked out or overexpressed in mice, the basal 2-AG levels in the brain are elevated or reduced, respectively (Chanda et al., 2010; Schlosburg et al., 2010; Jung et al., 2012). To examine whether the NRG1-mediated enhancement of MGL expression decreases constitutive 2-AG effects, we assessed the degree of basal CB1R activation by measuring an increase in eIPSC amplitude caused by SR141716. If the basal 2-AG level was lowered in NRG1-treated slices, SR141716 would increase the eIPSC to a lesser degree than in control cells. SR141716 (1 μm) increased the eIPSC amplitude by 34 ± 6% (n = 5) in control cells and 30 ± 8% (n = 8) in NRG1-treated cells (Fig. 5A). The similar effects of SR141716 in the two groups (p = 0.72, t test) imply that the degree of basal CB1R activation was not altered by the treatment with NRG1.
Figure 5.

NRG1 treatment alters neither the constitutive eCB signaling nor basal GABAergic Pr. A, The tonic activation of CB1Rs was assessed as an increase in eIPSC amplitudes caused by bath-applied SR141716 (1 μm), a CB1R antagonist. The mean increase in eIPSC amplitude in NRG1-treated cells was not different from that in control cells. B, The constitutive uptake of eCB was examined with bath-applied AM404 (20 μm), an eCB transporter inhibitor. The eIPSC reduction caused by AM404 in NRG1-treated cells was similar to that in control cells. A vehicle for AM404 (0.02% ethanol) was present before AM404 application. C, The short-term plasticity of eIPSC was measured by stimulating presynaptic axons three times at 20 Hz. The amplitudes of the second or third eIPSCs, after being normalized to the first amplitude, in NRG1-treated cells were not significantly different from their respective values in control cells. Error bars indicate SEM.
Next, we examined whether the constitutive uptake of basal eCB was affected by NRG1 treatment. If the tonic uptake of basal eCB is enhanced, a blockade of eCB transporter would suppress eIPSC amplitude to a greater extent (Kim and Alger, 2010) because AM404 can cause the accumulation of basal eCB in the extracellular space. AM404 (20 μm) that was applied to the bath solution slightly suppressed eIPSC amplitudes to a similar extent in both NRG1-treated (10.3 ± 3.9% suppression; n = 5) and control cells (8.9 ± 0.9% suppression; n = 5) (p = 0.73, t test; Fig. 5B). This result suggests that the tonic uptake of basal eCB is unaffected by NRG1 treatment. Together, these data indicate that the chronic elevation of NRG1 level appears to modulate the phasic signaling of 2-AG but not the constitutive signaling of eCB.
Because CB1R activation reduces the probability of release (Pr), any reduction in the tonic level of eCB would increase the basal Pr at CB1R-positive GABAergic synapses (Kim and Alger, 2010). We assayed relative Pr by measuring the short-term plasticity of IPSCs that were evoked three times at 20 Hz (Fig. 5C) because Pr is inversely proportional to the paired-pulse ratio (PPR) of synaptic responses (Thomson, 2000). When normalized to the first IPSC amplitude, the amplitudes of the second (referred to as “PPR2”) and third (referred to as “PPR3”) IPSCs in NRG1-treated cells (PPR2, 0.80 ± 0.01; PPR3, 0.73 ± 0.01) (n = 65) were not significantly different from the control values (PPR2, 0.78 ± 0.01; PPR3, 0.70 ± 0.01) (n = 68) (p = 0.10, two-way ANOVA; Fig. 5C). As expected from the unaltered tonic level of eCB (Fig. 5A), the chronic NRG1 treatment did not change the basal Pr at CB1R-positive GABAergic terminals, leaving the dynamic range for additional Pr modulation, e.g., by CB1R agonists, unaffected.
iLTD is impaired by treatment with NRG1
Our next focus was the functional significance of the NRG1-mediated changes in MGL and 2-AG signaling. When the stimulation of mGluRs drives the production of 2-AG from CA1 pyramidal cells, presynaptic iLTD is induced by the 2-AG-mediated prolonged activation of CB1Rs (Chevaleyre and Castillo, 2003; Castillo et al., 2012). We tested the hypothesis that the shortened lifetime of 2-AG after NRG1 treatment might impair the induction of 2-AG-mediated iLTD (Fig. 6A–C). iLTD was induced by a 10 min application of 50 μm (S)-3,5-DHPG, a group I mGluR agonist. The magnitude of the iLTD (47–52 min after the DHPG onset) was 60 ± 8% in control slices (n = 6) but only 26 ± 5% in NRG1-treated slices (n = 5) (p = 0.008, t test; Fig. 6A). We additionally probed the changes in Pr during the iLTD using the changes in 1/PPR3 because PPR3 is inversely proportional to Pr. In the iLTD experiment, eIPSCs were elicited three times at 20 Hz. In both control and NRG1-treated groups, 1/PPR3 during the iLTD was reduced compared with the respective pre-iLTD values, as predicted from the presynaptic nature of the iLTD expression, but to a lesser degree in NRG1-treated cells (−13 ± 3%) than in control cells (−28 ± 4%) (p = 0.015, t test; Fig. 6C). This result suggests that the impaired iLTD in the NRG1-treated slices is the result of a smaller depression of Pr.
If the iLTD reduction could be attributed to the elevated function of MGL, then JZL184 would equalize the difference in iLTD between control and NRG1-treated cells. We tested this hypothesis by measuring the iLTD in JZL184 (0.2 μm), which was applied to the bath solution ∼20 min before the application of DHPG. In accord with our hypothesis, the MGL inhibitor restored the iLTD in NRG1-treated cells (58 ± 5%; n = 6) to the control level (46 ± 6%; n = 6) (p = 0.14, t test; Fig. 6B). In the presence of JZL184, an iLTD-induced decrease in 1/PPR3 in NRG1-treated cells (−31 ± 4%) was also similar to that in control slices (−26 ± 4%) (p = 0.36, t test; Fig. 6C). These results suggest that the enhanced activity of MGL contributes to the NRG1-mediated impairment in iLTD.
A potential concern about our iLTD experiment with JZL184 was that a long-term blockade of MGL might increase the basal level of 2-AG (Chanda et al., 2010; Schlosburg et al., 2010; Pan et al., 2011), which would cause a decrease in GABAergic Pr. We examined the effect of JZL184 on GABAergic Pr (i.e., PPR) in control slices (i.e., without NRG1). When 0.2 μm JZL184 was applied for 89 ± 3 min (range, 75–105 min), the PPR3 (0.78 ± 0.04; n = 8) was not significantly different from the PPR3 obtained without JZL184 (0.70 ± 0.01; Fig. 5C) (p = 0.47, Bonferroni t test after ANOVA; Fig. 6D). This similarity implies that a JZL184-induced (0.2 μm, ∼90 min) increase in basal 2-AG, if any, was not sufficient to reduce GABAergic Pr effectively. Therefore, the JZL184-mediated rescue of iLTD in the NRG1 group (Fig. 6B) cannot be attributed to the gradual suppression of Pr. At a higher concentration (1 μm), JZL184 applied for 32 ± 3 min (range, 20–49 min) had no effect on PPR3 (0.70 ± 0.02; n = 10) (p = 1.0, Bonferroni t test vs non-JZL184), whereas its extended application for 96 ± 4 min (range, 75–120 min) increased PPR3 to 0.99 ± 0.05 (n = 10) (p < 0.005, Bonferroni t test vs other groups; Fig. 6D). Because JZL184 was applied for 25–30 min in our DSI experiments (Fig. 4C,D), the interpretation of the DSI data could not have been complicated by unwanted effects of JZL184 on GABAergic Pr.
NRG1 treatment weakens the mGluR-mediated suppression of IPSC
If the iLTD impairment (Fig. 6A) is the result of the curtailed action of 2-AG, then it is possible that the NRG1 treatment reduces the strength of mGluR-mediated 2-AG signaling, which can be assayed as eIPSC suppression. However, a high dose of DHPG (50 μm) had similar effects on eIPSC amplitudes in control and NRG1-treated cells (p = 0.61, t test at 4–9 min of DHPG; Fig. 6A). To avoid a potential ceiling effect of 50 μm DHPG, we used a low dose (0.4 μm) of DHPG. (S)-3,5-DHPG (0.4 μm) suppressed the eIPSC amplitude in NRG1-treated cells by 13 ± 4% (n = 5; Fig. 7A), which was less than half the effect in control cells (29 ± 1%; n = 5) (p = 0.0037, t test; Fig. 7A). This difference in the efficacy of DHPG at a low concentration was equalized by JZL184. In the presence of 1 μm JZL184, 0.4 μm DHPG reduced the eIPSC amplitude by 62 ± 8% (n = 5) in NRG1-treated cells and 57 ± 5% (n = 5) in control cells (p = 0.64, t test; Fig. 7B). These results indicate that a low dose of DHPG has a weaker effect in NRG1-treated cells than in control cells only when MGL is kept active and thus imply that the enhancement of MGL may contribute to the difference in the DHPG effect.
Figure 7.

A low concentration (0.4 μm) of DHPG has a weaker effect on the eIPSC amplitudes in NRG1-treated cells than in control cells. A, The mean reduction in eIPSC amplitudes caused by 0.4 μm DHPG in NRG1-teated cells was significantly smaller than that in control cells at 12–15 min. *p = 0.0037 (t test). B, In the presence of 1 μm JZL184, 0.4 μm DHPG reduced the eIPSC amplitude to a similar extent in both NRG1-treated and control cells. At the onset of DHPG, JZL184 had been present in the bath solution for 20–27 min. Error bars indicate SEM.
Discussion
Our results show that the chronic treatment of hippocampal slice cultures with NRG1 increases the expression of MGL, resulting in the facilitated degradation of 2-AG and, hence, a shortened duration of DSI and impaired iLTD (Fig. 8). Because NRG1-ErbB4 hyperfunction is a risk factor for schizophrenia, our study implies that the eCB system, especially MGL, might be involved in the NRG1-mediated pathophysiology of schizophrenia.
Figure 8.

A schematic diagram of the NRG1-mediated enhancement of MGL and the consequent reduction in 2-AG effects. The chronic treatment of the hippocampus with NRG1 increases the expression of MGL, which is a degradative enzyme for 2-AG. This increase facilitates the degradation of 2-AG, resulting in the shortened duration of DSI and the impairment of iLTD. The location and biochemical cascade of ErbB-to-MGL signaling that is responsible for the NRG1-mediated 2-AG curtailment must be determined.
Memory deficits are found in various strains of NRG1-ErbB4 transgenic mice, including NRG1-overexpressing mice (Kato et al., 2010; Deakin et al., 2012; Yin et al., 2013), NRG1 heterozygotes (Ehrlichman et al., 2009; Duffy et al., 2010), and ErbB4 knock-outs (KOs) (Chen et al., 2010), as well as in human patients with schizophrenia (Stone and Hsi, 2011). Investigations on the mechanisms of memory impairment in NRG1-ErbB4 transgenic mice have focused on the effects of NRG1 and/or ErbB4 on long-term synaptic plasticity. Indeed, NRG1 directly interrupts the LTP of excitatory synapses; LTP is inhibited by acutely applied NRG1 or enhanced by ErbB4 KO (Kwon et al., 2005; Pitcher et al., 2008, 2011; Chen et al., 2010; Shamir et al., 2012). However, some types of memory decline caused by NRG1 cannot be explained by the direct inhibition of LTP. For instance, NRG1 overexpression in mice has no effect on LTP (Deakin et al., 2012); ErbB4 KO in PV interneurons alters LTP but not fear memory (Shamir et al., 2012), indicating that fear memory in these mice is independent of LTP (but also see Chen et al., 2010).
Our data suggest that a reduction in iLTD might be an additional mechanism by which NRG1 can modulate long-term synaptic plasticity. By strengthening excitation-spike coupling, iLTD can lower the threshold for LTP induction at excitatory synapses (Chevaleyre and Castillo, 2003, 2004). Therefore, in addition to the NRG1-dependent direct inhibition of LTP, the 2-AG-mediated metaplasticity of LTP induction might also negatively influence LTP. This type of metaplasticity involves CB1R (or CCK)-positive GABAergic synapses possibly in the following ways. First, iLTD increases the chance for LTP induction via enhanced disinhibition (Chevaleyre and Castillo, 2004; Zhu and Lovinger, 2007). Therefore, an NRG1-mediated decrease in iLTD would have a negative effect on LTP. Second, 2-AG can promote LTP induction through DSI. DSI provides a short temporal window of disinhibition to allow a reduction in the threshold for LTP induction (Carlson et al., 2002). Thus, a decrease in DSI duration (e.g., by NRG1) would impair LTP induction. Third, 2-AG (and JZL184) can facilitate LTP induction without iLTD when distinct excitatory inputs in CA1 are simultaneously stimulated (Xu et al., 2012). Thus, MGL upregulation might reduce this type of associativity of LTP. These 2-AG-dependent mechanisms suggest that a broader range of network components (e.g., various types of inhibitory interneurons) should be considered for the accurate determination of the role of LTP in NRG1 pathology. In summary, the prolonged elevation of NRG1 may be involved in memory disruption in individuals suffering from schizophrenia by interfering directly with LTP and/or inducing the 2-AG-dependent metaplasticity of LTP.
The levels of NRG1 and/or ErbB4 are also increased in the hippocampus and CSF of patients with Alzheimer's disease (Chaudhury et al., 2003; Pankonin et al., 2009). Given the observation that MGL expression is also elevated in the postmortem brains of Alzheimer's disease patients (Mulder et al., 2011), our study may be relevant to Alzheimer's disease symptoms; rises in NRG1 and MGL, and hence the disruption of 2-AG-mediated metaplasticity of LTP, may contribute to the memory decline associated with Alzheimer's disease.
Although NRG1-expressing neurons are widely distributed in the brain, not all neurons express NRG1 (Law et al., 2004; Bernstein et al., 2006; Bare et al., 2011; Liu et al., 2011). Therefore, despite the diffusible nature of NRG1, it is possible that some neurons may not be exposed to high levels of NRG1 in a condition in which NRG1 is overexpressed in vivo (e.g., the brains of patients with schizophrenia). For this reason, the extrapolation of our data to in vivo conditions must be made carefully, even though our study provides a novel insight into the role of eCBs in the NRG1-mediated pathology. In addition, not all patients with schizophrenia have abnormal NRG1-ErbB signaling. Therefore, the role of MGL in schizophrenia pathology, as implicated by our finding, cannot be generalized for all types of schizophrenic patients.
Changes in the eCB degradation may alter the basal GABAergic Pr by shifting the resting levels of eCB (Kim and Alger, 2010; Pan et al., 2011). However, in contrast to the reduced 2-AG levels in MGL-overexpressing mice (Jung et al., 2012), the NRG1-induced upregulation of MGL did not change the tonic activation of CB1Rs in our experimental conditions (Fig. 5). Possible reasons for this discrepancy are as follows: First, a 20% increase in MGL expression (Fig. 4A,B) may have little effect on the basal 2-AG tone because even a >10-fold increase in MGL expression reduces the basal 2-AG level by only 50% (Jung et al., 2012). Second, the amount of 2-AG that is collected from whole tissue (Jung et al., 2012) can be different from the amount of 2-AG that is constitutively activating CB1Rs, perhaps because of the existence of different pools of 2-AG (Min et al., 2010a; Alger and Kim, 2011; Zhang et al., 2011). Third, it is controversial whether the tonic activation of CB1R is mediated by 2-AG or anandamide (Hashimotodani et al., 2007; Kim and Alger, 2010; Alger, 2012). If 2-AG were to constitutively activate CB1Rs, a blockade or KO of diacylglycerol lipase, a 2-AG synthase, would be expected to increase neurotransmitter release (or Pr) by reducing the basal 2-AG levels, but this effect has not been observed (Hashimotodani et al., 2007; Min et al., 2010b; Tanimura et al., 2010; Zhang et al., 2011). Therefore, it is possible that 2-AG is constitutively produced, but does not substantially stimulate, CB1Rs because of limitations of quantity and/or physical access. The NRG1-mediated enhancement of MGL appears to selectively weaken the phasic or acute action of 2-AG without a significant disruption of the GABAergic inhibitory tone.
The distinct modulation of tonic and phasic eCB signaling can also be found in other conditions. The mutation or deletion of neuroligin-3 reduces tonic eCB signaling while preserving its phasic signaling (e.g., DSI) and iLTD in the hippocampus (Földy et al., 2013). Therefore, their study implies that the modulatory mechanism for tonic eCB signaling could be different from that for DSI and iLTD. Another example of differential modulation is the effect of chronic inactivity on the eCB system (Kim and Alger, 2010). The long-term deprivation of neuronal activity decreases the tonic activation of CB1R in a FAAH-dependent manner but has no effect on DSI (Kim and Alger, 2010). All of these studies, including our present data, suggest that tonic and phasic eCB signaling can be separately regulated. It would be interesting to investigate how only one type of eCB signaling can be selectively modulated without affecting the other type and what the functional roles for the distinct eCB actions in physiological and pathological conditions are.
The interaction between NRG1 and the eCB system is supported by many studies (Boucher et al., 2007a, b, 2011; Long et al., 2010, 2012, 2013; Stadelmann et al., 2010; Spencer et al., 2013). Most of these studies are based on hypomorphic NRG1 in rodents, and the detailed patterns of interaction vary with developmental stages, gender, and the duration of cannabinoid action. Our results additionally show that hypermorphic NRG1 can also modulate eCB function. In particular, the changes in the DSI time course in our study are caused by a chronic increase in NRG1 levels but not by a reduction in the basal activity of NRG1. Therefore, hypermorphic NRG1 appears to have cellular effects that are distinct from those of hypomorphic NRG1, although both situations can result in schizophrenic behavioral phenotypes in vivo. In the future, a detailed investigation of the pathological mechanisms underlying how bidirectional changes in NRG1-ErbB signaling cause similar phenotypes will be an important task.
Footnotes
This work was supported by the National Institute on Aging Grant R01AG036794. We thank Drs. Lin Mei and Il-Man Kim and members of the J.K. laboratory for advice and technical help and the National Institute on Drug Abuse for providing SR141716.
The authors declare no competing financial interests.
References
- Abe Y, Namba H, Kato T, Iwakura Y, Nawa H. Neuregulin-1 signals from the periphery regulate AMPA receptor sensitivity and expression in GABAergic interneurons in developing neocortex. J Neurosci. 2011;31:5699–5709. doi: 10.1523/JNEUROSCI.3477-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE. Endocannabinoids at the synapse a decade after the dies mirabilis (29 March 2001): what we still do not know. J Physiol. 2012;590:2203–2212. doi: 10.1113/jphysiol.2011.220855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE, Kim J. Supply and demand for endocannabinoids. Trends Neurosci. 2011;34:304–315. doi: 10.1016/j.tins.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Macdonald ML, Borgmann-Winter KE, Hahn CG. Neuregulin 1-erbB4 pathway in schizophrenia: from genes to an interactome. Brain Res Bull. 2010;83:132–139. doi: 10.1016/j.brainresbull.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat A, Dean B, Scarr E, Evin G. Decreased Neuregulin 1 C-terminal fragment in Brodmann's area 6 of patients with schizophrenia. Schizophr Res. 2010;124:200–207. doi: 10.1016/j.schres.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Bare DJ, Becker-Catania SG, DeVries GH. Differential localization of neuregulin-1 type III in the central and peripheral nervous system. Brain Res. 2011;1369:10–20. doi: 10.1016/j.brainres.2010.10.092. [DOI] [PubMed] [Google Scholar]
- Bernstein HG, Lendeckel U, Bertram I, Bukowska A, Kanakis D, Dobrowolny H, Stauch R, Krell D, Mawrin C, Budinger E, Keilhoff G, Bogerts B. Localization of neuregulin-1α (heregulin-α) and one of its receptors, ErbB-4 tyrosine kinase, in developing and adult human brain. Brain Res Bull. 2006;69:546–559. doi: 10.1016/j.brainresbull.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Bertram I, Bernstein HG, Lendeckel U, Bukowska A, Dobrowolny H, Keilhoff G, Kanakis D, Mawrin C, Bielau H, Falkai P, Bogerts B. Immunohistochemical evidence for impaired neuregulin-1 signaling in the prefrontal cortex in schizophrenia and in unipolar depression. Ann N Y Acad Sci. 2007;1096:147–156. doi: 10.1196/annals.1397.080. [DOI] [PubMed] [Google Scholar]
- Boucher AA, Hunt GE, Karl T, Micheau J, McGregor IS, Arnold JC. Heterozygous neuregulin 1 mice display greater baseline and Δ9-tetrahydrocannabinol-induced c-Fos expression. Neuroscience. 2007a;149:861–870. doi: 10.1016/j.neuroscience.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Boucher AA, Arnold JC, Duffy L, Schofield PR, Micheau J, Karl T. Heterozygous neuregulin 1 mice are more sensitive to the behavioural effects of Δ9-tetrahydrocannabinol. Psychopharmacology (Berl) 2007b;192:325–336. doi: 10.1007/s00213-007-0721-3. [DOI] [PubMed] [Google Scholar]
- Boucher AA, Hunt GE, Micheau J, Huang X, McGregor IS, Karl T, Arnold JC. The schizophrenia susceptibility gene neuregulin 1 modulates tolerance to the effects of cannabinoids. Int J Neuropsychopharmacol. 2011;14:631–643. doi: 10.1017/S146114571000091X. [DOI] [PubMed] [Google Scholar]
- Buonanno A. The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits. Brain Res Bull. 2010;83:122–131. doi: 10.1016/j.brainresbull.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda PK, Gao Y, Mark L, Btesh J, Strassle BW, Lu P, Piesla MJ, Zhang MY, Bingham B, Uveges A, Kowal D, Garbe D, Kouranova EV, Ring RH, Bates B, Pangalos MN, Kennedy JD, Whiteside GT, Samad TA. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol Pharmacol. 2010;78:996–1003. doi: 10.1124/mol.110.068304. [DOI] [PubMed] [Google Scholar]
- Chaudhury AR, Gerecke KM, Wyss JM, Morgan DG, Gordon MN, Carroll SL. Neuregulin-1 and erbB4 immunoreactivity is associated with neuritic plaques in Alzheimer disease brain and in a transgenic model of Alzheimer disease. J Neuropathol Exp Neurol. 2003;62:42–54. doi: 10.1093/jnen/62.1.42. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Zhang M, Yin DM, Wen L, Ting A, Wang P, Lu YS, Zhu XH, Li SJ, Wu CY, Wang XM, Lai C, Xiong WC, Mei L, Gao TM. ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc Natl Acad Sci U S A. 2010;107:21818–21823. doi: 10.1073/pnas.1010669107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/S0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Chong VZ, Thompson M, Beltaifa S, Webster MJ, Law AJ, Weickert CS. Elevated neuregulin-1 and ErbB4 protein in the prefrontal cortex of schizophrenic patients. Schizophr Res. 2008;100:270–280. doi: 10.1016/j.schres.2007.12.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin IH, Nissen W, Law AJ, Lane T, Kanso R, Schwab MH, Nave KA, Lamsa KP, Paulsen O, Bannerman DM, Harrison PJ. Transgenic overexpression of the type I isoform of neuregulin 1 affects working memory and hippocampal oscillations but not long-term potentiation. Cereb Cortex. 2012;22:1520–1529. doi: 10.1093/cercor/bhr223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza DC, Abi-Saab WM, Madonick S, Forselius-Bielen K, Doersch A, Braley G, Gueorguieva R, Cooper TB, Krystal JH. Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biol Psychiatry. 2005;57:594–608. doi: 10.1016/j.biopsych.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Duffy L, Cappas E, Lai D, Boucher AA, Karl T. Cognition in transmembrane domain neuregulin 1 mutant mice. Neuroscience. 2010;170:800–807. doi: 10.1016/j.neuroscience.2010.07.042. [DOI] [PubMed] [Google Scholar]
- Ehrlichman RS, Luminais SN, White SL, Rudnick ND, Ma N, Dow HC, Kreibich AS, Abel T, Brodkin ES, Hahn CG, Siegel SJ. Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Res. 2009;1294:116–127. doi: 10.1016/j.brainres.2009.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron. 2013;78:498–509. doi: 10.1016/j.neuron.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida A, Leweke FM, Gerth CW, Schreiber D, Koethe D, Faulhaber J, Klosterkötter J, Piomelli D. Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology. 2004;29:2108–2114. doi: 10.1038/sj.npp.1300558. [DOI] [PubMed] [Google Scholar]
- Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, Bakshi K, Kamins J, Borgmann-Winter KE, Siegel SJ, Gallop RJ, Arnold SE. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Straub RE, Weickert CS, Hyde TM, Kleinman JE, Weinberger DR. Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia. Mol Psychiatry. 2004;9:299–307. doi: 10.1038/sj.mp.4001434. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J Neurosci. 2007;27:1211–1219. doi: 10.1523/JNEUROSCI.4159-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KM, Clapper JR, Fu J, D'Agostino G, Guijarro A, Thongkham D, Avanesian A, Astarita G, DiPatrizio NV, Frontini A, Cinti S, Diano S, Piomelli D. 2-Arachidonoylglycerol signaling in forebrain regulates systemic energy metabolism. Cell Metab. 2012;15:299–310. doi: 10.1016/j.cmet.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Kasai A, Mizuno M, Fengyi L, Shintani N, Maeda S, Yokoyama M, Ozaki M, Nawa H. Phenotypic characterization of transgenic mice overexpressing neuregulin-1. PLoS One. 2010;5:e14185. doi: 10.1371/journal.pone.0014185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci. 2004;7:697–698. doi: 10.1038/nn1262. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE. Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci. 2010;13:592–600. doi: 10.1038/nn.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon OB, Longart M, Vullhorst D, Hoffman DA, Buonanno A. Neuregulin-1 reverses long-term potentiation at CA1 hippocampal synapses. J Neurosci. 2005;25:9378–9383. doi: 10.1523/JNEUROSCI.2100-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law AJ, Shannon Weickert C, Hyde TM, Kleinman JE, Harrison PJ. Neuregulin-1 (NRG-1) mRNA and protein in the adult human brain. Neuroscience. 2004;127:125–136. doi: 10.1016/j.neuroscience.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Law AJ, Lipska BK, Weickert CS, Hyde TM, Straub RE, Hashimoto R, Harrison PJ, Kleinman JE, Weinberger DR. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc Natl Acad Sci U S A. 2006;103:6747–6752. doi: 10.1073/pnas.0602002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law AJ, Kleinman JE, Weinberger DR, Weickert CS. Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Hum Mol Genet. 2007;16:129–141. doi: 10.1093/hmg/ddl449. [DOI] [PubMed] [Google Scholar]
- Li KX, Lu YM, Xu ZH, Zhang J, Zhu JM, Zhang JM, Cao SX, Chen XJ, Chen Z, Luo JH, Duan S, Li XM. Neuregulin 1 regulates excitability of fast-spiking neurons through Kv1.1 and acts in epilepsy. Nat Neurosci. 2012;15:267–273. doi: 10.1038/nn.3006. [DOI] [PubMed] [Google Scholar]
- Liu X, Bates R, Yin DM, Shen C, Wang F, Su N, Kirov SA, Luo Y, Wang JZ, Xiong WC, Mei L. Specific regulation of NRG1 isoform expression by neuronal activity. J Neurosci. 2011;31:8491–8501. doi: 10.1523/JNEUROSCI.5317-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long LE, Chesworth R, Arnold JC, Karl T. A follow-up study: acute behavioural effects of Δ9-THC in female heterozygous neuregulin 1 transmembrane domain mutant mice. Psychopharmacology (Berl) 2010;211:277–289. doi: 10.1007/s00213-010-1896-6. [DOI] [PubMed] [Google Scholar]
- Long LE, Chesworth R, Huang XF, Wong A, Spiro A, McGregor IS, Arnold JC, Karl T. Distinct neurobehavioural effects of cannabidiol in transmembrane domain neuregulin 1 mutant mice. PLoS One. 2012;7:e34129. doi: 10.1371/journal.pone.0034129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long LE, Chesworth R, Huang XF, McGregor IS, Arnold JC, Karl T. Transmembrane domain Nrg1 mutant mice show altered susceptibility to the neurobehavioural actions of repeated THC exposure in adolescence. Int J Neuropsychopharmacol. 2013;16:163–175. doi: 10.1017/S1461145711001854. [DOI] [PubMed] [Google Scholar]
- Makara JK, Mor M, Fegley D, Szabó SI, Kathuria S, Astarita G, Duranti A, Tontini A, Tarzia G, Rivara S, Freund TF, Piomelli D. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci. 2005;8:1139–1141. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- Marco EM, García-Gutierrez MS, Bermúdez-Silva FJ, Moreira FA, Guimarães F, Manzanares J, Viveros MP. Endocannabinoid system and psychiatry: in search of a neurobiological basis for detrimental and potential therapeutic effects. Front Behav Neurosci. 2011;5:63. doi: 10.3389/fnbeh.2011.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min R, Di Marzo V, Mansvelder HD. DAG lipase involvement in depolarization-induced suppression of inhibition: does endocannabinoid biosynthesis always meet the demand? Neuroscientist. 2010a;16:608–613. doi: 10.1177/1073858410373281. [DOI] [PubMed] [Google Scholar]
- Min R, Testa-Silva G, Heistek TS, Canto CB, Lodder JC, Bisogno T, Di Marzo V, Brussaard AB, Burnashev N, Mansvelder HD. Diacylglycerol lipase is not involved in depolarization-induced suppression of inhibition at unitary inhibitory connections in mouse hippocampus. J Neurosci. 2010b;30:2710–2715. doi: 10.1523/JNEUROSCI.BC-3622-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder J, Zilberter M, Pasquaré SJ, Alpár A, Schulte G, Ferreira SG, Köfalvi A, Martín-Moreno AM, Keimpema E, Tanila H, Watanabe M, Mackie K, Hortobágyi T, de Ceballos ML, Harkany T. Molecular reorganization of endocannabinoid signalling in Alzheimer's disease. Brain. 2011;134:1041–1060. doi: 10.1093/brain/awr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neddens J, Buonanno A. Selective populations of hippocampal interneurons express ErbB4 and their number and distribution is altered in ErbB4 knockout mice. Hippocampus. 2010;20:724–744. doi: 10.1002/hipo.20675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl) piperidine-1-carboxylate (JZL184) enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Zhong P, Blankman JL, Cravatt BF, Liu QS. Alterations of endocannabinoid signaling, synaptic plasticity, learning, and memory in monoacylglycerol lipase knock-out mice. J Neurosci. 2011;31:13420–13430. doi: 10.1523/JNEUROSCI.2075-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankonin MS, Sohi J, Kamholz J, Loeb JA. Differential distribution of neuregulin in human brain and spinal fluid. Brain Res. 2009;1258:1–11. doi: 10.1016/j.brainres.2008.12.047. [DOI] [PubMed] [Google Scholar]
- Patenaude C, Chapman CA, Bertrand S, Congar P, Lacaille JC. GABAB receptor- and metabotropic glutamate receptor-dependent cooperative long-term potentiation of rat hippocampal GABAA synaptic transmission. J Physiol. 2003;553:155–167. doi: 10.1113/jphysiol.2003.049015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryshen TL, Middleton FA, Kirby A, Aldinger KA, Purcell S, Tahl AR, Morley CP, McGann L, Gentile KL, Rockwell GN, Medeiros HM, Carvalho C, Macedo A, Dourado A, Valente J, Ferreira CP, Patterson NJ, Azevedo MH, Daly MJ, Pato CN, et al. Support for involvement of neuregulin 1 in schizophrenia pathophysiology. Mol Psychiatry. 2005;10:366–374. 328. doi: 10.1038/sj.mp.4001608. [DOI] [PubMed] [Google Scholar]
- Pitcher GM, Beggs S, Woo RS, Mei L, Salter MW. ErbB4 is a suppressor of long-term potentiation in the adult hippocampus. Neuroreport. 2008;19:139–143. doi: 10.1097/WNR.0b013e3282f3da10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher GM, Kalia LV, Ng D, Goodfellow NM, Yee KT, Lambe EK, Salter MW. Schizophrenia susceptibility pathway neuregulin 1-ErbB4 suppresses Src upregulation of NMDA receptors. Nat Med. 2011;17:470–478. doi: 10.1038/nm.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico B, Marín O. Neuregulin signaling, cortical circuitry development and schizophrenia. Curr Opin Genet Dev. 2011;21:262–270. doi: 10.1016/j.gde.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamir A, Kwon OB, Karavanova I, Vullhorst D, Leiva-Salcedo E, Janssen MJ, Buonanno A. The importance of the NRG-1/ErbB4 pathway for synaptic plasticity and behaviors associated with psychiatric disorders. J Neurosci. 2012;32:2988–2997. doi: 10.1523/JNEUROSCI.1899-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M, Komi E, Wang R, Kato T, Watanabe Y, Sakai M, Ozaki M, Someya T, Nawa H. Measurement and comparison of serum neuregulin 1 immunoreactivity in control subjects and patients with schizophrenia: an influence of its genetic polymorphism. J Neural Transm. 2010;117:887–895. doi: 10.1007/s00702-010-0418-3. [DOI] [PubMed] [Google Scholar]
- Spencer JR, Darbyshire KM, Boucher AA, Kashem MA, Long LE, McGregor IS, Karl T, Arnold JC. Novel molecular changes induced by Nrg1 hypomorphism and Nrg1-cannabinoid interaction in adolescence: a hippocampal proteomic study in mice. Front Cell Neurosci. 2013;7:15. doi: 10.3389/fncel.2013.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmann AM, Roser P, Arning L, Gallinat J, Epplen JT, Juckel G. Acute effects of Δ9-tetrahydrocannabinol on the auditory evoked mismatch negativity are modulated by the NRG1 gene. Pharmacopsychiatry. 2010;43:194–195. doi: 10.1055/s-0030-1254088. [DOI] [PubMed] [Google Scholar]
- Stone WS, Hsi X. Declarative memory deficits and schizophrenia: problems and prospects. Neurobiol Learn Mem. 2011;96:544–552. doi: 10.1016/j.nlm.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Tan GH, Liu YY, Hu XL, Yin DM, Mei L, Xiong ZQ. Neuregulin 1 represses limbic epileptogenesis through ErbB4 in parvalbumin-expressing interneurons. Nat Neurosci. 2012;15:258–266. doi: 10.1038/nn.3005. [DOI] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase α mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/S0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Vigano D, Guidali C, Petrosino S, Realini N, Rubino T, Di Marzo V, Parolaro D. Involvement of the endocannabinoid system in phencyclidine-induced cognitive deficits modelling schizophrenia. Int J Neuropsychopharmacol. 2009;12:599–614. doi: 10.1017/S1461145708009371. [DOI] [PubMed] [Google Scholar]
- Vullhorst D, Neddens J, Karavanova I, Tricoire L, Petralia RS, McBain CJ, Buonanno A. Selective expression of ErbB4 in interneurons, but not pyramidal cells, of the rodent hippocampus. J Neurosci. 2009;29:12255–12264. doi: 10.1523/JNEUROSCI.2454-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Lu YS, Zhu XH, Li XM, Woo RS, Chen YJ, Yin DM, Lai C, Terry AV, Jr, Vazdarjanova A, Xiong WC, Mei L. Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc Natl Acad Sci U S A. 2010;107:1211–1216. doi: 10.1073/pnas.0910302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/S0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Woo RS, Li XM, Tao Y, Carpenter-Hyland E, Huang YZ, Weber J, Neiswender H, Dong XP, Wu J, Gassmann M, Lai C, Xiong WC, Gao TM, Mei L. Neuregulin-1 enhances depolarization-induced GABA release. Neuron. 2007;54:599–610. doi: 10.1016/j.neuron.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Xu JY, Zhang J, Chen C. Long-lasting potentiation of hippocampal synaptic transmission by direct cortical input is mediated via endocannabinoids. J Physiol. 2012;590:2305–2315. doi: 10.1113/jphysiol.2011.223511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau HJ, Wang HF, Lai C, Liu FC. Neural development of the neuregulin receptor ErbB4 in the cerebral cortex and the hippocampus: preferential expression by interneurons tangentially migrating from the ganglionic eminences. Cereb Cortex. 2003;13:252–264. doi: 10.1093/cercor/13.3.252. [DOI] [PubMed] [Google Scholar]
- Yin DM, Chen YJ, Lu YS, Bean JC, Sathyamurthy A, Shen C, Liu X, Lin TW, Smith CA, Xiong WC, Mei L. Reversal of behavioral deficits and synaptic dysfunction in mice overexpressing neuregulin 1. Neuron. 2013;78:644–657. doi: 10.1016/j.neuron.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanettini C, Panlilio LV, Alicki M, Goldberg SR, Haller J, Yasar S. Effects of endocannabinoid system modulation on cognitive and emotional behavior. Front Behav Neurosci. 2011;5:57. doi: 10.3389/fnbeh.2011.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang M, Bisogno T, Di Marzo V, Alger BE. Endocannabinoids generated by Ca2+ or by metabotropic glutamate receptors appear to arise from different pools of diacylglycerol lipase. PLoS One. 2011;6:e16305. doi: 10.1371/journal.pone.0016305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Persistent synaptic activity produces long-lasting enhancement of endocannabinoid modulation and alters long-term synaptic plasticity. J Neurophysiol. 2007;97:4386–4389. doi: 10.1152/jn.01228.2006. [DOI] [PubMed] [Google Scholar]