

Abstract
It is increasingly apparent that treatment with a variety of anticancer agents often is associated with adverse neurological consequences. Clinical studies indicate that exposure even to tamoxifen (TMX), a putatively benign antihormonal agent widely used in breast cancer treatment, causes cognitive dysfunction and changes in CNS metabolism, hippocampal volume, and brain structure. We found that TMX is toxic for a variety of CNS cell populations in vitro and also increased cell death in the corpus callosum and reduced cell division in the mouse subventricular zone, the hippocampal dentate gyrus, and the corpus callosum. We further discovered that MEK1/2 inhibition selectively rescued primary glial progenitors from TMX toxicity in vitro while enhancing TMX effects on MCF7 luminal human breast cancer cells. In vivo, MEK1/2 inhibition prevented TMX-induced cell death in systemically treated mice. Our results demonstrate unexpected cytotoxicity of this putatively benign antihormonal agent and offer a potential strategy for rescuing CNS cells from adverse effects of TMX.
Introduction
Injury to the CNS from systemic chemotherapy is increasingly recognized as a neurological problem affecting many individuals (Dietrich, 2010; Monje and Dietrich, 2012). This problem is particularly well studied in breast cancer survivors, for whom current data suggest that 18% of individuals receiving standard-dose chemotherapy manifest cognitive defects on post-treatment evaluation (Meyers and Abbruzzese, 1992). For those patients receiving high-dose chemotherapy, over 30% of individuals examined 2 years after treatment exhibit detectable cognitive impairment (van Dam et al., 1998; Schagen et al., 1999), a more than eightfold increase over age-matched control patients. Moreover, imaging studies reveal CNS white matter changes in up to 70% of treated individuals (Stemmer et al., 1994; Brown et al., 1998). Thus, it is particularly important to understand the basis for these toxicities and how to protect from them without compromising cancer treatments themselves.
A particularly important anticancer agent for which evidence of adverse neurological effects is emerging is tamoxifen (TMX). TMX is used both before and after breast cancer surgery and may be applied for multiple years in women at increased genetic risk for breast cancer. Although TMX is considered to be one of the most benign cancer treatments, women treated with TMX nonetheless exhibit subtle but significant changes in neuropsychological function and brain structure (Espeland et al., 2010; Schilder et al., 2010). TMX-related toxicities include cerebral and cerebellar toxicities, irritability, confusion, and ocular toxicities (Bender et al., 2001), hippocampal atrophy, and widespread areas of hypometabolism in the inferior and dorsal lateral frontal lobes (Eberling et al., 2004). Moreover, several studies suggest an association between TMX exposure and cognitive dysfunction (Palmer et al., 2008; Schilder et al., 2010).
Despite increasing evidence that systemic TMX administration is associated with adverse neurological consequences, there is little biological understanding on the effects of this substance on cells of the CNS, or on strategies for protecting against potential toxicities.
Materials and Methods
Cell culture.
All CNS cell populations, including oligodendrocyte-type-2 astrocyte progenitor cells/oligodendrocyte precursor cells (O-2A/OPCs), glial-restricted precursor (GRP) cells, astrocytes, neuroepithelial stem cells (NSCs), and oligodendrocytes were purified and grown as described previously (Dietrich et al., 2006; Han et al., 2008). Human glial progenitors (obtained from Clonetics) were propagated as described previously (Dietrich et al., 2002). MCF7 cells were obtained from ATCC and maintained in DMEM plus 10% FBS.
Cell viability.
Cell number and death of O-2A/OPCs were analyzed by Calcein-AM and propidium iodide (PI) staining, respectively, in live culture using a Celigo adherent cell cytometer as described previously (Chen et al., 2013). MCF7 cell viability was determined using AlamarBlue and measurement of absorbance at 570 nm, as their ability to grow in three-dimensional aggregates compromises Celigo-based analyses.
In vivo examination.
All procedures were approved by the University of Rochester Committee on Animal Resources. Female 6- to 8-week-old CBA mice receiving TMX and/or AZD6244 were killed 1 d after completion of treatment and perfused with 4% paraformaldehyde. Analysis of cell death and cell division was as described previously (Dietrich et al., 2006; Han et al., 2008).
Results
Multiple CNS cell populations are sensitive to TMX
To identify CNS cell types vulnerable to TMX, we examined effects of this agent on NSCs, astrocytes, oligodendrocytes, O-2A/OPCs, GRP cells, and human glial progenitor cells in vitro. After 48 h of TMX exposure at clinically relevant concentrations, cell viability was measured.
O-2A/OPCs, GRP cells, and oligodendrocytes were most vulnerable to TMX (Fig. 1A). In O-2A/OPCs, exposure to even 100 nm TMX caused>30% reductions in cell number. TMX at 1 μm reduced the number of O-2A/OPCs, oligodendrocytes, and human glial progenitor cells by >75%. In O-2A/OPCs, apoptosis examined by TUNEL analyses was significantly induced after 12 h at TMX concentrations ≥1 μm (Fig. 1B). In contrast with other cell types, astrocytes were resistant to TMX, even at levels of 10 μm TMX. Sublethal exposure to TMX also suppressed division of O-2A/OPCs. Exposure to 50 nm TMX for 48 h caused an ∼14% reduction in the proportion of O-2A/OPCs incorporating BrdU, with greater reductions caused by TMX concentrations of 500 nm and 1 μm (∼29 and ∼43%, respectively; p < 0.01; Fig. 1C).
Figure 1.

Normal CNS cells are vulnerable to TMX. A, Different primary CNS cells were exposed to TMX for 48 h. Cell viability was determined by Calcein-AM staining and quantitative analysis with a Celigo adherent cell cytometer. B, C, In O-2A/OPCs exposed to TMX for 12 h, the percentage of TUNEL+ cells (B) and BrdU+ nuclei (C) from A2B5+ cells with intact nuclei (determined by DAPI staining) were counted in each condition. Data represent the mean ± SEM; N ≥ 3. *p < 0.05; **p < 0.01 (Student's t test).
The above dosages were within clinically relevant exposures for TMX, which readily crosses the blood–brain barrier. As used in long-term breast cancer treatment, TMX exposure results in average steady-state plasma concentrations of ∼200 nm. In comparison, concentrations of TMX and its metabolites achieved with standard dosages can be up to 2 μm in tumors and several-fold higher in brain tissue (Lien et al., 1991a,b; Iusuf et al., 2011). These low micromolar concentrations appear to be critical in enabling TMX to induce death of cancer cells (Chen et al., 2013).
An unbiased drug screening identifies pharmacological agents that reduce TMX toxicity on O-2A/OPCs
To identify agents potentially useful in protecting against TMX-induced toxicity, we screened the NINDS-II library of 1040 compounds containing drugs approved for human use or in clinical trials. O-2A/OPCs exposed to 1 μm TMX for 48 h, which killed >80% of cells, were also exposed to NINDS-II library members at concentrations of 1 μm. We found 27 compounds that protected against O-2A/OPC cell death, in some cases providing almost complete protection. Not surprisingly, treatment with multiple estrogen analogs prevented TMX toxicity, as shown for β-estradiol, which reduced the proportion of PI+ cells by more than half (Fig. 2A). Protection also was provided by the antioxidants α-tocopherol and resveratrol, and a variety of other substances.
Figure 2.

MEK1/2 inhibition suppresses TMX toxicity on O-2A/OPCs. A, Protective agents were identified in cultures of O-2A/OPCs coexposed to 1 μm TMX and an individual drug from the NINDS-II drug library for 48 h. Cell death was measured by percentage of PI+ cells in the total cell pool. B, C, O-2A/OPCs treated with DMSO or 1 μm TMX were coexposed to increasing concentrations of U0126 (B) or AZD6244 (C) for 48 h. Viable cell number was determined by Calcein-AM+ cell counting with a Celigo adherent cell cytometer. Error bars indicate SEM.
Of particular interest as protective agents were inhibitors of the Raf/MEK/ERK pathway (including L779450 (2-Chloro-5-[2-Phenyl-5-(4-pyridinyl-)-1H-imidazol-4-yl]phenol), U0126 (1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene), GW5074 (3-(3,5-Dibromo-4-hydroxy-benzylidene)-5-iodo-1,3-dihydro-indol-2-one); Fig. 2A), due to existing interest in this pathway as a potential anticancer target (Roberts and Der, 2007; Montagut and Settleman, 2009). This pathway was also of interest in light of findings that although genetic deficiency of ERK2 delays early stages of differentiation of O-2A/OPCs into oligodendrocytes, it has no apparent negative impacts on progenitor cells (Fyffe-Maricich et al., 2011; Guardiola-Diaz et al., 2012).
Coexposure to TMX plus 1μm L779450, U0126, or GW5074 reduced TMX-induced cell death to <40, <30, and <5% PI+ cells, respectively (Fig. 2A), leading us to also examine AZD6244 (also referred to as ARRY-142886/Selumetinib; a selective MEK1/2 inhibitor currently in Phase-II clinical trials for cancer treatment; Bodoky et al., 2012; Kirkwood et al., 2012). Exposure of O-2A/OPCs to 1 μm TMX in combination with UO126 or AZD6244 also attenuated TMX-induced reductions in cell number. UO126 was particularly effective at concentrations of 0.5–2.5 μm, and AZD6244 was particularly effective at a concentration of 2.5 μm, at which they essentially eliminated effects of TMX. Although rescue was also seen at higher concentrations, cell numbers at these higher concentrations were lower, as one would predict from the importance of MEK1/2 in cell division (Fig. 2B,C). Thus, these results also suggest optimal protection requires a particular dosage range.
MEK1/2 inhibition increases sensitivity of breast cancer cells to TMX
Agents that protect normal cells are of little use if they also protect cancer cells, making the possible utility of AZD6244 as an antineoplastic agent of particular interest. As TMX is used primarily as a treatment for luminal breast cancer, we examined effects of MEK1/2 inhibition on the response of MCF7 cells (the most widely studied TMX-sensitive breast cancer cells) to TMX.
Although MEK1/2 inhibition protected O-2A/OPCs from TMX toxicity, these inhibitors simultaneously enhanced the efficacy of TMX in suppressing MCF7 cell growth. Exposure to 1 μm TMX (i.e., an intratumor concentrations routinely achieved clinically; Kisanga et al., 2004; Gjerde et al., 2012) for 4 d reduced viability by ∼37% compared to undosed controls, while concurrent treatment with 2.5 μm U0126 or AZD6244 caused an additional 20% reduction in viability (Fig. 3A). These outcomes are in agreement with previous findings (Reddy and Glaros, 2007) that the antitumor efficacy of TMX was not compromised by the MEK1/2 inhibitor CI-1040 (2-(2-Chloro-4-iodo-phenylamino)-N-cyclopropylmethoxy-3,4-difluoro-benzamide) in vivo, and with other studies in which combined use of AZD6244 with standard chemotherapeutic agents enhances tumor suppression in melanoma patients with BRAF mutations (Patel et al., 2013) and in xenograft models of colorectal tumors (Holt et al., 2012). In contrast, β-estradiol suppressed the TMX toxicity for MCF7 cells (Fig. 3B,C), but these differential effects were not seen for O-2A/OPCs. In these progenitor cells, both AZD6244 and ß-estradiol protected against adverse effects of TMX.
Figure 3.

Differential effects of MEK1/2 inhibition on O-2A/OPCs and breast cancer cells. A, MCF7 cells were coexposed to TMX and 2.5 μm U0126, 2.5 μm AZD6244, or 1 μm β-estradiol for 4 d. AlamarBlue was used to measure viability of MCF7 cells as their ability to grow as aggregates precluded accurate Celigo-based analysis with Calcein-AM and PI. B, C, O-2A/OPCs were dosed with 1 μm TMX ± 2.5 μm U0126, 2.5 μm AZD6244, or 1 μm β-estradiol for 48 h, after which live cell number (B) and cell death (C) were determined by Calcein-AM and PI analyses, respectively. One-way ANOVA followed by Bonferroni pairwise comparison was performed. Data represent the mean ± SEM, normalized to control, N ≥ 3. *p < 0.05; **p < 0.01; ***p < 0.001. NS, Not significant.
AZD6244 rescues O-2A/OPCs from systemic TMX administration in vivo
To further examine potential concerns that TMX can cause CNS toxicity, and the potential utility of MEK1/2 inhibition in protecting against TMX toxicity, we next examined effects of TMX and AZD6244 in vivo. Animals were treated daily for 14 d with 5 mg/kg TMX plus 20 mg/kg AZD6244 (the dosage used in multiple human cancer xenograft mouse models and which shows marked antineoplastic effects; Davies et al., 2007; Yeh et al., 2007).
TMX treatment caused increased cell death and decreased cell division in the corpus callosum (CC). TMX-treated mice showed a more than threefold increase in TUNEL+ cells in the CC and a >60% reduction in BrdU+ cells (Fig. 4). TMX also caused an ∼34% reduction in the number of O-2A/OPCs in the CC (from 4.1 to 2.7% of total DAPI+ nuclei; p < 0.01), as determined by coexpression of the Olig2 transcriptional regulator and platelet-derived growth factor receptor-α (PDGFRα). In contrast, numbers of DAPI+ nuclei, oligodendrocyte-lineage cells (defined as Olig2+ cells), and differentiated oligodendrocytes (identified as PDGFRα−/Olig2+ cells) were not significantly altered. Although TMX did not cause increases in TUNEL+ cells in the subventricular zone (SVZ) or the hippocampal dentate gyrus (DG), two other zones of the adult CNS in which precursor cells are found, it did significantly suppress cell division in the CC, SVZ, and DG.
Figure 4.
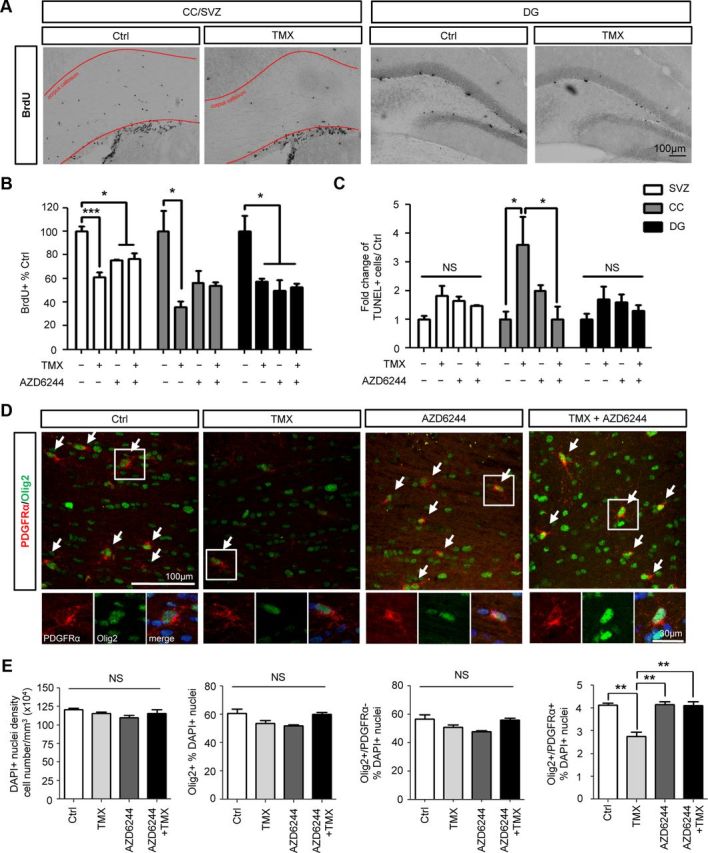
AZD6244 rescues O-2A/OPCs from TMX toxicity in vivo. Animals were treated with daily TMX (5 mg/kg/d), AZD6244 (20 mg/kg/d), or both for 14 d. One day after completing treatment, cell division and death in the SVZ, DG, and CC were examined by TUNEL and BrdU analyses, respectively. A, Representative images of BrdU+ cells in the SVZ, CC, and DG illustrate the reduction seen with TMX treatment. B, C, Quantitative analysis of BrdU and TUNEL labeling are presented in B and C, respectively. D, E, More detailed analysis of numbers of Olig2+PDGFRα+ O-2A/OPCs in the CC. Data represent the mean ± SEM, normalized to control; N = 3. *p < 0.05; **p < 0.01; ***p < 0.001 (as determined by one-way ANOVA followed by Bonferroni pairwise comparison). NS, Not significant. Arrows indicate cells coexpressing PDGFRα and Olig2, which are O-2A/OPCs.
Concurrent AZD6244 treatment effectively suppressed TMX-induced cell death in the CC (Fig. 4C). In mice cotreated with TMX plus AZD6244, cell death in the CC was restored to that seen in untreated animals (Fig. 4C). Cotreatment with TMX plus AZD6244 also prevented TMX-induced reductions in numbers of Olig2+/PDGFRα+ cells in the CC (Fig. 4D,E). Although treatment with AZD6244 alone suppressed cell division in the CNS, as expected for a MEK1/2 inhibitor, it neither increased nor decreased the suppression of division in the CC, SVZ or DG caused by systemic TMX treatment.
Discussion
We have discovered that TMX is toxic for precursor cells of the CNS at clinically relevant exposure levels, causes increased death of cells in the CC, and suppresses cell division in the CC, SVZ, and DG. Attempts to identify means of preventing TMX toxicity led to the discovery of multiple protective agents, some of them already approved for human use. Of particular interest among these agents were MEK1/2 inhibitors due to interest in such compounds as anticancer treatments. This led us to examine the compound AZD6244, which is currently in Phase-II clinical trials (Bodoky et al., 2012; Kirkwood et al., 2012). We found that AZD6244 protected O-2A/OPCs, but not MCF7 breast cancer cells from TMX toxicity in vitro, and further found that administration of AZD6244 in vivo prevented TMX-induced increases in cell death and reductions in the number of O-2A/OPCs in the CC.
Our findings present a possible cellular biological basis for the adverse neurological consequences that sometimes occur in patients receiving long-term TMX treatment. The increases in apoptosis in the CC and reductions in cell division in the SVZ, DG, and CC are of a nature that could lead to changes in neurological function and structure. Just as suppression of hippocampal neurogenesis by irradiation may be relevant to understanding cognitive alterations associated with this cancer treatment (Monje et al., 2002), similar suggestions apply to suppression of neuronal progenitor division in the hippocampus of animals treated with chemotherapeutic agents (Dietrich et al., 2006; Han et al., 2008; Janelsins et al., 2010; Mondie et al., 2010). The toxic effects of multiple chemotherapeutic agents on myelin-forming oligodendrocytes and their progenitors (Dietrich et al., 2006; Han et al., 2008), which are essential for normal axonal impulse conduction, suggests that neuron-related toxicities may represent a partial view of the complexity of this damage. In this context, it is intriguing that changes in white matter integrity offer one of the strongest correlates of cognitive decline in aging and of reduced intelligence in association with stroke (Silbert et al., 2008; Gläscher et al., 2010), raising the possibility that myelin damage is of importance in understanding adverse neurological effects of systemic chemotherapy. It is important to note, however, that although TMX was toxic, it was less so than such other cancer treatments as carmustine [1,3-Bis(2-chloroethyl)-1-nitrosourea (BCNU)], cisplatin, cytarabine, and 5-fluorouracil. Our previous studies showed these agents all caused marked cell death in the CC, DG, and SVZ (Dietrich et al., 2006; Han et al., 2008), as well as exhibiting the division-suppressing effects of TMX.
The observations that astrocytes were more resistant than precursor cells to TMX in vitro (as also observed for other chemotherapeutic agents; Dietrich et al., 2006; Han et al., 2008) was intriguing in light of the sensitivity of GFAP-expressing neuroprogenitor cells of the hippocampus to irradiation (Encinas et al., 2008). Whether this is due to higher glutathione content in astrocytes as compared with CNS precursor cells (Thorburne and Juurlink, 1996; Dringen, 2000) is not known, but observations that irradiation did not increase cell death outside of the neurogenic zone of the hippocampus (Encinas et al., 2008) suggest that GFAP+ stem cells may differ from astrocytes themselves in this aspect of their biology. It also will be of importance in future studies to define in detail which specific precursor cell populations outside of O-2A/OPCs are affected in their division by TMX treatment, and to discover means of protecting from this effect.
The strategy of screening agents already approved for human usage or in clinical trials was surprisingly successful in identifying potential protective agents. While the protective properties of estrogenic compounds was not surprising, and the findings that TMX expresses pro-oxidant activities (Gundimeda et al., 1996; Ferlini et al., 1999; Chen et al., 2013) makes it not surprising that we found antioxidants with protective activity, neither of these classes of agents would be suitable for combatting CNS toxicity in vivo due to potential rescue also of tumor cells. MEK1/2 inhibitors, in contrast, have the particularly interesting property of protecting normal cells while enhancing toxicity of TMX for MCF7 cells.
Our studies on AZD6244 offer the apparently novel demonstration that this agent seems able to cross the blood–brain barrier and is of potential use in reducing toxicity of TMX. AZD6244 has been shown previously to suppress tumor progression and to sensitize cancer cells to other therapeutic agents by inhibiting MEK1/2 activation (Holt et al., 2012; Patel et al., 2013). It was of interest in this regard that AZD6244 rendered MCF7 luminal breast cancer cells more sensitive to TMX (Fig. 3A), raising the possibility that AZD6244 could be used as an adjunct breast cancer therapy with TMX. Moreover, the protective properties of AZD6244 in the CNS, where TMX monotherapy causes apparent damage, makes this agent a particularly attractive candidate for possible use with TMX clinically. While previous studies have not examined AZD6244 in the context of either breast cancer or effects on the CNS, previous studies (Reddy and Glaros, 2007) showed that the antitumor efficacy of TMX was not compromised by coexposure to MEK1/2 inhibition (by CI-1040) in vivo. It will be of interest to optimize dosage in future experiments, to obtain maximum protection with minimum suppression of division of primary progenitor cells in vivo. Nonetheless, the identification of TMX-induced CNS toxicity and of a means of providing protection with an agent that at the same time does not protect, and may enhance, suppression of cancer cell growth is a promising first step in understanding and preventing the adverse neurological consequences of TMX exposure.
Footnotes
This work was supported by the National Institutes of Health (CA131385), the Department of Defense (W81XWH-07-1-060), the Susan G. Komen Race for the Cure Foundation, and the Carlson Stem Cell Fund. We thank Christoph Pröschel, Margot Mayer-Pröschel, Helene McMurray, Brett Stevens, and Craig Jordan for multiple helpful suggestions and critical discussions. We are also grateful to members of the Department of Biostatistics, in particular Olivier Hyrrien, for advice on all statistical analyses.
The authors declare no competing financial interests.
References
- Bender CM, Paraska KK, Sereika SM, Ryan CM, Berga SL. Cognitive function and reproductive hormones in adjuvant therapy for breast cancer: a critical review. J Pain Symptom Manage. 2001;21:407–424. doi: 10.1016/S0885-3924(01)00268-8. [DOI] [PubMed] [Google Scholar]
- Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, Tebbutt NC. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30:1216–1223. doi: 10.1007/s10637-011-9687-4. [DOI] [PubMed] [Google Scholar]
- Brown MS, Stemmer SM, Simon JH, Stears JC, Jones RB, Cagnoni PJ, Sheeder JL. White matter disease induced by high-dose chemotherapy: longitudinal study with MR imaging and proton spectroscopy. Am J Neuroradiol. 1998;19:217–221. [PMC free article] [PubMed] [Google Scholar]
- Chen HY, Yang YM, Stevens BM, Noble M. Inhibition of redox/Fyn/c-Cbl pathway function by Cdc42 controls tumour initiation capacity and tamoxifen sensitivity in basal-like breast cancer cells. EMBO Mol Med. 2013;5:723–736. doi: 10.1002/emmm.201202140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6:2209–2219. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- Dietrich J. Chemotherapy associated central nervous system damage. Adv Exp Med Biol. 2010;678:77–85. doi: 10.1007/978-1-4419-6306-2_11. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Noble M, Mayer-Proschel M. Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia. 2002;40:65–77. doi: 10.1002/glia.10116. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Han R, Yang Y, Mayer-Pröschel M, Noble M. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J Biol. 2006;5:22. doi: 10.1186/jbiol50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/S0301-0082(99)00060-X. [DOI] [PubMed] [Google Scholar]
- Eberling JL, Wu C, Tong-Turnbeaugh R, Jagust WJ. Estrogen- and tamoxifen-associated effects on brain structure and function. Neuroimage. 2004;21:364–371. doi: 10.1016/j.neuroimage.2003.08.037. [DOI] [PubMed] [Google Scholar]
- Encinas JM, Vazquez ME, Switzer RC, Chamberland DW, Nick H, Levine HG, Scarpa PJ, Enikolopov G, Steindler DA. Quiescent adult neural stem cells are exceptionally sensitive to cosmic radiation. Exp Neurol. 2008;210:274–279. doi: 10.1016/j.expneurol.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeland MA, Shumaker SA, Limacher M, Rapp SR, Bevers TB, Barad DH, Coker LH, Gaussoin SA, Stefanick ML, Lane DS, Maki PM, Resnick SM. Relative effects of tamoxifen, raloxifene, and conjugated equine estrogens on cognition. J Womens Health (Larchmt) 2010;19:371–379. doi: 10.1089/jwh.2009.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlini C, Scambia G, Marone M, Distefano M, Gaggini C, Ferrandina G, Fattorossi A, Isola G, Benedetti Panici P, Mancuso S. Tamoxifen induces oxidative stress and apoptosis in oestrogen receptor-negative human cancer cell lines. Br J Cancer. 1999;79:257–263. doi: 10.1038/sj.bjc.6690042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyffe-Maricich SL, Karlo JC, Landreth GE, Miller RH. The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J Neurosci. 2011;31:843–850. doi: 10.1523/JNEUROSCI.3239-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjerde J, Gandini S, Guerrieri-Gonzaga A, Haugan Moi LL, Aristarco V, Mellgren G, Decensi A, Lien EA. Tissue distribution of 4-hydroxy-N-desmethyltamoxifen and tamoxifen-N-oxide. Breast Cancer Res Treat. 2012;134:693–700. doi: 10.1007/s10549-012-2074-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gläscher J, Rudrauf D, Colom R, Paul LK, Tranel D, Damasio H, Adolphs R. Distributed neural system for general intelligence revealed by lesion mapping. Proc Natl Acad Sci U S A. 2010;107:4705–4709. doi: 10.1073/pnas.0910397107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardiola-Diaz HM, Ishii A, Bansal R. Erk1/2 MAPK and mTOR signaling sequentially regulates progression through distinct stages of oligodendrocyte differentiation. Glia. 2012;60:476–486. doi: 10.1002/glia.22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundimeda U, Chen ZH, Gopalakrishna R. Tamoxifen modulates protein kinase C via oxidative stress in estrogen receptor-negative breast cancer cells. J Biol Chem. 1996;271:13504–13514. doi: 10.1074/jbc.271.23.13504. [DOI] [PubMed] [Google Scholar]
- Han R, Yang YM, Dietrich J, Luebke A, Mayer-Pröschel M, Noble M. Systemic 5-fluorouracil treatment causes a syndrome of delayed myelin destruction in the central nervous system. J Biol. 2008;7:12. doi: 10.1186/jbiol69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt SV, Logié A, Odedra R, Heier A, Heaton SP, Alferez D, Davies BR, Wilkinson RW, Smith PD. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer. 2012;106:858–866. doi: 10.1038/bjc.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iusuf D, Teunissen SF, Wagenaar E, Rosing H, Beijnen JH, Schinkel AH. P-glycoprotein (ABCB1) transports the primary active tamoxifen metabolites endoxifen and 4-hydroxytamoxifen and restricts their brain penetration. J Pharmacol Exp Ther. 2011;337:710–717. doi: 10.1124/jpet.110.178301. [DOI] [PubMed] [Google Scholar]
- Janelsins MC, Roscoe JA, Berg MJ, Thompson BD, Gallagher MJ, Morrow GR, Heckler CE, Jean-Pierre P, Opanashuk LA, Gross RA. IGF-1 partially restores chemotherapy-induced reductions in neural cell proliferation in adult C57BL/6 mice. Cancer Invest. 2010;28:544–553. doi: 10.3109/07357900903405942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, Middleton M, Cantarini M, Zazulina V, Kemsley K, Dummer R. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–567. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanga ER, Gjerde J, Guerrieri-Gonzaga A, Pigatto F, Pesci-Feltri A, Robertson C, Serrano D, Pelosi G, Decensi A, Lien EA. Tamoxifen and metabolite concentrations in serum and breast cancer tissue during three dose regimens in a randomized preoperative trial. Clin Cancer Res. 2004;10:2336–2343. doi: 10.1158/1078-0432.CCR-03-0538. [DOI] [PubMed] [Google Scholar]
- Lien EA, Solheim E, Ueland PM. Distribution of tamoxifen and its metabolites in rat and human tissues during steady-state treatment. Cancer Res. 1991a;51:4837–4844. [PubMed] [Google Scholar]
- Lien EA, Wester K, Lønning PE, Solheim E, Ueland PM. Distribution of tamoxifen and metabolites into brain tissue and brain metastases in breast cancer patients. Br J Cancer. 1991b;63:641–645. doi: 10.1038/bjc.1991.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers CA, Abbruzzese JL. Cognitive functioning in cancer patients: effect of previous treatment. Neurology. 1992;42:434–436. doi: 10.1212/WNL.42.2.434. [DOI] [PubMed] [Google Scholar]
- Mondie CM, Vandergrift KA, Wilson CL, Gulinello ME, Weber ET. The chemotherapy agent, thioTEPA, yields long-term impairment of hippocampal cell proliferation and memory deficits but not depression-related behaviors in mice. Behav Brain Res. 2010;209:66–72. doi: 10.1016/j.bbr.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje M, Dietrich J. Cognitive side effects of cancer therapy demonstrate a functional role for adult neurogenesis. Behav Brain Res. 2012;227:376–379. doi: 10.1016/j.bbr.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med. 2002;8:955–962. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283:125–134. doi: 10.1016/j.canlet.2009.01.022. [DOI] [PubMed] [Google Scholar]
- Palmer JL, Trotter T, Joy AA, Carlson LE. Cognitive effects of Tamoxifen in pre-menopausal women with breast cancer compared to healthy controls. J Cancer Surviv. 2008;2:275–282. doi: 10.1007/s11764-008-0070-1. [DOI] [PubMed] [Google Scholar]
- Patel SP, Lazar AJ, Papadopoulos NE, Liu P, Infante JR, Glass MR, Vaughn CS, LoRusso PM, Cohen RB, Davies MA, Kim KB. Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. Cancer. 2013;119:799–805. doi: 10.1002/cncr.27790. [DOI] [PubMed] [Google Scholar]
- Reddy KB, Glaros S. Inhibition of the MAP kinase activity suppresses estrogen-induced breast tumor growth both in vitro and in vivo. Int J Oncol. 2007;30:971–975. [PubMed] [Google Scholar]
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Schagen SB, van Dam FS, Muller MJ, Boogerd W, Lindeboom J, Bruning PF. Cognitive deficits after postoperative adjuvant chemotherapy for breast carcinoma. Cancer. 1999;85:640–650. doi: 10.1002/(SICI)1097-0142(19990201)85:3<640::AID-CNCR14>3.0.CO%3B2-G. [DOI] [PubMed] [Google Scholar]
- Schilder CM, Seynaeve C, Beex LV, Boogerd W, Linn SC, Gundy CM, Huizenga HM, Nortier JW, van de Velde CJ, van Dam FS, Schagen SB. Effects of tamoxifen and exemestane on cognitive functioning of postmenopausal patients with breast cancer: results from the neuropsychological side study of the tamoxifen and exemestane adjuvant multinational trial. J Clin Oncol. 2010;28:1294–1300. doi: 10.1200/JCO.2008.21.3553. [DOI] [PubMed] [Google Scholar]
- Silbert LC, Nelson C, Howieson DB, Moore MM, Kaye JA. Impact of white matter hyperintensity volume progression on rate of cognitive and motor decline. Neurology. 2008;71:108–113. doi: 10.1212/01.wnl.0000316799.86917.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer SM, Stears JC, Burton BS, Jones RB, Simon JH. White matter changes in patients with breast cancer treated with high-dose chemotherapy and autologous bone marrow support. Am J Neuroradiol. 1994;15:1267–1273. [PMC free article] [PubMed] [Google Scholar]
- Thorburne SK, Juurlink BH. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J Neurochem. 1996;67:1014–1022. doi: 10.1046/j.1471-4159.1996.67031014.x. [DOI] [PubMed] [Google Scholar]
- van Dam FS, Schagen SB, Muller MJ, Boogerd W, vd Wall E, Droogleever Fortuyn ME, Rodenhuis S. Impairment of cognitive function in women receiving adjuvant treatment for high-risk breast cancer: high-dose versus standard-dose chemotherapy. J Natl Cancer Inst. 1998;90:210–218. doi: 10.1093/jnci/90.3.210. [DOI] [PubMed] [Google Scholar]
- Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, Marlow A, Hurley B, Lyssikatos J, Lee PA, Winkler JD, Koch K, Wallace E. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]