

Abstract
Systemic inflammatory events often precipitate acute cognitive dysfunction in elderly and demented populations. Delirium is a highly prevalent neuropsychiatric syndrome that is characterized by acute inattention and cognitive dysfunction, for which prior dementia is the major predisposing factor and systemic inflammation is a frequent trigger. Inflammatory mechanisms of delirium remain unclear. We have modeled aspects of delirium during dementia by exploiting progressive neurodegeneration in the ME7 mouse model of prion disease and by superimposing systemic inflammation induced by the bacterial endotoxin lipopolysaccharide (LPS). Here, we have used this model to demonstrate that the progression of underlying disease increases the incidence, severity, and duration of acute cognitive dysfunction. This increasing susceptibility is associated with increased CNS expression of cyclooxygenase (COX)-1 in microglia and perivascular macrophages. The COX-1-specific inhibitor SC-560 provided significant protection against LPS-induced cognitive deficits, and attenuated the disease-induced increase in hippocampal and thalamic prostaglandin E2, while the COX-2-specific inhibitor NS-398 was ineffective. SC-560 treatment did not alter levels of the proinflammatory cytokines interleukin (IL)-1β, tumor necrosis factor-α, IL-6, or C-X-C chemokine ligand 1 in blood or brain, but systemic IL-1RA blocked LPS-induced cognitive deficits, and systemic IL-1β was sufficient to induce similar deficits in the absence of LPS. Furthermore, the well tolerated COX inhibitor ibuprofen was protective against IL-1β-induced deficits. These data demonstrate that progressive microglial COX-1 expression and prostaglandin synthesis can underpin susceptibility to cognitive deficits, which can be triggered by systemic LPS-induced IL-1β. These data contribute to our understanding of how systemic inflammation and ongoing neurodegeneration interact to induce cognitive dysfunction and episodes of delirium.
Introduction
Systemic inflammation can induce acute cognitive impairment in the vulnerable brain (Barrientos et al., 2006; Chen et al., 2008; Murray et al., 2012). The clearest evidence for this in clinical populations is the frequent precipitation of delirium by surgical and inflammatory insults (MacLullich et al., 2008). Delirium is a profound acute disturbance of consciousness and cognitive function that is very common in the aged and demented populations, with a prevalence of 22–89% (Fick et al., 2002). Episodes of delirium are associated with significant lengthening of hospital stays, subsequent cognitive decline (MacLullich et al., 2009; Davis et al., 2012), acceleration of dementia (Kat et al., 2008; Fong et al., 2009), and earlier permanent institutionalization (Witlox et al., 2010). Despite this considerable medical and economic burden, delirium is poorly understood (MacLullich and Hall, 2011), and a lack of animal models has contributed to this. Aging and dementia are the major risk factors for delirium, and, on this background of prior cognitive impairment, systemic inflammation is among the most common triggers (MacLullich et al., 2008).
We have exploited these etiological factors to create an animal model to study aspects of delirium induced by systemic inflammation superimposed on prevalent dementia (Murray et al., 2012). Key neurobiological features of dementia were modeled using the ME7 model of prion disease, which shows extracellular amyloidosis, synaptic loss, neuronal loss, and a well characterized sequence of affective, cognitive, and neurological changes (Guenther et al., 2001; Cunningham et al., 2003). By challenging normal and ME7 animals with the low-dose bacterial endotoxin lipopolysaccharide (LPS; 100 μg/kg) to mimic Gram-negative bacterial infection, we demonstrated that systemic inflammation induces acute and transient working memory deficits in animals with prior neurodegenerative disease without effect in normal animals. LPS also impaired learning of a Y-maze task in these animals but did not impair memory of spatial information learned before LPS treatment (Cunningham et al., 2009). These data are consistent with clinical data showing impairments in tasks requiring attention to and memory for trial-specific details but relative preservation of crystallized memory (Brown et al., 2011). Inattention and short-term memory deficits of acute onset and transient course are key features of DSM-IV (American Psychiatric Association, 1994) and ICD-10 (World Health Organization, 1992) definitions of delirium. Thus, this animal model has validity for performing mechanistic studies relevant to understanding how systemic inflammation and underlying neurodegeneration interact to produce delirium.
Consistent with a neuroinflammatory hypothesis of delirium (Cunningham and Maclullich, 2013), mediators such as interleukin (IL)-6 and IL-8 are associated with postsurgical delirium (Beloosesky et al., 2007; van Munster et al., 2008; MacLullich et al., 2011), but specific inflammatory mediators cannot be demonstrated as being causal in clinical association studies. Here, using the previously described model system, we hypothesized that underlying neurodegenerative disease would lead to changes in prostaglandins and that this would progressively predispose these animals to acute cognitive dysfunction in response to systemic inflammation. Furthermore, we hypothesized that cyclooxygenase (COX) inhibition using selective nonsteroidal anti-inflammatory drugs (NSAIDs) would protect against the acute cognitive deficits induced by systemic LPS, via inhibition of prostaglandin synthesis.
Materials and Methods
Animals and stereotaxic surgery.
Female C57BL/6 mice at 8–12 weeks of age (Harlan Olac Ltd) were housed in cages containing five mice at 21°C with a 12 h light/dark cycle, and food and water available ad libitum. They were anesthetized with intraperitoneal injection of Avertin (2,2,2-tribromoethanol 50% w/v in tertiary amyl alcohol, diluted 1:40 in H2O; 20 ml/kg, i.p.) and stereotactically injected bilaterally with 1 μl of a 10% w/v scrapie (ME7 strain)-infected C57BL/6 brain homogenate [or 10% w/v normal brain homogenate (NBH)] at bregma (anteroposterior, −2.0 mm; mediolateral, ±1.6 mm; dorsoventral, −1.7 mm) using a Hamilton microsyringe. All animal procedures were performed in strict accordance with Irish Department of Health and Children regulations, after approval by the Trinity College Animal Research Ethics Committee.
Intraperitoneal challenges.
Experimental groups with ME7 or NBH were injected intraperitoneally with 100 μg/kg LPS (Salmonella Equine abortus; L5886, Sigma) in saline (10 ml/kg injection volume) at 12 or 15–16 weeks postinoculation. This dose mimics a mild-to-moderate infection, producing small changes (<1°C) in core body temperature, and has been previously shown to produce acute working memory deficits in ME7 animals (Murray et al., 2012). Control animals were administered nonpyrogenic saline in each case. In subsequent experiments, piroxicam (10 mg/kg in 0.2 m Tris-HCl, pH 8), NS-398 (8 mg/kg in 33% DMSO), SC-560 (30 mg/kg in 24% DMSO), and ibuprofen (30 mg/kg in saline) were administered intraperitoneally 60 min before LPS, and human recombinant IL-1RA (10 mg/kg in saline; Peprotech) was administered intraperitoneally at the same time as LPS. Piroxicam has previously been shown to effect robust inhibition of systemic prostaglandin E2 (PGE2) levels (Teeling et al., 2010), while NS-398 and SC-560 are specific inhibitors of COX-2 and COX-1, respectively. A subset of naive C57BL/6 mice were administered NS-398 (4 mg/kg, i.p.) and challenged with LPS (100 μg/kg) to test the ability of NS-398 to provide appropriate anti-inflammatory action in a well described paradigm. Another group of ME7 animals were challenged with recombinant mouse IL-1β (Escherichia coli derived; 401-ML/CF, R&D Systems) at a dose of 15 μg/kg, i.p., in saline. The dose was chosen based on previous work in the laboratory, which indicated it could induce a systemic and CNS inflammatory response with similar hypothermic effects as the LPS dose used in the current study (data not shown).
T-maze alternation: working memory.
We assessed working memory (3–8 h post-LPS administration) using alternation behavior in our previously described “escape from shallow water” T-maze task to allow assessment of working memory performance in animals experiencing sickness behavior (Murray et al., 2012). The T-maze was constructed of black Perspex with the following dimensions: long axis, 67 cm; short axis, 38 cm; depth, 20 cm; and arm width, 7 cm. There was a single 40-mm-diameter hole at the end of each choice arm, 2 cm from the floor. Black exit tubes were inserted into these holes (these may also be blocked to prevent exit). A “guillotine” door was inserted to prevent access to one or other choice arm. This maze was filled with water at 20°C to a depth of 2 cm to motivate mice to leave the maze by “paddling” or walking “on tip-toe” to an exit tube. Animals were taken with their cage mates to a holding cage. Each mouse was placed in the start arm of the maze with one arm blocked such that they were forced to make a left (or right) turn, selected in a pseudo-random sequence (equal numbers of left and right turns, no more than two consecutive runs to the same arm). On making this turn, the mouse could escape from the water by entering the small tube, and then a transit tube, in which it was carried to another holding cage. The mouse was held here for 25 s (intratrial interval), during which time the guillotine door was removed and the exit tube was switched to the alternate arm. The mouse was then replaced in the start arm and could choose either arm. The mouse must alternate from its original turn to escape. On choosing correctly, mice escape to the transit tube as before and are returned to their home cage. On choosing incorrectly, the mice were allowed to self-correct to find the correct exit arm. Animals were trained for blocks of 10 trials (intertrial interval of 20 min) before other experimental manipulations are performed. If animals developed a side preference early in training, a correction strategy could be used, as described in Murray et al. (2012). This correction strategy was never used within 2 d of testing for the memory-disruptive effects of substances. Only animals maintaining a criterion performance of ≥80%, and not showing any evidence of a side preference, were subsequently challenged with LPS.
RNA isolation, cDNA synthesis, and quantitative PCR.
The isolation of total RNA, the synthesis of cDNA, and the analysis of transcription by quantitative PCR have been performed essentially as previously described (Cunningham et al., 2007). Briefly, treated animals were transcardially perfused with heparinized saline, and the region of known prion pathology encompassing the hippocampus and thalamus was punched out of coronal brain sections of ∼2 mm thickness. Tissue was snap frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted from brain samples using Qiagen RNeasy Plus mini kits according to the manufacturer's instructions. Contaminating genomic DNA was removed using the Qiagen RNase-free DNase I enzyme. RNA yields were determined by spectrophotometry at 260 and 280 nm, and were stored at −80°C until cDNA synthesis and PCR assay. Using a High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems), cDNA was synthesized from total RNA. Two hundred nanograms of total RNA were reverse transcribed in a 10 μl reaction volume and 1 μl of the reverse transcription (RT) reaction was used for PCR. Reagents were supplied by Applied Biosystems (SYBR Green PCR Master Mix) and Roche [FastStart Universal Probe Master (Rox)]. Assays were designed using the published sequences for the genes of interest, and Primer Express software and primer pairs were checked for specificity by standard RT-PCR using Promega PCR reagents. Assays were quantified using a relative standard curve, as previously described (Cunningham et al., 2007), constructed from cDNA synthesized from 1 μg of total RNA isolated from mice showing upregulation of all target transcripts of interest in this study.
ELISA for plasma cytokines.
Under terminal anesthesia, the thoracic cavity was opened and blood was collected in heparinized tubes directly from the right atrium of the heart. Whole blood was centrifuged at 3000 rpm for 15 min at 4°C to remove cells, and the remaining plasma was aliquoted and stored at −20°C. These samples were then analyzed for IL-1β, tumor necrosis factor-α (TNF-α), IL-6, and chemokine C-X-C ligand-1 (CXCL-1). TNF-α, IL-6, and CXCL-1 were quantified using R&D Systems DuoSet sandwich-type ELISA, while IL-1β was analyzed using a Quantikine kit (R&D Systems). A standard protocol was followed as previously described (Murray et al., 2011) except for IL-1β, which was used according to the manufacturer's instructions with minor modification. Standards ranged from 500 to 7.8 pg/ml. Samples for other cytokine assays were serially diluted as follows: TNF-α, 1:2 and 1:4; IL-6, 1:12, 1:36, and 1:108; and CXCL1, 1:9, 1:81, and 1:243. Standards were prepared in the range of 1000–8 pg/ml, and samples were quantified only if the absorbance fell on the linear portion of the standard curve.
ELISA for brain PGE2.
PGE2 levels in hippocampal and hypothalamic tissue were quantified using a PGE2 EIA kit [Enzo Life Sciences (previously manufactured by Assay Designs)]. Standards were in the range of 2500–39.1 pg/ml. Briefly, hippocampal tissue was homogenized in 200 μl of homogenization buffer (0.1 m phosphate buffer, pH 7.4, containing 1 mm EDTA and 10 μm indomethacin), mixed with 1 ml of 100% ethanol, and kept on ice for 30 min. Samples were centrifuged at 3000 rpm for 10 min at 4°C and transferred to an empty tube, and the ethanol was evaporated under a constant stream of nitrogen. Samples were then resuspended in 250 μl of assay buffer, vortexed, and then allowed to rest for 5 min. This was repeated twice more. PGE2 levels were then measured according to manufacturer's instructions. Sample PGE2 concentrations were then normalized to the wet weight of the piece of tissue analyzed.
Immunohistochemistry.
ME7 and NBH animals at 12 and 16 weeks postinoculation and at 3 h post-LPS/saline administration were perfused transcardially with heparinized saline (5 U/ml) followed by 10% formalin, and were paraffin wax embedded for immunohistochemistry. Sections were labeled with antibodies against COX-1 (catalog #160109, Cayman Chemicals), COX-2 (sc-1747, Santa Cruz Biotechnology), and ionized calcium-binding adapter molecule-1 (IBA-1; Abcam). The IBA-1 protocol was adapted to include a 20 min pepsin pretreatment, as described below. Primary antibody against IBA-1 was used at 1:2000. In the case of COX-1, nonspecific peroxidase activity was eliminated by incubating sections in 1 ml of 30% H2O2/100 ml of methanol for 10 min before antigen retrieval was performed by microwaving in citrate buffer, pH 6, for 2 × 5 min. Sections were washed and treated with pepsin for 10 min (prepared at 0.4% in 0.01 m HCl and diluted 1:8). This pretreatment was also used with IBA-1. After washing, sections were blocked with normal sera and incubated overnight with primary antibodies at 1:600. Labeling was completed using the appropriate biotinylated secondary antibody ABC complex and diaminobenzidine as chromagen before hematoxylin counterstaining.
Double labeling and confocal microscopy.
Double-labeling experiments were performed using the primary antibody MR5D3 (AbD, Serotec) against CD206, a marker of perivascular macrophages (PVMs), and the COX-1 and IBA-1 primary antibodies described above. The Alexa Fluor (Invitrogen Biosciences) secondary reagents were as follows: goat anti-rabbit 488 (COX-1), chicken anti-rat 594 (CD206), and donkey anti-goat 546 (IBA-1) used at 1:400. Sections were treated for 20 min with 0.04% pepsin in 0.1 m HCl. For COX-1 and CD206, sections were blocked in 10% normal goat serum plus 10% normal chicken serum in PBS. CD206 primary antibody was incubated overnight (1:1500). The following day, sections were incubated in ant-rat 594 antibody and treated overnight with the primary antibody against COX-1 (1:600). Following the addition of the anti-rabbit 488, sections were mounted in ProLong Gold (Invitrogen Biosciences) and coverslipped. For COX-1 and IBA-1, sections were blocked in 10% normal donkey serum. IBA-1 antibody was incubated overnight (1:2000). The following day, sections were incubated in anti-goat 546 antibody and treated overnight with the primary antibody against COX-1 (1:600). Following the addition of the anti-rabbit 488, sections were mounted in ProLong Gold (Invitrogen Biosciences) and coverslipped. Double-labeled sections were visualized and captured on an Olympus FV1000 confocal microscope using sequential excitation at 488 and 594 (or 546) excitation with a 60× oil objective (numerical aperture, 1.35). Images shown for COX-1 and CD206 are single sections of 0.36 μm extracted from Z-stacks of 10 μm sections.
Statistical analyses.
Behavioral data were compared by repeated-measures ANOVA with Bonferroni post hoc analyses performed after any significant main effect. All PCR and ELISA data were compared by two-way ANOVA, with Bonferroni post hoc analyses performed after any significant main effect, with the exception of hippocampal PGE2 in Figure 4, which was analyzed by one-way ANOVA with Bonferroni post hoc analyses. All data were analyzed using GraphPad Prism 5.0 for Mac OS X.
Figure 4.

Nonspecific COX inhibition protects against LPS-induced working memory deficits and attenuates disease-associated increases in hippocampal PGE2. a, Working memory was assessed by alternation in the paddling T-maze. ME7 animals were challenged with LPS (100 μg/kg, n = 11) or saline (n = 7) in the presence of the nonspecific COX inhibitor piroxicam administered at 10 mg/kg, i.p., 60 min before LPS injection. Control ME7 animals (ME7+LPS, n = 12) were challenged with LPS 60 min post-vehicle injection (0.2 m Tris-HCl). Data were analyzed by two-way ANOVA, and Bonferroni post hoc analyses followed significant main effects: **p < 0.01, *p < 0.05 with respect to ME7+piroxicam (PIROX); +p < 0.05 with respect to ME7+LPS+PIROX. b, Hippocampal and thalamic PGE2 was measured in NBH and ME7 animals 2 h postchallenge with LPS or saline, in the presence or absence of piroxicam. After a significant main effect by one-way ANOVA, Bonferroni post hoc tests revealed significant differences as denoted by *p < 0.05 with respect to NBH+saline, and +++p < 0.001 with respect to ME7+saline. NBH+saline, n = 7; NBH+LPS, n = 5; ME7+saline, n = 9; ME7+LPS, n = 9; ME7+LPS+piroxicam, n = 4. All data have been presented as the mean ± SEM.
Results
Progression of disease increases the incidence, severity, and duration of acute exacerbations of cognitive function
To assess how the progression of disease influenced susceptibility to systemic inflammation-induced working memory deficits, we administered LPS (100 μg/kg, i.p.) to ME7 animals at 12 weeks or 15–16 weeks postinoculation. Regardless of the time point in disease, we challenged only ME7 animals that were still performing the task above the criterion (see Materials and Methods). As can be observed in Figure 1, ME7 animals at 12 or 15–16 weeks used in this part of the study showed equivalent performance before their challenge with LPS, and those challenged with saline maintained good performance across the entire experiment. NBH animals challenged with LPS (100 μg/kg, i.p.) did not show any impairment in working memory performance (alternation) between 3 and 8 h, but there were clear effects of LPS in ME7 animals and these were different at 12 and 15–16 weeks (Fig. 1). Repeated-measures ANOVA showed a main effect of treatment (p < 0.0001, F(3,48) = 18.43), a main effect of time (p < 0.0001, F(6,288) = 5.945), and an interaction between these two factors (p = 0.0037, F(18,288) = 2.201). ME7 animals at 12 weeks postinoculation showed decreased alternation at 5 h post-LPS (p < 0.05 vs NBH+LPS), although robust impairment did not occur in all animals and deficits had resolved by 24 h. By contrast, LPS-challenged ME7 animals at 15–16 weeks postinoculation showed robustly decreased alternation performance, and this impairment was more prolonged, peaking at 7 h and remaining incompletely resolved at 24 h (p < 0.05 with respect to ME7+saline). Bonferroni post hoc analyses show that the working memory of ME7+LPS animals at 15 weeks remained impaired with respect to NBH+LPS controls between 3 and 8 h (p < 0.01 at 3 h; p < 0.001 at 5 and 7 h) and was also significantly impaired with respect to ME7+LPS animals (12 weeks) at 7 h (p < 0.05). Thus, LPS has more profound effects at 15–16 weeks of disease compared with 12 weeks postinoculation. These data demonstrate that progression of disease predicts increased incidence, severity, and duration of acute working memory deficits induced by systemic inflammation.
Figure 1.

Disease progression increases the incidence, severity, and duration of acute exacerbations of cognitive dysfunction. Working memory was assessed by alternation in the paddling T-maze. All animals were assessed 24 h before acute challenge, tested for 5–6 h postchallenge, and reassessed 24 h after challenge. T-maze alternation of NBH animals (n = 9) versus ME7 animals at 12 weeks (n = 16) or 15–16 weeks (n = 15) postinoculation challenged with LPS (100 μg/kg) and ME7 animals at 15–16 weeks challenged with sterile saline (n = 12) were compared by two-way ANOVA. Significant post hoc differences between ME7+LPS (12 weeks and 15–16 weeks) and NBH+LPS are denoted as follows: *p < 0.05; **p < 0.01; and ***p < 0.001. Significant post hoc differences between ME7+LPS (15–16 weeks) and ME7+LPS (12 weeks) animals are denoted as follows: +p < 0.05. All data have been presented as the mean ± SEM. sal, Saline; w, week; hr, hour.
Prostaglandin biosynthetic enzymes during disease progression and after LPS challenge
Brain tissue punches, which encompassed the hippocampus and thalamus (area of maximum pathology in ME7 model; Fig. 2b, dotted line) were taken from NBH and ME7 animals. The transcription of mRNA for the biosynthetic enzymes COX-1, COX-2, microsomal PGE synthase 1 (mPGES)-1, mPGES-2, and cytosolic PGES (cPGES) was then examined as a function of disease progression (Fig. 2c) and in response to LPS challenge (Fig. 2d) in these hippocampal/thalamic punches. COX-1 mRNA expression was increased in ME7 animals with progressing disease. Analysis by two-way ANOVA revealed that there was a main effect of disease (p < 0.0001, F(1,16) = 110.7), a main effect of time (p = 0.0002, F(1,16) = 22.76), and an interaction of disease and time (p = 0.0021, F(1,16) = 13.41). Post hoc Bonferroni tests revealed a significant increase in COX-1 expression in ME7 at both 12 and 16 weeks versus NBH control (p < 0.001) and a further increase at 16 weeks versus ME7 at 12 weeks (p < 0.001). COX-2 mRNA expression also showed a disease-associated increase, although this was less marked and was not yet obvious at 12 weeks. There was a main effect of disease (p = 0.0034, F(1,16) = 11.75), a main effect of time (p = 0.0241, F(1,16) = 6.202), and an interaction between these two factors (p = 0.0241, F(1,16) = 6.206). Bonferroni post hoc tests revealed a significant increase in COX-2 expression in ME7 at 16 weeks with respect to NBH control and ME7 at 12 weeks. mPGES-1 was also induced by disease progression, and there were main effects of disease (p < 0.0001, F(1,15) = 38.21) and of time (p = 0.1609, F(1,15) = 6.094). Bonferroni post hoc analyses revealed a significant increase in mPGES-1 expression at both 12 and 16 weeks with respect to NBH control (p < 0.05, p < 0.001 respectively), with a further increase in ME7 at 16 weeks versus ME7 at 12 weeks (p < 0.05). Expression of mPGES2 and cPGES mRNA was not significantly altered by disease progression.
Figure 2.
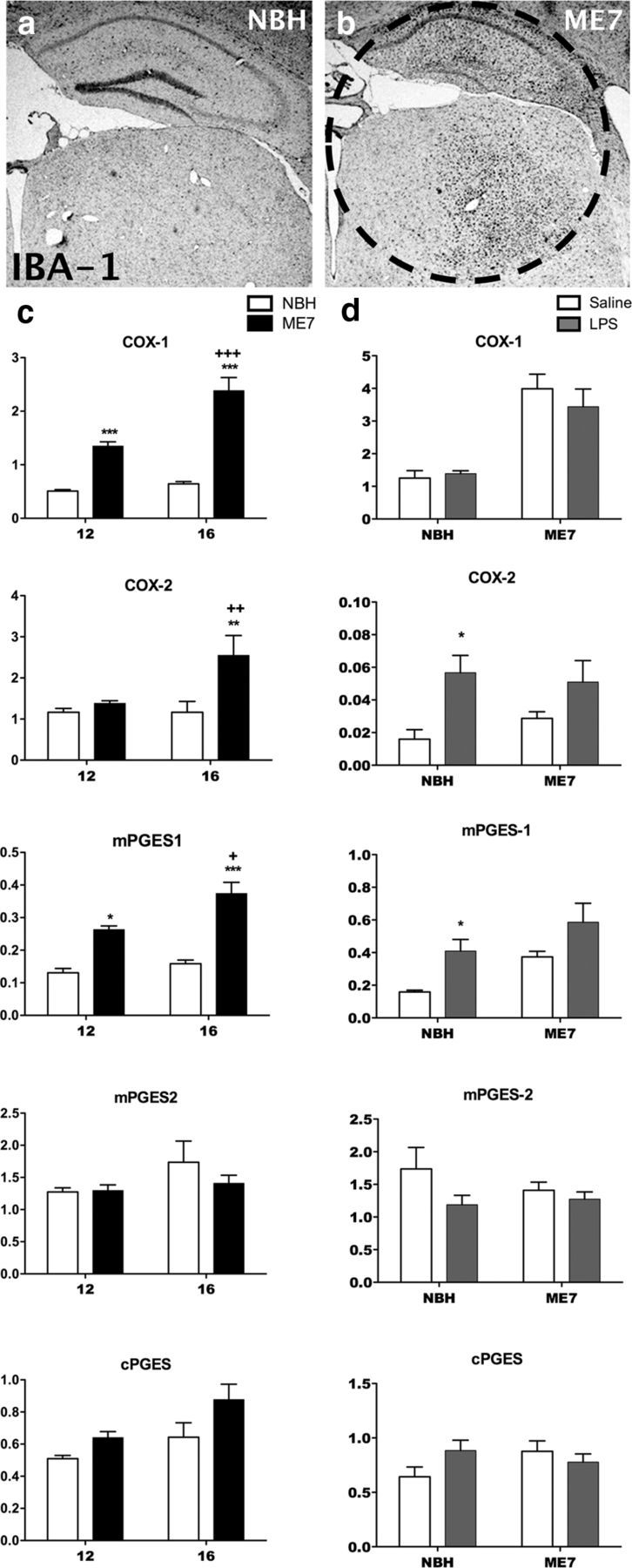
Expression of prostaglandin biosynthetic enzymes during disease progression and post-LPS challenge. a, b, Low-magnification (2.5×) images of IBA-1 immunohistochemistry showing the pattern of microgliosis in the hippocampus and thalamus of NBH (a) and ME7 (16 week; b) animals. The circled region is the hippocampal and thalamic “punch” dissected from 2 mm coronal sections and used for PCR analysis. The transcription of mRNA species for the biosynthetic enzymes COX-1, COX-2, mPGES-1, mPGES-2, and cPGES were assessed in the hippocampal and thalamic region of the brain using quantitative PCR. c, NBH at 12 and 16 weeks postinoculation is shown by white bars; ME7 at 12 and 16 weeks is shown by black bars. Expression was assessed by two-way ANOVA with significant post hoc Bonferroni analyses denoted by **p < 0.01, ***p < 0.001 with respect to NBH controls and ++p < 0.01, +++ p < 0.001 with respect to ME7 animals at 12 weeks; n = 4 for all groups except for ME7 at 12 weeks (n = 8) for COX-1 and COX-2; and ME7 at 16 weeks (n = 7) for the PGE synthases. d, The effect of saline or LPS injection on NBH and ME7 at 16 weeks is shown by white and gray bars, respectively. Expression was assessed by two-way ANOVA with post hoc Bonferroni analyses: *p < 0.05 with respect to saline-treated control. n = 7 for all groups except NBH+saline (n = 6) for COX-1 and COX-2; NBH+saline (n = 4), NBH+LPS (n = 5), ME7+saline (n = 7), and ME7+LPS (n = 4) for the PGE synthases. All data have been presented as the mean ± SEM.
The effect of LPS challenge on the transcription of mRNA for these biosynthetic enzymes was also assessed in ME7 animals, 16 weeks postinoculation (Fig. 2d). At 2 h postchallenge with LPS (100 μg/kg),there was a main effect of LPS on COX-2 mRNA (p = 0.0029, F(1,21) = 11.33) and on mPGES-1 expression (p = 0.0023, F(1,16) = 13.06). Expression of COX-1, mPGES2, and cPGES was not altered by LPS challenge. These data demonstrate increased expression of prostaglandin biosynthetic enzymes COX-1, COX-2, and mPGES-1 with the progression of disease and inducible expression of COX-2 and mPGES-1 with systemic inflammatory challenge.
Immunohistochemical detection of microglia, COX-1, and COX-2
We used IBA-1 to show the distribution of microglial cells in the hippocampus and thalamus (Fig. 3), and counted the number of IBA-1-positive cells per field of view at 20× magnification (counts were performed in four dorsal hippocampal fields of view per animal). These cells can be seen to increase from NBH to ME7 at 12 weeks (NBH: n = 3, 20 ± 1 cells; ME7 at 12 weeks: n = 4, 60 ± 5 cells) and to increase in number to ME7 at 16 weeks (n = 3, 80 ± 17 cells; Fig. 3a–c). Neither microglial cell density nor numbers significantly change after LPS challenge (n = 3, 88 ± 16 cells; Fig. 3d), as reported in the initial description of microglial priming (Cunningham et al., 2005). Cells were too densely packed in the thalamus to reliably count. COX-1-positive cells showed similarly increased numbers as disease progressed (Fig. 3e–h). These cells are clearly morphologically identifiable as microglia (Fig. 3g, inset) and were confirmed, using double-labeling immunofluorescence for COX-1 and IBA-1, to be microglia (Fig. 3i–k). There were also a number of vascular-associated cells that show a perinuclear labeling pattern and can be clearly distinguished from the unlabeled endothelial cell layer (Fig. 3h, inset). These cells were confirmed, using double-labeling immunofluoresence for COX-1 and CD206, to be perivascular macrophages (Fig. 3l–n). Thus, COX-1 is strongly expressed in both microglia and PVMs. COX-2 was increased in ME7 with respect to NBH (Fig. 3o–r). However, COX-2 labeling is induced at the endothelium following LPS challenge (Fig. 3r, inset). Preabsorption of the COX-2 antibody with a blocking peptide completely ablates the well described post-LPS COX-2 labeling of the endothelium (Fig. 3p, inset) and the constitutive neuronal COX-2-positive labeling. It also blocks the observed microglial COX-2 labeling without effect on COX-1 labeling. Collectively, these data illustrate reliable and specific COX-2 labeling using this antibody.
Figure 3.

Immunohistochemical detection of microglia and cyclooxygenases. a–d, The distribution of microglia in the CA1 hippocampal region of normal and diseased brains was examined using the microglial marker IBA-1 (20×). e–h, Expression of COX-1 in the CA1 hippocampal region of normal and diseased brains shows increased density of positive cells at 16 weeks > 12 weeks > NBH (40×; scale bar, 100 μm), but no further increase post-LPS. g, Inset, The COX-1-positive cells show clear microglial morphology. h, Inset, Hippocampal blood vessels show a population of COX-1-positive cells that are clearly distinguishable from the endothelial layer. i–k, Double-immunofluoresence labeling for IBA-1 (i, red) and COX-1 (j, green) shows double-labeled microglia (k). l–n, Double-immunofluoresence labeling for CD206 (l, red) and COX-1 (m, green) shows double-labeled perivascular macrophages (n). Scale bar, 10 μm. o–r, Expression of COX-2 in the CA1 hippocampal region with inset magnifications of the endothelium. COX-2 increases at the endothelium following LPS (r, inset). COX-2 antibody preabsorbed with COX-2 peptide blocks LPS-induced endothelial labeling (p, inset).
Effects of cyclooxygenase inhibition on LPS-induced working memory deficits and brain prostaglandin levels
Since there were both disease-associated and LPS-induced changes in the expression of prostaglandin biosynthetic enzymes, we assessed the ability of COX inhibition to prevent the acute working memory deficits induced by LPS in ME7 animals. We have demonstrated that LPS has no effect on T-maze alternation in NBH animals (Fig. 1), and similarly, we have also demonstrated that saline injection has no effect on working memory in ME7 animals (Fig. 1); therefore, we do not repeat these groups in subsequent cognitive experiments. The nonspecific cyclooxygenase inhibitor piroxicam afforded significant protection against the acute working memory deficits induced by LPS (Fig. 4a). There was a main effect of treatment (p < 0.0001, F(2,27) = 17.11) and a main effect of time (p < 0.0001, F(6,162) = 6.12). Bonferroni post hoc tests revealed a decrease in T-maze alternation in the ME7+LPS at 3 and 5 h (p < 0.01) and at 7 h (p < 0.05), and a significant difference between ME7+LPS and ME7+LPS+piroxicam at 3 h postchallenge (p < 0.05), indicating that piroxicam prevented LPS-induced decrease in T-maze alternation. As piroxicam was effective in attenuating LPS-induced working memory deficits, we also assessed its impact on PGE2 levels in the hippocampus and thalamus, which were elevated in ME7 animals (Fig. 4b). There was a significant main effect by one-way ANOVA (p = 0.0001, F(4,29) = 8.848), and post hoc Bonferroni analyses revealed a significant disease-associated increase in hippocampal and thalamic PGE2 levels. ME7+saline had markedly higher levels of PGE2 relative to NBH+saline animals (p < 0.05, t = 3.441) but was not different to ME7+LPS (p > 0.05). Piroxicam completely ablated the synthesis of PGE2 in ME7 animals challenged with LPS relative to ME7+LPS (p < 0.05, t = 3.819), suggesting inhibition of both isoforms of cyclooxygenase.
Since systemic administration of LPS induced COX-2 expression at the brain endothelium, we next assessed the ability of a COX-2-specific inhibitor to prevent LPS-induced working memory deficits (Fig. 5a). Pretreatment of animals with the specific COX-2 inhibitor NS-398 did not protect against LPS-induced working memory deficits: there were significant effects of time (p < 0.0001, F(6,138) = 8.906) and treatment (p = 0.0307, F(2,138) = 4.067), reflecting the LPS-induced deficit in working memory, but there was no difference between ME7+LPS and ME7+LPS+NS-398, indicating that NS-398 could not prevent the LPS-induced decrease in T-maze alternation. Moreover, NS-398 did not significantly attenuate the disease-induced increase in hippocampal and thalamic PGE2 (Fig. 5b). There was a significant effect of disease (p = 0.0157, F(1,12) = 7.901), reflecting the increase in hippocampal and thalamic PGE2 in ME7 animals but no effect of NS-398 treatment, supporting the hypothesis that the disease-induced increase in hippocampal PGE2 is primarily COX-1 dependent. To control for the efficacy of NS-398 per se, we assessed the ability of NS-398 pretreatment to attenuate LPS-induced hypothalamic PGE2 (Fig. 5c). There was a main effect of LPS (p = 0.0349, F(1,14) = 5.454) and of NS-398 (p = 0.0332, F(1.14) = 5.578). Bonferroni post hoc analyses revealed a significant induction of hypothalamic PGE2 as a result of LPS challenge (p < 0.05) as expected, which was blocked by NS-398 treatment (NS-398+LPS vs vehicle+LPS, p < 0.05), confirming the efficacy of the drug. We also found that pretreatment with the COX-2-selective drug nimesulide (10 mg/kg, i.p.) was not protective against the LPS-induced deficits (data not shown).
Figure 5.

COX-2-specific inhibition fails to protect against LPS-induced working memory deficits, and does not affect disease-associated elevated hippocampal and thalamic PGE2, but blocks LPS-induced increases in hypothalamic PGE2. a, Working memory was assessed by alternation in the paddling T-maze. ME7 animals were challenged with LPS (100 μg/kg, i.p.; n = 9) or saline (n = 9) in the presence of the COX-2-specific inhibitor NS-398 administered at 8 mg/kg, i.p., 60 min before LPS injection. Control ME7 animals (ME7+LPS, n = 8) were challenged with LPS 60 min post-vehicle injection (33% DMSO). Data were analyzed by two-way ANOVA, and Bonferroni post hoc analyses followed significant main effects: *p < 0.05 with respect to saline challenged animals (ME7+NS-398). b, Hippocampal and thalamic PGE2 levels were measured in NBH and ME7 animals in the presence or absence of NS-398 (8 mg/kg, i.p.). There was a significant main effect of disease by two-way ANOVA but no effect of COX-2 inhibition on PGE2 concentrations (NBH+vehicle, n = 3; NBH+NS-398, n = 4; ME7+vehicle, n = 4; ME7+NS-398, n = 5). c, Hypothalamic PGE2 was measured 2 h postchallenge with LPS or saline, in the presence (n = 5 per group) or absence (n = 4 per group) of NS-398 (4 mg/kg, i.p.). After a significant main effect by two-way ANOVA, Bonferroni post hoc tests revealed significant differences, as denoted by *p < 0.05 with respect to vehicle+saline, and +p < 0.05 with respect to vehicle+LPS. All data have been presented as the mean ± SEM.
In light of these results, we assessed the ability of COX-1-specific inhibition to protect against inflammation-induced cognitive deficits using the COX-1-specific inhibitor SC-560 (Fig. 6a). SC-560 showed protection against LPS-induced deficits. There was a main effect of time (p < 0.0001, F(6,204) = 7.955), a main effect of treatment (p = 0.0286, F(2,34) = 3.952), and a significant interaction between these two factors (p = 0.0105, F(12,204) = 2.259). Bonferroni post hoc analyses revealed a significant LPS-induced deficit in T-maze alternation peaking at 5 h postchallenge (p < 0.01 with respect to saline-challenged controls), which was blocked by treatment with SC-560 (p < 0.05), indicating that COX-1-specific inhibition provides significant protection against inflammation-induced cognitive deficits in diseased animals. Moreover, SC-560 decreased the concentrations of PGE2 in the hippocampus and thalamus of ME7 animals (Fig. 6b). Analysis by two-way ANOVA revealed a significant effect of treatment (p < 0.0001, F(1,15) = 43.73) but no main effect of challenge, indicating that PGE2 in the hippocampus and thalamus was reduced by SC-560 treatment in both ME7+saline (p < 0.001) and ME7+LPS (p < 0.01) groups relative to vehicle-treated controls. However, a direct comparison of the two SC-560-treated groups by Student's t test indicated that in the absence of COX-1-mediated PGE2, LPS still induces a detectable increase in hippocampal and thalamic PGE2 (p = 0.0297, t = 2.722; Fig. 6b). These data suggest that COX-1-specific inhibition can reduce disease-elevated prostaglandins in the brain but has no impact on LPS-induced prostaglandin production, which is mediated by COX-2. Despite this lack of effect against COX-2-mediated PGE2 synthesis, SC-560 is nonetheless protective against LPS-induced deficits.
Figure 6.

COX-1-specific inhibition provides clear protection against LPS-induced working memory deficits, and decreases hippocampal and thalamic prostaglandin E2 concentrations. a, Working memory was assessed by alternation in the paddling T-maze. ME7 animals were challenged with LPS (100 μg/kg, i.p.; n = 13) or saline (n = 8) in the presence of the COX-1-specific inhibitor SC-560 administered at a dose of 30 mg/kg, i.p., 60 min before LPS injection. Control ME7 animals (ME7+LPS, n = 16) were challenged with LPS 60 min post-vehicle injection (24% DMSO). Data were analyzed by two-way ANOVA, and Bonferroni post hoc analyses followed significant main effects: **p < 0.01 denotes a significant decrease in ME7+LPS alternation versus ME7+SC-560; and +p < 0.05 versus ME7+LPS+SC-560. b, Hippocampal and thalamic PGE2 levels were measured in ME7 animals 2 h postchallenge with LPS or saline, in the presence or absence of SC-560 (30 mg/kg, i.p.). After a significant main effect by two-way ANOVA, Bonferroni post hoc tests revealed significant differences, as denoted by ++p < 0.01 and +++p < 0.001 with respect to vehicle-treated controls; n = 5 per group with the exception of ME7+saline+SC-560 (n = 4). All data have been presented as the mean ± SEM.
Effects of SC-560 on systemic and CNS proinflammatory cytokines
To assess whether SC-560 may have exerted its effects via inhibition of cytokine synthesis, systemic proinflammatory cytokine synthesis was also examined in the plasma of ME7 animals 2 h post-LPS challenge with or without SC-560 pretreatment (Fig. 7). Data were analyzed by two-way ANOVA, and significant main effects were followed by post hoc Bonferroni analyses. There was a significant main effect of LPS challenge on IL-1β concentration in the plasma (p < 0.001, F(1,15) = 41.96) but no effect of SC-560 treatment, indicating an LPS-induced increase in plasma IL-1β, which was not affected by COX-1 inhibition. Similarly for plasma IL-6, TNFα, and CXCL-1, LPS induced a significant increase in plasma concentrations of these proinflammatory cytokines (main effect of challenge, respectively: p < 0.0001, F(1,15) = 37.64; p = 0.0039, F(1,14) = 11.88; p < 0.0001, F(1,15) = 155.8), but SC-560 treatment had no effect. Thus, systemically administered SC-560 has no impact on the induction of plasma cytokine concentrations induced by systemic LPS.
Figure 7.

COX-1-specific inhibition does not attenuate inflammatory responses to LPS challenge. Expression of IL-1β, TNF-α, IL-6, and CXCL-1 was measured in the plasma and the hippocampus and thalamus of ME7 animals 2 h after challenge with saline or LPS (100 μg/kg), in the presence or absence of SC-560 (30 mg/kg). Plasma levels of proinflammatory cytokines were measured using DuoSet ELISA kits and brain transcription of proinflammatory cytokine transcripts using quantitative PCR. There was a significant main effect of challenge by two-way ANOVA, with post hoc Bonferroni tests indicating a significant effect of LPS: *p < 0.05, **p < 0.01, ***p < 0.001 with respect to saline-challenged control, n = 5 per group with the exception of ME7+saline+SC-560 (n = 4). All data have been presented as the mean ± SEM.
Quantitative PCR for these proinflammatory cytokines was also examined in the hippocampus and thalamus (Fig. 7). Systemic LPS induced robust transcription of the mRNA species for IL-1β, IL-6, TNF-α, and CXCL-1 in the hippocampus and thalamus of ME7 animals compared with saline-treated ME7 animals. Pretreatment with SC-560 did not inhibit the transcription of any of these cytokines. Two-way ANOVAs revealed the main effects of challenge (for IL-1β, IL-6, TNF-α, and CXCL-1, respectively: p < 0.0001, F(1,15) = 111.4; p < 0.0001, F(1,15) = 91.21; p = 0.0099, F(1,15) = 8.704; p < 0.0001 F(1,15) = 92.74) but no effect of treatment. Thus, systemically administered SC-560 has no impact on the induction of CNS inflammatory cytokine transcripts by LPS. Similar to the results for the SC-560 study, piroxicam did not attenuate the LPS-induced inflammatory cytokine response (data not shown) at both the plasma and brain transcript level.
LPS induces working memory deficits via IL-1β, and ibuprofen pretreatment blocks these
Although both piroxicam and SC-560 block LPS-induced working memory deficits in the absence of any effect on systemic or central cytokines, proinflammatory cytokines have been implicated in clinical delirium (Beloosesky et al., 2007; van Munster et al., 2008; MacLullich et al., 2011). We hypothesized that systemic IL-1β might be sufficient to induce similar cognitive deficits to LPS and that systemic IL-1RA would be protective against the LPS-induced working memory deficits. Human recombinant IL-1RA (10 mg/kg, i.p.) attenuated the LPS-induced deficit in T-maze performance (Fig. 8a). Two-way ANOVA revealed significant main effects of time (p = 0.0013, F(1,17) = 14.85) and treatment (p = 0.0306, F(2,17) = 4.309), and a significant interaction (p = 0.0149, F(2,17) = 5.437). Bonferroni post hoc analysis showed a significant impairment in T-maze alternation postchallenge in the ME7+LPS-treated group with respect to ME7+IL-1RA (p < 0.001). Cotreatment with recombinant IL-1RA was protective (p < 0.05 vs ME7+LPS), implicating systemic IL-1 in the LPS-induced deficits in working memory.
Figure 8.

Systemic IL-1RA blocks LPS-induced impairments in T-maze alternation, systemic IL-1β induces working memory deficits, and ibuprofen is protective. a, ME7 animals were challenged with LPS (100 μg/kg, n = 7) or saline (n = 5) in the presence of human recombinant IL-1RA at a dose of 10 mg/kg, i.p. Control ME7 animals (ME7+LPS, n = 8) were challenged with LPS and placebo. Data were analyzed by two-way ANOVA, and Bonferroni post hoc analyses followed significant main effects: ***p < 0.001 denotes a significant decrease in ME7+LPS with respect to ME7+IL-1RA; +p < 0.05 with respect to ME7+LPS postchallenge. b, Working memory was assessed by alternation in the paddling T-maze at the earlier time point of 1 h post-IL-1 challenge. ME7 animals were challenged with recombinant IL-1β (15 μg/kg, i.p.; n = 10) in the presence of ibuprofen administered at (30 mg/kg, i.p.), 60 min before IL-1β injection. Control NBH and ME7 animals (NBH+IL-1, n = 14; ME7+IL-1, n = 15) were challenged with IL-1β or saline (ME7+Saline, n = 14) 60 min after vehicle (Saline) injection. Data were analyzed by two-way ANOVA, and Bonferroni post hoc analyses followed significant main effects: ***p < 0.001 denotes a significant decrease in ME7+IL-1 with respect to ME7+saline; ++p < 0.01 denotes a significant protection with respect to ME7+IL-1 postchallenge. All data have been presented as the mean ± SEM.
Given these data, we further hypothesized that a systemic IL-1β challenge (15 μg/kg, i.p.) would replicate the effects of LPS on working memory performance in diseased animals. To test this hypothesis, working memory was assessed by alternation in the paddling T-maze at the earlier time point of 1 h post-IL-1 challenge (Fig. 8b). Furthermore, given that NS-398 and SC-560 are not licensed drugs for use in patient populations, we tested the ability of the commonly used and well tolerated nonselective COX inhibitor ibuprofen (30 mg/kg, i.p.) to protect against IL-1-induced cognitive deficits. Two-way ANOVA revealed significant main effects of time (p < 0.0001, F(1,49) = 19.25) and treatment (p < 0.0001, F(3,49) = 15.42), and a significant interaction (p = 0.0012, F(3,49) = 6.22). Bonferroni post hoc analysis showed a significant impairment in T-maze alternation postchallenge in the IL-1-treated group with respect to ME7+saline (p < 0.001). Pretreatment with ibuprofen protected against this IL-1-induced impairment, indicating that COX inhibition with a more clinically acceptable, albeit nonselective, drug is also protective against inflammation-induced cognitive deficits occurring via systemic IL-1.
Discussion
We have shown here that as neurodegenerative disease progresses, the risk, severity, and duration of acute cognitive dysfunction induced by systemic inflammation increase. This occurs in parallel with significant upregulation of the prostaglandin biosynthetic enzyme COX-1 in microglia and PVMs. Systemic challenge with LPS causes marked working memory deficits that can be blocked by COX-1-specific inhibition but not by COX-2-specific inhibition. Furthermore, it is COX-1 rather than COX-2 that primarily drives disease-associated PGE2 increases in the hippocampus and thalamus. We have also shown that systemic IL-1β can mimic and IL-1RA can block the effect of systemic LPS, and that upon IL-1β challenge the widely used NSAID ibuprofen affords significant protection against the cognitive deficits.
The inflammatory route to delirium
Despite the enormous medical and economic imperatives of understanding delirium, very little is known about its pathophysiology. However, there is evidence for an inflammatory component in episodes triggered by systemic infection and surgery. We investigated mechanisms of inflammation-induced cognitive dysfunction in the current study. Given that COX-2 is robustly induced at the brain endothelium in response to systemic inflammatory stimuli (Yamagata et al., 2001) and that COX-2 derived prostaglandins have been demonstrated to have roles in the generation of fever (Li et al., 2001) and sickness behavior (Dunn et al., 2006), it has been the pharmacological target of choice against neuroinflammation in several clinical trials. Here, we used the COX-2-specific inhibitor NS-398 with a view to blocking LPS-induced effects on cognitive function. However, COX-2 inhibition was not protective. Importantly, NS-398 successfully blocked the well described LPS-induced increase in hypothalamic PGE2, demonstrating efficacy at its specific target. Moreover, neuronal COX-2 is known to have a role in hippocampal memory function (Sang et al., 2005), and its inhibition can impair learning, while COX-1 inhibition does not (Cowley et al., 2008). Therefore, COX-1 may be a better pharmacological target.
It has recently become clear that COX-1 has an important role mediating the neuroinflammatory response in multiple settings (Choi et al., 2009). COX-1 expression is increased in microglia in Alzheimer's disease (AD; Yermakova et al., 1999; Hoozemans et al., 2001), and its inhibition was beneficial in AD studies in mice and humans (Gómez-Isla et al., 2008; Coma et al., 2010). Here, we found clear microglial expression of COX-1. We demonstrated that piroxicam significantly protected against LPS-induced cognitive deficits and diseased-associated brain prostaglandin elevations. Piroxicam is a nonspecific COX inhibitor, with reported selectivity for COX-1 (Smith et al., 1994a, b; Mitchell et al., 1995; Blanco et al., 1999). The complete ablation of PGE2 synthesis observed with piroxicam suggests that at the current dose, it actually inhibits both isoforms. However, the COX-1-specific inhibitor SC-560 was also protective against acute cognitive dysfunction, and reduced hippocampal and thalamic PGE2 concentrations, while the COX-2-specific inhibitor NS-398 did not afford protection against the LPS-induced working memory deficits and did not reduce the disease-induced elevation in brain PGE2 levels. Recent studies inducing chronic hippocampal overexpression of IL-1β also showed that COX-1 was chronically elevated (≥2 months) and was involved in inflammation-mediated cognitive change (Matousek et al., 2010), and it is known that direct infusion of PGE2 into the brain can induce working memory deficits (Matsumoto et al., 2004) and impair contextual fear conditioning (Hein et al., 2007). The results presented here suggest that levels of COX-1-dependent prostaglandins are elevated in diseased animals and cause a vulnerability to working memory deficits when subsequently exposed to mild-to-moderate inflammation. Neither the chronic COX-1 elevation nor the acute inflammatory stimulation alone is sufficient to induce these deficits.
It is important to stress that the PGE2 levels in the hippocampus and thalamus were robustly elevated by disease per se and were not significantly further elevated upon LPS treatment. IL-1β is known to induce PGE2 synthesis (Dinarello et al., 1983), with direct consequences for brain function (Engblom et al., 2003). Here, in the diseased brain, COX-1-dependent prostaglandins dominate, and the small LPS-induced, COX-2-mediated PGE2 is visible only during COX-1 inhibition (Fig. 6b). Furthermore, NS-398 inhibition affected hypothalamic PGE2 levels, but not hippocampal and thalamic levels (Fig. 5b,c). Although COX-2 is robustly expressed in microglia during acute inflammation (Levi et al., 1998; Tomimoto et al., 2000), there is evidence that COX-1 predominates in chronic neurodegeneration (Hoozemans et al., 2001) and in the M2 phenotype (Martinez et al., 2006; Ajmone-Cat et al., 2013). Given the dominance of COX-1 in the generation of PGE2 observed here, it is not surprising that the deleterious effects of prostaglandins on working memory are mediated by COX-1 rather than COX-2.
While pre-existing chronically elevated levels of prostaglandins appear important for the deficits observed, there is also a role for acutely elevated IL-1β. Although there is evidence for direct effects of IL-1β on hippocampal neurons (Cunningham et al., 1996) and these apparently mediate deficits in the contextual fear-conditioning paradigm (Cibelli et al., 2010; Frank et al., 2010; Terrando et al., 2010), we have shown here that systemically administered IL-1β induces working memory deficits in diseased animals and that systemically administered IL-1RA protects against LPS-induced deficits, suggesting a systemic role for IL-1β. The contextual fear-conditioning task is reliant on slow protein synthesis-dependent consolidation of memory, and inflammation-induced impairment is associated with late-phase long-term potentiation disruption (Chapman et al., 2010). This is manifestly different in nature to the T-maze alternation deficits shown here, which reflect a failure of working memory or short-term habituation and attentional processes (Sanderson et al., 2010). These tasks are therefore predicted to rely on different neural processes, and the current data are consistent with distinct inflammatory mechanisms.
Given these results and the known neuronal expression of a number of different prostaglandin E receptors (Sugimoto and Narumiya, 2007), prostaglandins may be stronger candidates for direct effects on neuronal function in the working memory paradigm. EP3 has been hypothesized to be involved in acute cognitive impairments via decreased brain-derived neurotrophic factor transcription (Hein and O'Banion, 2009). EP1 and EP3 have documented important roles in acute stress responses, which could play a role in the observed cognitive deficits (Matsuoka et al., 2003). There are also data demonstrating a key role for EP3 in the generation of Aβ peptides and synaptic degeneration in the APP/PS1 model of Alzheimer's disease (Shi et al., 2012). Although we have shown robust PGE2 expression, levels of multiple prostanoids would be affected by COX-1 inhibition, and it is therefore plausible that other prostanoids may contribute to the deficits observed. Nonetheless, the COX-1 dependence reported here is robust and likely to generalize to neurodegenerative disease more broadly, since COX-1-positive microglia are a feature of AD (Yermakova et al., 1999; Hoozemans et al., 2001) and many other neurodegenerative diseases (Lima et al., 2012).
Clinical evidence for inflammation in delirium
We have shown protection against the acute working memory deficits induced by LPS using a drug that blocks prostaglandin synthesis but having no effect on blood or brain cytokine synthesis. Thus, SC-560 is acting to inhibit prostaglandin production specifically and not through other anti-inflammatory pathways ascribed to NSAIDs, such as peroxisome proliferator-activated receptor-γ-mediated inhibition of proinflammatory gene transcription (Landreth and Heneka, 2001, 2007). The cytokines IL-6 and IL-8 have been associated with delirium in a number of post-hip fracture cohorts (Beloosesky et al., 2007; van Munster et al., 2008; MacLullich et al., 2011), but it is not clear whether these cytokines represent measures of the severity of the systemic inflammatory insult or are causal in delirium. The current studies suggest that IL-1β is causative when superimposed upon disease-associated, chronically elevated, brain prostaglandins. Although speculative, it seems plausible that elevated brain levels of prostaglandins create a permissive state that is not sufficient to induce cognitive dysfunction, but upon subsequent inflammatory stimulation, a synergism between prostanoids and additional inflammatory mediators triggers the acute dysfunction. Our previous work has argued that microglial priming and the consequent exaggerated CNS inflammatory response may be a key contributor to deleterious cognitive consequences of systemic inflammation in the aged and demented (Cunningham et al., 2005; Cunningham and Maclullich, 2013). The current data begin to elucidate how microglial expression of COX-1 creates a predisposition to acute cognitive deficits, which can then be triggered by acute systemic inflammatory insult. Systemic IL-1β is sufficient to act as this trigger, but there remain significant questions to answer about how this interaction occurs.
Conclusion
We have demonstrated a role for COX-1 in acute and transient working memory deficits resembling delirium, and provide clear empirical evidence that progressing features of neurodegenerative disease increase the risk, duration, and severity of episodes of acute cognitive dysfunction. In addition to microglial COX-1 elevation, it is highly likely that there are additional factors that predispose the neurodegenerative brain to delirium, which merit further study. We propose that COX-1-generated prostaglandins constitute an important part of the inflammatory cascade leading to delirium during dementia (Cunningham and Skelly, 2012). We have highlighted the ability of a well tolerated, widely used NSAID to offer protection in this model, suggesting that in appropriate populations ibuprofen might be investigated as a potential prophylactic treatment or intervention for delirium of inflammatory etiology, such as that associated with systemic infection or surgery, particularly on a background of existing dementia.
Footnotes
This work was supported by Wellcome Trust Grant SRF 090907/Z/09/A. D.T.S. was supported by the Health Research Board of Ireland 4 year postgraduate program. The authors thank Gavin McManus for technical assistance with confocal microscopy.
References
- Ajmone-Cat MA, Mancini M, De Simone R, Cilli P, L., M Microglial polarization and plasticity: evidence from organotypic hippocampal slice cultures. Glia. 2013 doi: 10.1002/glia.22550. doi: 10.1002/glia.22550. Advance online publication. Retrieved August 20, 2013. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Ed 4. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- Barrientos RM, Higgins EA, Biedenkapp JC, Sprunger DB, Wright-Hardesty KJ, Watkins LR, Rudy JW, Maier SF. Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging. 2006;27:723–732. doi: 10.1016/j.neurobiolaging.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Beloosesky Y, Hendel D, Weiss A, Hershkovitz A, Grinblat J, Pirotsky A, Barak V. Cytokines and C-reactive protein production in hip-fracture-operated elderly patients. J Gerontol A Biol Sci Med Sci. 2007;62:420–426. doi: 10.1093/gerona/62.4.420. [DOI] [PubMed] [Google Scholar]
- Blanco FJ, Guitian R, Moreno J, de Toro FJ, Galdo F. Effect of antiinflammatory drugs on COX-1 and COX-2 activity in human articular chondrocytes. J Rheumatol. 1999;26:1366–1373. [PubMed] [Google Scholar]
- Brown LJ, Ferner HS, Robertson J, Mills NL, Pessotto R, Deary IJ, MacLullich AM. Differential effects of delirium on fluid and crystallized cognitive abilities. Arch Gerontol Geriatr. 2011;52:153–158. doi: 10.1016/j.archger.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Chapman TR, Barrientos RM, Ahrendsen JT, Maier SF, Patterson SL. Synaptic correlates of increased cognitive vulnerability with aging: peripheral immune challenge and aging interact to disrupt theta-burst late-phase long-term potentiation in hippocampal area CA1. J Neurosci. 2010;30:7598–7603. doi: 10.1523/JNEUROSCI.5172-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;22:301–311. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Aid S, Bosetti F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol Sci. 2009;30:174–181. doi: 10.1016/j.tips.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibelli M, Fidalgo AR, Terrando N, Ma D, Monaco C, Feldmann M, Takata M, Lever IJ, Nanchahal J, Fanselow MS, Maze M. Role of interleukin-1beta in postoperative cognitive dysfunction. Ann Neurol. 2010;68:360–368. doi: 10.1002/ana.22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coma M, Serenó L, Da Rocha-Souto B, Scotton TC, España J, Sánchez MB, Rodríguez M, Agulló J, Guardia-Laguarta C, Garcia-Alloza M, Borrelli LA, Clarimón J, Lleó A, Bacskai BJ, Saura CA, Hyman BT, Gómez-Isla T. Triflusal reduces dense-core plaque load, associated axonal alterations and inflammatory changes, and rescues cognition in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2010;38:482–491. doi: 10.1016/j.nbd.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley TR, Fahey B, O'Mara SM. COX-2, but not COX-1, activity is necessary for the induction of perforant path long-term potentiation and spatial learning in vivo. Eur J Neurosci. 2008;27:2999–3008. doi: 10.1111/j.1460-9568.2008.06251.x. [DOI] [PubMed] [Google Scholar]
- Cunningham AJ, Murray CA, O'Neill LA, Lynch MA, O'Connor JJ. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Maclullich AM. At the extreme end of the psychoneuroimmunological spectrum: delirium as a maladaptive sickness behaviour response. Brain Behav Immun. 2013;28:1–13. doi: 10.1016/j.bbi.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Skelly DT. Non-steroidal anti-inflammatory drugs and cognitive function: are prostaglandins at the heart of cognitive impairment in dementia and delirium? J Neuroimmune Pharmacol. 2012;7:60–73. doi: 10.1007/s11481-011-9312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Deacon R, Wells H, Boche D, Waters S, Diniz CP, Scott H, Rawlins JN, Perry VH. Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur J Neurosci. 2003;17:2147–2155. doi: 10.1046/j.1460-9568.2003.02662.x. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Teeling J, Felton L, Perry VH. The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C) Brain Behav Immun. 2007;21:490–502. doi: 10.1016/j.bbi.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, Rawlins JN, Perry VH. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65:304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DH, Muniz Terrera G, Keage H, Rahkonen T, Oinas M, Matthews FE, Cunningham C, Polvikoski T, Sulkava R, MacLullich AM, Brayne C. Delirium is a strong risk factor for dementia in the oldest-old: a population-based cohort study. Brain. 2012;135:2809–2816. doi: 10.1093/brain/aws190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, Marnoy SO, Rosenwasser LJ. Role of arachidonate metabolism in the immunoregulatory function of human leukocytic pyrogen/lymphocyte-activating factor/interleukin 1. J Immunol. 1983;130:890–895. [PubMed] [Google Scholar]
- Dunn AJ, Swiergiel AH, Zhang H, Quan N. Reduced ingestion of sweetened milk induced by interleukin-1 and lipopolysaccharide is associated with induction of cyclooxygenase-2 in brain endothelia. Neuroimmunomodulation. 2006;13:96–104. doi: 10.1159/000096291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom D, Saha S, Engström L, Westman M, Audoly LP, Jakobsson PJ, Blomqvist A. Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat Neurosci. 2003;6:1137–1138. doi: 10.1038/nn1137. [DOI] [PubMed] [Google Scholar]
- Fick DM, Agostini JV, Inouye SK. Delirium superimposed on dementia: a systematic review. J Am Geriatr Soc. 2002;50:1723–1732. doi: 10.1046/j.1532-5415.2002.50468.x. [DOI] [PubMed] [Google Scholar]
- Fong TG, Jones RN, Shi P, Marcantonio ER, Yap L, Rudolph JL, Yang FM, Kiely DK, Inouye SK. Delirium accelerates cognitive decline in Alzheimer disease. Neurology. 2009;72:1570–1575. doi: 10.1212/WNL.0b013e3181a4129a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Hein AM, Biedenkapp JC, Watkins LR, Maier SF. IL-1RA blocks E. coli-induced suppression of Arc and long-term memory in aged F344xBN F1 rats. Brain Behav immun. 2010;24:254–262. doi: 10.1016/j.bbi.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Isla T, Blesa R, Boada M, Clarimón J, Del Ser T, Domenech G, Ferro JM, Gómez-Ansón B, Manubens JM, Martínez-Lage JM, Muñoz D, Peña-Casanova J, Torres F. A randomized, double-blind, placebo controlled-trial of triflusal in mild cognitive impairment: the TRIMCI study. Alzheimer Dis Assoc Disord. 2008;22:21–29. doi: 10.1097/WAD.0b013e3181611024. [DOI] [PubMed] [Google Scholar]
- Guenther K, Deacon RM, Perry VH, Rawlins JN. Early behavioural changes in scrapie-affected mice and the influence of dapsone. Eur J Neurosci. 2001;14:401–409. doi: 10.1046/j.0953-816x.2001.01645.x. [DOI] [PubMed] [Google Scholar]
- Hein AM, O'Banion MK. Neuroinflammation and memory: the role of prostaglandins. Mol Neurobiol. 2009;40:15–32. doi: 10.1007/s12035-009-8066-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein AM, Stutzman DL, Bland ST, Barrientos RM, Watkins LR, Rudy JW, Maier SF. Prostaglandins are necessary and sufficient to induce contextual fear learning impairments after interleukin-1 beta injections into the dorsal hippocampus. Neuroscience. 2007;150:754–763. doi: 10.1016/j.neuroscience.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Landreth GE, Hüll M. Drug insight: effects mediated by peroxisome proliferator-activated receptor-gamma in CNS disorders. Nat Clin Pract Neurol. 2007;3:496–504. doi: 10.1038/ncpneuro0586. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, Rozemuller AJ, Janssen I, De Groot CJ, Veerhuis R, Eikelenboom P. Cyclooxygenase expression in microglia and neurons in Alzheimer's disease and control brain. Acta Neuropathol. 2001;101:2–8. doi: 10.1007/s004010000251. [DOI] [PubMed] [Google Scholar]
- Kat MG, Vreeswijk R, de Jonghe JF, van der Ploeg T, van Gool WA, Eikelenboom P, Kalisvaart KJ. Long-term cognitive outcome of delirium in elderly hip surgery patients. A prospective matched controlled study over two and a half years. Dement Geriatr Cogn Disord. 2008;26:1–8. doi: 10.1159/000140611. [DOI] [PubMed] [Google Scholar]
- Landreth GE, Heneka MT. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer's disease. Neurobiol Aging. 2001;22:937–944. doi: 10.1016/S0197-4580(01)00296-2. [DOI] [PubMed] [Google Scholar]
- Levi G, Minghetti L, Aloisi F. Regulation of prostanoid synthesis in microglial cells and effects of prostaglandin E2 on microglial functions. Biochimie. 1998;80:899–904. doi: 10.1016/S0300-9084(00)88886-0. [DOI] [PubMed] [Google Scholar]
- Li S, Ballou LR, Morham SG, Blatteis CM. Cyclooxygenase-2 mediates the febrile response of mice to interleukin-1beta. Brain Res. 2001;910:163–173. doi: 10.1016/S0006-8993(01)02707-X. [DOI] [PubMed] [Google Scholar]
- Lima IV, Bastos LF, Limborço-Filho M, Fiebich BL, de Oliveira AC. Role of prostaglandins in neuroinflammatory and neurodegenerative diseases. Mediators Inflamm. 2012;2012:946813. doi: 10.1155/2012/946813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLullich AM, Hall RJ. Who understands delirium? Age Ageing. 2011;40:412–414. doi: 10.1093/ageing/afr062. [DOI] [PubMed] [Google Scholar]
- MacLullich AM, Ferguson KJ, Miller T, de Rooij SE, Cunningham C. Unravelling the pathophysiology of delirium: a focus on the role of aberrant stress responses. J Psychosom Res. 2008;65:229–238. doi: 10.1016/j.jpsychores.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLullich AM, Beaglehole A, Hall RJ, Meagher DJ. Delirium and long-term cognitive impairment. Int Rev Psychiatry. 2009;21:30–42. doi: 10.1080/09540260802675031. [DOI] [PubMed] [Google Scholar]
- MacLullich AM, Edelshain BT, Hall RJ, de Vries A, Howie SE, Pearson A, Middleton SD, Gillies F, Armstrong IR, White TO, Cunningham C, de Rooij SE, van Munster BC. Cerebrospinal fluid interleukin-8 levels are higher in people with hip fracture with perioperative delirium than in controls. J Am Geriatr Soc. 2011;59:1151–1153. doi: 10.1111/j.1532-5415.2011.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- Matousek SB, Hein AM, Shaftel SS, Olschowka JA, Kyrkanides S, O'Banion MK. Cyclooxygenase-1 mediates prostaglandin E(2) elevation and contextual memory impairment in a model of sustained hippocampal interleukin-1beta expression. J Neurochem. 2010;114:247–258. doi: 10.1111/j.1471-4159.2010.06759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Yamaguchi T, Watanabe S, Yamamoto T. Involvement of arachidonic acid cascade in working memory impairment induced by interleukin-1 beta. Neuropharmacology. 2004;46:1195–1200. doi: 10.1016/j.neuropharm.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Furuyashiki T, Bito H, Ushikubi F, Tanaka Y, Kobayashi T, Muro S, Satoh N, Kayahara T, Higashi M, Mizoguchi A, Shichi H, Fukuda Y, Nakao K, Narumiya S. Impaired adrenocorticotropic hormone response to bacterial endotoxin in mice deficient in prostaglandin E receptor EP1 and EP3 subtypes. Proc Natl Acad Sci U S A. 2003;100:4132–4137. doi: 10.1073/pnas.0633341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Larkin S, Williams TJ. Cyclooxygenase-2: regulation and relevance in inflammation. Biochem Pharmacol. 1995;50:1535–1542. doi: 10.1016/0006-2952(95)00212-X. [DOI] [PubMed] [Google Scholar]
- Murray CL, Skelly DT, Cunningham C. Exacerbation of CNS inflammation and neurodegeneration by systemic LPS treatment is independent of circulating IL-1beta and IL-6. J Neuroinflammation. 2011;8:50. doi: 10.1186/1742-2094-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C, Sanderson DJ, Barkus C, Deacon RM, Rawlins JN, Bannerman DM, Cunningham C. Systemic inflammation induces acute working memory deficits in the primed brain: relevance for delirium. Neurobiol Aging. 2012;33:603–616.e603. doi: 10.1016/j.neurobiolaging.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson DJ, McHugh SB, Good MA, Sprengel R, Seeburg PH, Rawlins JN, Bannerman DM. Spatial working memory deficits in GluA1 AMPA receptor subunit knockout mice reflect impaired short-term habituation: evidence for Wagner's dual-process memory model. Neuropsychologia. 2010;48:2303–2315. doi: 10.1016/j.neuropsychologia.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N, Zhang J, Marcheselli V, Bazan NG, Chen C. Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J Neurosci. 2005;25:9858–9870. doi: 10.1523/JNEUROSCI.2392-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang Q, Johansson JU, Liang X, Woodling NS, Priyam P, Loui TM, Merchant M, Breyer RM, Montine TJ, Andreasson K. Inflammatory prostaglandin E2 signaling in a mouse model of Alzheimer disease. Ann Neurol. 2012;72:788–798. doi: 10.1002/ana.23677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WL, Meade EA, DeWitt DL. Pharmacology of prostaglandin endoperoxide synthase isozymes-1 and -2. Ann N Y Acad Sci. 1994a;714:136–142. doi: 10.1111/j.1749-6632.1994.tb12037.x. [DOI] [PubMed] [Google Scholar]
- Smith WL, Meade EA, DeWitt DL. Interactions of PGH synthase isozymes-1 and -2 with NSAIDs. Ann N Y Acad Sci. 1994b;744:50–57. doi: 10.1111/j.1749-6632.1994.tb52723.x. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Teeling JL, Cunningham C, Newman TA, Perry VH. The effect of non-steroidal anti-inflammatory agents on behavioural changes and cytokine production following systemic inflammation: implications for a role of COX-1. Brain Behav Immun. 2010;24:409–419. doi: 10.1016/j.bbi.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrando N, Rei Fidalgo A, Vizcaychipi M, Cibelli M, Ma D, Monaco C, Feldmann M, Maze M. The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit Care. 2010;14:R88. doi: 10.1186/cc9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimoto H, Akiguchi I, Wakita H, Lin JX, Budka H. Cyclooxygenase-2 is induced in microglia during chronic cerebral ischemia in humans. Acta Neuropathol. 2000;99:26–30. doi: 10.1007/PL00007402. [DOI] [PubMed] [Google Scholar]
- van Munster BC, Korevaar JC, Zwinderman AH, Levi M, Wiersinga WJ, De Rooij SE. Time-course of cytokines during delirium in elderly patients with hip fractures. J Am Geriatr Soc. 2008;56:1704–1709. doi: 10.1111/j.1532-5415.2008.01851.x. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Diagnostic criteria for research. Geneva, Switzerland: World Health Organization; 1992. The ICD-10 classification of mental and behavioural disorders. [Google Scholar]
- Witlox J, Eurelings LS, de Jonghe JF, Kalisvaart KJ, Eikelenboom P, van Gool WA. Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta-analysis. JAMA. 2010;304:443–451. doi: 10.1001/jama.2010.1013. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, Sugiura H, Cao C, Watanabe Y, Kobayashi S. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J Neurosci. 2001;21:2669–2677. doi: 10.1523/JNEUROSCI.21-08-02669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yermakova AV, Rollins J, Callahan LM, Rogers J, O'Banion MK. Cyclooxygenase-1 in human Alzheimer and control brain: quantitative analysis of expression by microglia and CA3 hippocampal neurons. J Neuropathol Exp Neurol. 1999;58:1135–1146. doi: 10.1097/00005072-199911000-00003. [DOI] [PubMed] [Google Scholar]