

Abstract
Type 2 diabetes is characterized by a deficit in β-cell function and mass, and its incidence increases with age. Autophagy is a highly regulated intracellular process for degrading cytoplasmic components, particularly protein aggregates and damaged organelles. Impaired or deficient autophagy is believed to cause or contribute to aging and age-related disease. Autophagy may be necessary to maintain structure, mass, and function of pancreatic β-cells. In this study, we investigated the effects of age on β-cell function and autophagy in pancreatic islets of 4-month-old (young), 14-month-old (adult), and 24-month-old (old) male Wistar rats. We found that islet β-cell function decreased gradually with age. Protein expression of the autophagy markers LC3/Atg8 and Atg7 exhibited a marked decline in aged islets. The expression of Lamp-2, a good indicator of autophagic degradation rate, was significantly reduced in the islets of old rats, suggesting that autophagic degradation is decreased in the islets of aged rats. However, protein expression of beclin-1/Atg6, which plays an important role in the induction and formation of the pre-autophagosome structure by associating with a multimeric complex of autophagy regulatory proteins (Atg14, Vps34/class 3 PI3 kinase, and Vps15), was most prominent in the islets of adult rats, and was higher in 24-month-old islets than in 4-month-old islets. The levels of p62/SQSTM1 and polyubiquitin aggregates, representing the functions of autophagy and proteasomal degradation, were increased in aging islets. 8-Hydroxydeoxyguanosine, a marker of mitochondrial and nuclear DNA oxidative damage, exhibited strong immunostaining in old islets. Analysis by electron microscopy demonstrated swelling and disintegration of cristae in the mitochondria of aged islets. These results suggest that β-cell and autophagic function in islets decline simultaneously with increasing age in Wistar rats, and that impaired autophagy in the islets of older rats may cause accumulation of misfolded and aggregated proteins and reduce the removal of abnormal mitochondria in β-cells, leading to reduced β-cell function. Dysfunctional autophagy in islets during the aging process may be an important mechanism leading to the development of type 2 diabetes.
Keywords: Autophagy, Islet, Aging, Mitochondria, Oxidative damage
Introduction
Macroautophagy (here referred to as autophagy) is a process of sequestration and removal of damaged organelles/proteins to recycle their constituents and nutrients. It also removes apoptotic cells and provides genomic stability (Levine and Kroemer 2008). Hence, autophagy is generally considered a protector of cells against various types of injury or continuous cellular degradation. Autophagy is expected to play a protective role in diverse types of cellular stress. This may be of particular importance during the aging process.
Aging denotes a post-maturational deterioration of cells and organisms with the passage of time, an increased vulnerability to challenges and age-associated diseases, and a decreased ability to survive (Marino and Lopez-Otin 2008; Rajawat et al. 2009). The pancreatic islet is a target organ of age-associated tissue damage, and the increased incidence of type 2 diabetes (T2D) in the elderly is a health problem worldwide (Cowie et al. 2006; Yang et al. 2010). The prevalence of T2D increases with age due to changes in or insufficient compensation of the β-cell functional mass in the face of increasing insulin resistance, but the underlying mechanism(s) within the dysfunctional β-cells have not been fully elucidated. Apoptosis, oxidative stress, mitochondrial dysfunction, and endoplasmic reticulum (ER) stress responses have been suggested as mechanisms of pancreatic β-cell dysfunction in T2D. However, mitochondria and ER, which play crucial roles in β-cell survival, death, and insulin secretion, primarily rely on autophagy for proper function (Laybutt et al. 2007; Petersen et al. 2004). Furthermore, several lines of evidence have shown that autophagy is involved in aging and is an essential part of the caloric restriction anti-aging mechanism (Rajawat and Bossis 2008; Rajawat et al. 2009; Wohlgemuth et al. 2007). Recently, it has been shown that pancreatic β-cell-specific Atg7-knockout mice develop hypoinsulinemia and hyperglycemia. β-Cell mass is reduced in autophagy-deficient mice because of increased apoptosis and decreased β-cell proliferation (Jung et al. 2008). These results suggest that autophagy is necessary to maintain the structure, mass, and function of pancreatic β-cells. Dysfunctional autophagy of islets may be an important mechanism behind the increasing prevalence of T2D in the elderly.
Many studies have demonstrated that age-related changes result from the accumulation of reactive oxygen species (ROS) and oxidative damage (Beckman and Ames 1998; Sohal and Weindruch 1996). The increase in cytoplasmic ROS levels is a causative event of aging (Angelopoulou et al. 2009; Blagosklonny 2008), and mitochondria are a key source of ROS in many cell types, including β-cells (Lenaz 2001; Newsholme et al. 2007; Turrens 2003). Autophagy is the only intracellular degradative mechanism for removing damaged mitochondria. Growing evidence indicates that ROS play a pivotal role in β-cell dysfunction in T2D by reducing insulin secretory capability and enhancing β-cell apoptosis (Drews et al. 2010; Sakai et al. 2003; Sakuraba et al. 2002). Autophagy-deficient β-cells show increased ROS content and enhanced apoptosis (Marsh et al. 2007). Thus, we speculate that impaired autophagy with age may lead to mitochondrial dysfunction and increased ROS content in β-cells. However, it is unknown whether age-related changes in autophagy occur in the islets. In the present study, we investigated the effect of age on the expression of autophagy-related genes and 8-oxo-2′-deoxyguanosine (8-OHdG), a marker of mitochondrial and nuclear DNA injury and oxidative stress (Shigenaga et al. 1989), in the islets of 4-, 14-, and 24-month-old male Wistar rats.
Materials and methods
Animals
Male Wistar rats were purchased from Vital River Laboratory Animals (Beijing, China) and were maintained under specific pathogen-free conditions of 22 ± 1 °C, 40 % humidity, a 12/12-h light/dark cycle, five males per cage, and free access to food at the animal center of the General Hospital of the Chinese People’s Liberation Army (PLA), Beijing. All rats were fed standard laboratory chow (containing 5 % fat). The experimental protocol was approved by the Animal Research Protection Committee of the General Hospital of the PLA. Rats were anesthetized with an intraperitoneal injection of sodium pentobarbital (40 mg/kg). Three age groups of rats were used: 4 months (n = 10), 14 months (n = 10), and 24 months old (n = 10), corresponding to young, adult, and old rats, respectively.
Oral glucose tolerance test (OGTT)
Rats were fasted overnight and orally administered glucose at 2 g/kg body weight. Their blood samples were collected from the angular vein, and blood glucose concentrations were measured at approximately 0, 10, 30, 60, and 120 min post-glucose challenge.
Isolation of pancreatic islets
Islets of Langerhans were isolated from the rats using in situ pancreatic collagenase infusion and were separated by density gradient centrifugation at 800 × g, according to a modified protocol described previously (Kinasiewicz et al. 2004).
Glucose-stimulated insulin release
Isolated islets were cultured in RPMI 1640 medium with 10 % fetal bovine serum overnight, and aliquots (50/well) of islets were incubated in triplicate in RPMI medium containing 2.8 mM glucose at 37 °C for 30 min. Subsequently, the islets were incubated in RPMI medium containing 2.8 mM glucose and then 16.7 mM glucose at 37 °C for 1 h. The supernatants were harvested to measure immunoreactive insulin (IRI) by enzyme immunoassay (Linco, St. Charles, MO, USA). The values of released insulin were normalized to islet protein content and expressed as mU IRI/ng protein. The islet protein content was measured using a BCA kit, according to the manufacturer’s instructions (Thermo Scientific, Rockford, IL, USA).
Senescence-associated β-galactosidase staining
The pancreatic tissues were removed from each rat and a sample was immersed in OCT compound (Tissue-Tek; Sakura Finetek, Torrance, CA, USA) and stored at −80 °C. Cryostat sections (4 μm) were mounted onto glass slides and fixed in 0.2 % glutaraldehyde and 2 % formaldehyde at room temperature for 15 min. Sections were washed in phosphate-buffered saline (PBS) and incubated in freshly prepared senescence-associated β-galactosidase (SA-β-gal) staining solution (1 mg/ml X-gal, 40 mM citric acid/sodium phosphate [pH 6.0], 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl2) overnight at 37 °C without CO2. Tissue sections were counterstained with eosin and examined under a microscope.
Western blotting
Freshly isolated islets from the three groups of rats were lysed in RIPA buffer (50 mM Tris-Cl [pH 7.6], 150 mM NaCl, 1 % NP-40, 0.1 % SDS, 0.5 % deoxycholic acid, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 0.5 mM phenylmethylsulfonyl fluoride) for 30 min on ice prior to centrifugation at 12,000 rpm for 30 min at 4 °C. Protein concentration was determined with the Pierce BCA Assay kit (lot JK126465; Thermo Fisher Scientific). Total protein (60–100 μg) was separated by 10–16 % SDS–PAGE, transferred to a nitrocellulose membrane, blocked with 5 % skim milk for 1 h at room temperature, and probed with the following primary antibodies at 4 °C overnight: rabbit polyclonal anti-LC3 antibody (1:2,000; Sigma), mouse monoclonal anti-p16 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal anti-Atg7 (1:1,000; Beyotime Institute of Biotechnology, Beijing, China), rabbit polyclonal anti-beclin-1 (1:2,000; Abcam Ltd, Hong Kong, China), rabbit monoclonal anti-Lamp2 (1:1,000; Epitomics, Inc., CA, USA), mouse monoclonal anti-ubiquitin (1:1,000; MABtech, Nacka Strand, Sweden), and mouse monoclonal anti-p62 (1:1,000; Abcam Inc., Cambridge, MA, USA). Blots were subsequently probed with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (Santa Cruz Biotechnology) (1:1,000). Immunoreactive bands were visualized by enhanced chemiluminescence, and densitometry was performed using Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
Immunohistochemistry staining
Pancreatic tissue samples were fixed in 10 % formaldehyde at 4 °C overnight and then embedded in paraffin according to standard procedures. Sections of 3-μm thickness were prepared. For immunohistochemical analysis, tissue sections were subjected to antigen retrieval by trypsinization for 20 min in 10 mM sodium citrate buffer (pH 6.0). Endogenous peroxidase was blocked by incubation with 3 % hydrogen peroxide for 15 min. Sections were washed in PBS and incubated with 1.5 % normal goat serum for 20 min, followed by incubation with a 1:50 dilution of guinea pig polyclonal anti-insulin (Abcam) and mouse monoclonal anti-LC3 (MBL Ltd, Tokyo, Japan) for double immunohistochemistry staining, or rabbit polyclonal anti-beclin-1 (1:1,000), mouse monoclonal anti-p62 (1:500), or mouse monoclonal anti-8-OHdG antibody (Santa Cruz Biotechnology) overnight at 4 °C. Sections were washed three times with PBS and incubated with biotin-conjugated goat anti-guinea pig IgG (Santa Cruz Biotechnology), anti-mouse IgG (Invitrogen, Carlsbad, CA, USA), or anti-rabbit IgG (Invitrogen) for 30 min at room temperature. Sections were washed again in PBS and incubated with labeled streptavidin-biotin horseradish peroxidase (Invitrogen) or labeled streptavidin-biotin alkaline phosphatase (Dako, Carpentaria, CA, USA) for 30 min at room temperature. Sections were washed a final time in PBS, incubated with DAB (brown) and NBT/BCIP (blue) sequentially for double staining of insulin and LC3, incubated again with DAB (brown) for beclin-1, p62, and 8-OHdG, and then examined by microscopy.
Electron microscopy
Pancreatic tissues were post-fixed with 2 % osmium tetroxide after fixation in 2.5 % glutaraldehyde in 0.01 mol/l phosphate buffer at 4 °C and washing in PBS. The tissues were dehydrated in a series of graded ethanol solutions. Propylene oxide was substituted for ethanol, and the tissues were embedded in epoxy resin. Ultrathin sections were double-stained with uranyl acetate and lead and examined under a JEM1200EX transmission electron microscope (JEOL, Tokyo, Japan) at 80 kV.
Statistical analysis
All data analyses were performed using SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA). Data are expressed as the mean±SD. Comparisons among groups were made using analysis of variance. A P value <0.05 was considered statistically significant.
Results
Changes in islet β-cell function with aging
Although no significant differences were observed in the levels of fasting blood glucose among the rat groups, the results from OGTT revealed significant glucose intolerance in 24-month-old rats. Plasma glucose at 120 min post-oral glucose challenge (2hPG) and the area under the glucose curve throughout the 120-min period (AUCg) were significantly greater, and the plasma glucose peak significantly delayed, in old rats than in young and adult rats. The 2hPG and AUCg values were higher in adult rats than in young rats (Fig. 1a,b).
Fig. 1.

Changes in β-cell islet function with aging. a Plasma glucose curve from oral glucose tolerance test (OGTT) in young (4-month-old), adult (14-month-old) and old (24-month-old) Wistar rats (n = 6, 7, and 6, respectively). Rats were fasted overnight, and blood glucose levels were measured before and after a glucose challenge. b Area under the glucose curve (AUCg) throughout the 120-min OGTT in the three groups. c, d Insulin secretion ex vivo. Pancreatic islets from rats of different ages were incubated in RPMI 1640 medium containing 2.8 mM glucose for 1 h and then in RPMI 1640 medium containing 16.7 mM glucose for 1 h (n = 6/group). Data are expressed as the mean±SD for each group. *P < 0.05 vs. young rats; #P < 0.05 vs. adult rats
We then conducted a functional analysis of pancreatic islets, measuring insulin secretion ex vivo using the same number of viable primary islets of similar size, and found that basal insulin secretion was decreased in the islets of old rats compared to young rats, whereas no difference was observed between adult and old rats (Fig. 1c). High glucose-stimulated insulin secretion from primary islets was also reduced significantly in old rats compared to young and adult rats (Fig. 1d). These results indicate that β-cell function declines with increasing age in Wistar rats.
Changes in senescence markers in rat pancreatic islets with aging
We measured the expression of p16, a robust biomarker and a possible effector of mammalian aging (Krishnamurthy et al. 2004), in the rats’ pancreatic islets and found that it was significantly higher in 24-month-old rats (Fig. 2) than in the other age groups, and was higher in 14-month-old rats than in 4-month-old rats. We also determined the expression of another well-defined in vivo senescence marker, SA-β-gal, which has high-pH galactosidase activity in senescent cells and tissues (Dimri et al. 1995). As shown in Fig. 3, the positive rate of SA-β-gal staining was markedly elevated in the islets of 24-month-old rats compared to 4-month-old rats. Furthermore, it was observed predominantly in the senescent cells of islets and was hardly detected in aging exocrine pancreatic tissue. These results suggest that these aging-related changes were mainly limited to the pancreatic islets.
Fig. 2.

Expression of the p16 senescent biomarker in the pancreatic islets of young (4-month-old), adult (14-month-old) and old (24-month-old) Wistar rats. a Western blotting results for p16 protein. b Quantitative analysis of p16 band densities. Protein expression data are presented as the mean ± SD (n = 6). *P < 0.05 vs. young rats, #P < 0.05 vs. adult rats
Fig. 3.

Senescence-associated β-galactosidase staining results in pancreatic tissues from young (4-month-old), adult (14-month-old) and old (24-month-old) Wistar rats. Magnification: ×400. Blue precipitation in the cytoplasm was observed in the senescent islet with aging but was hardly detectable in pancreatic exocrine tissues. Scale bars: 100 μm
Examination of autophagy-related protein expression during the rat islet aging process
Autophagic function is usually measured by quantifying autophagy-related (Atg) proteins. Among the 31 Atg proteins identified, Atgs 1–10 are involved in autophagosome formation and are markers for isolation membrane formation. We evaluated changes in the expression of autophagy-related proteins (LC3/Atg8, beclin-1/Atg6, Atg7, Lamp-2, p62, and polyubiquitin aggregates) in autophagosome formation and lysosomal fusion (Fig. 4) during the aging of pancreatic islets.
Fig. 4.

Autophagy-related proteins (LC3/Atg8, beclin-1/Atg6, Atg7, Lamp-2, p62, and polyubiquitin aggregates) in autophagosome formation and lysosomal fusion: (1) Beclin 1-Vps34/class 3 PI3 kinase complex regulate autophagy initiation. (2) Expansion of the membrane to form early autophagosomes is modulated by LC3/Atg8 and Atg7, which control the size of the autophagosomes and the amplitude of autophagy. (3) p62 mediates the specific recognition of polyubiquitin aggregates (an intracellular protein aggregate) by binding to LC3, and is then scavenged by autophagy, (4) Lamp-2, a lysosomal transmembrane protein, is required for the proper fusion of lysosomes with autophagosomes in the late stage of the autophagic process. (5) Once autophagosomes fuse with lysosomes to form autolysosomes, autophagosomal contents are degraded by lysosomal enzymes to regenerate nutrients
LC3/Atg8
Microtubule-associated protein light-chain 3 (LC3; also known as Atg8) is the only Atg protein that remains associated with the completed autophagosome; hence, it serves as one of the few autophagy markers (Kabeya et al. 2000). Pre-LC3 is cleaved into its cytosolic form LC3-I by Atg4. LC3-I is then activated by Atg7 and converted into its membrane-bound form, LC3-II, localized in pre-autophagosomes and autophagosomes (Maiuri et al. 2007). An increased amount of LC3-II and the LC3II/I ratio correlates with increased autophagy, serving as a good indicator of autophagosome formation (Kadowaki and Karim 2009). Furthermore, changes in the intracellular localization of LC3 provide the best method for detecting autophagy. When autophagy is activated, LC3-I is converted into LC3-II, which translocates to autophagosomes to form LC3-II puncta structures. We first observed LC3 expression in the rat pancreas by immunohistochemistry. The navy blue punctae reflected aggregation of LC3-II dots in the histochemical images (Fig. 5a). We found that the number of LC3-II punctae formed in the rat islets decreased gradually with aging. LC3-II puncta formation was significantly reduced in 24-month-old rats compared to 4- and 14-month-old rats (Fig. 5a). Western blot analyses revealed that LC3-I expression did not change remarkably, and that LC3-II expression decreased significantly in 24-month-old rats compared to the other groups, leading to a significantly decreased LC3II/I ratio in the oldest rats. These results indicate that autophagy declines in the islets of aged rats (Fig. 5b,c).
Fig. 5.

a Dual immunohistochemistry staining results for the LC3 protein (blue) and insulin (brown), in the pancreatic islets of young (4-month-old), adult (14-month-old) and old (24-month-old) Wistar rats. LC3 levels decreased gradually with aging. Scale bars: 100 μm. b LC3-I and LC3-II protein expression in the pancreatic islets of young, adult and old Wistar rats was quantified by Western blotting. c Quantitative analysis of LC3-II band densities. d Ratio of the LC3-II band to the LC3-I band. Protein expression data are presented as mean ± SD (n = 6). *P < 0.05 vs. young rats, #P < 0.05 vs. adult rats
Atg7
Atg7 is required for the formation and expansion of autophagosomes, which it triggers by initiating the conjugation of Atg12 to Atg5 and of LC3 to phosphatidylethanolamine. Overexpression of Atg effectively induces basal autophagy with no detrimental effects on cell survival, suggesting that it can activate autophagy (Juhasz et al. 2007; Pattison et al. 2011). We examined Atg7 protein expression and found that it was significantly lower in the oldest rats (Fig. 6b,d), suggesting that autophagy is reduced in the islets of aged rats.
Fig. 6.

a Immunohistochemistry results for SQSTM1/p62 (brown) in the pancreatic islets of young (4-month-old), adult (14-month-old) and old (24-month-old) Wistar rats. Scale bars: 100 μm. b, c Expression of Atg7, Lamp-2, p62/SQSTM1, and polyubiquitin aggregate proteins in the pancreatic islets of young, adult, and old Wistar rats was quantified by Western blotting. d, e, f Quantitative analysis of Atg7, Lamp-2, and p62/SQSTM1 band densities. Protein expression data are presented as mean ± SD (n = 6). *P < 0.05 vs. young rats, #P < 0.05 vs. adult rats
Lamp-2
Lysosomal-associated membrane protein 2 (Lamp-2) is a ubiquitous lysosomal membrane protein that is required for the proper fusion of lysosomes with autophagosomes in the late stages of autophagy. Lamp-2 depletion results in the inhibition of autophagic degradation secondary to the failure of lysosomes and autophagosomes to fuse to each other (Eskelinen 2006; Eskelinen et al. 2002). Therefore, Lamp-2 is a good indicator of the autophagic degradation rate. We found that Lamp-2 expression was significantly reduced in the islets of old rats, suggesting that the autophagic degradation rate and autophagic flux were decreased in the islets of aged rats (Fig. 6b,e).
Beclin-1/Atg6
Beclin-1, the mammalian homolog of yeast Atg6, is part of an autophagy regulatory protein complex (Atg14, Vps34/class 3 PI3 kinase, and Vps15) (He and Klionsky 2009) that promotes the formation of autophagosomes in mammalian cells (Itakura et al. 2008). Beclin-1 is correlated with diverse biological processes, including aging, development, tumor suppression, and protection against certain cardiac and neurodegenerative diseases (Levine and Kroemer 2008). Previous studies have found that beclin-1 has a significant increase in the aged rat heart (Wohlgemuth et al. 2007) and skeletal muscle (Wohlgemuth et al. 2010), but a significant decrease in the liver of old rats (Wohlgemuth et al. 2007). Changes in beclin-1 are tissue and cell context dependent. We observed beclin-1 expression in pancreas tissues by immunohistochemical staining and found that its expression is most prominent in the islets of adult rats and least prominent in that of young rats (Fig. 7a). Western blot analyses revealed that beclin-1 expression in the islets was higher in old rats than in young rats but lower compared to adult rats (Fig. 7b,c). These results suggest that beclin-1 may function through direct interactions with proteins that can either promote or inhibit autophagy, rather than a change in its expression during the aging process. Moreover, beclin-1 expression changes with age may be in line with its various functions in the complex and interdependent regulation of autophagy, tumorigenesis and apoptosis, all of which undergo age-related changes.
Fig. 7.

a Immunohistochemistry results for beclin-1 (brown) in the pancreatic islets of young (4-month-old), adult (14-month-old) and old (24-month-old) Wistar rats. Scale bars: 100 μm. b Beclin-1 protein expression in the pancreatic islets of young, adult, and old Wistar rats was quantified by Western blotting. c Quantitative analysis of beclin-1 band densities. Protein expression data are presented as mean ± SD (n = 6). *P < 0.05 vs. young rats, #P < 0.05 vs. adult rats
SQSTM1/p62 and polyubiquitin aggregates
Sequestosome 1 (SQSTM1, also known as p62) mediates the specific recognition of ubiquitinated protein aggregates by binding to LC3, and is then scavenged by autophagy. Therefore, its accumulation is a good indicator of suppressed autophagy and can be used to monitor fluctuations in autophagy under certain conditions (Bjorkoy et al. 2005). Polyubiquitin, an intracellular protein aggregate, is a signal for degradation by the ubiquitin–proteasome system. The accumulation of polyubiquitin aggregates can be induced by dysfunction in protein proteasome degradation pathways and by autophagy (Bjorkoy et al. 2006). SQSTM1/p62 plays a major role in the degradation of polyubiquitin substrates (Seibenhener et al. 2004). Thus, we also investigated its expression in the islets by immunohistochemistry and found that it significantly accumulated in the islets of old rats, but was hardly detectable in young and adult rats (Fig. 6a). It probably accumulated because its degradation decreased as autophagic function declined with age (Komatsu et al. 2006). Western blot analyses revealed that the expression of SQSTM1/p62 and polyubiquitin aggregates was significantly increased in 24-month-old rat islets (Fig. 6b,c,f). These results suggest that impaired autophagy with age leads to the accumulation of polyubiquitin aggregates associated with SQSTM1/p62.
Analysis of mitochondrial oxidative damage in the islets of older rats
Impaired autophagy may result in the accumulation of oxidative damage, ultimately leading to aging. Therefore, oxidative damage in pancreatic islets was evaluated by analyzing 8-OHdG expression. We found strong 8-OHdG reactions in the islet cell cytoplasm of old rats. In contrast, islets in young rats did not show positive reactions, whereas those in the adult rats showed conspicuous staining of many endocrine cells (Fig. 8), indicating that oxidative damage in rat islets increased significantly during the aging process. We further observed changes in the mitochondrial structures using electron microscopy. The results showed that β-cells of 14-month-old rats showed little mitochondrial oxidative damage, whereas the mitochondria in the β-cells of the 24-month-old rats exhibited swelling and disintegration or disruption of mitochondrial cristae (Fig. 9).
Fig. 8.

Immunoreactive 8-hydroxy-2′-deoxyguanosine (8-OHdG) reactions (brown) in the pancreatic islets of young (4-month-old), adult (14-month-old) and old (24-month-old) Wistar rats. Scale bars: 100 μm. Islets in young rats did not show positive reactions (a). In contrast, many islet cells in adult rats showed conspicuous staining (b), and the islets of old rats showed strong 8-OHdG reactions (c)
Fig. 9.
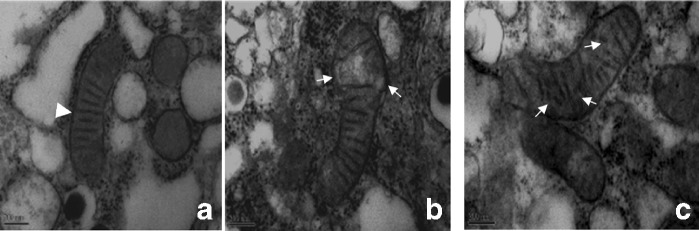
Electron microscopic analysis of ultrastructural changes in β-cells in young (4-month-old), adult (14-month-old) and old (24-month-old) Wistar rats. Arrowhead regular mitochondria; white arrows swelling of mitochondria and broken mitochondrial crests in the β-cells of old rats. Scale bars: 200 μm
Discussion
The incidence of and susceptibility to T2D increases with age, but the underlying mechanism(s) of this phenomenon are not yet clearly understood. It has been hypothesized that insulin resistance increases with age due to increased adiposity, decreased lean muscle mass, changes in dietary habits, and reduced physical activity (Scheen 2005). However, these factors alone do not account for age-related glucose intolerance. Studies of the effects of age on β-cells have shown that aging is negatively correlated with baseline β-cell proliferative activity and positively correlated with enhanced sensitivity to glucose-induced apoptosis (Gu et al. 2011; Maedler et al. 2006). The limited adaptive capacity of aging β-cells may contribute to the risk of developing T2D. A retrospective analysis of the European Group for the Study of Insulin Resistance database revealed a 25 % decline in the insulin delivery rate (calculated as the sum of the clamp-derived posthepatic insulin clearance rate and fasting plasma insulin concentration) from age 18 to 85 years (Iozzo et al. 1999). That study controlled for body mass index, fasting plasma glucose, and insulin sensitivity in both men and women. These results suggest that β-cell function declines with age. However, the underlying mechanism(s) of this decline have not been fully elucidated.
Pancreatic β-cell-specific Atg7-knockout mice show increased apoptosis and decreased proliferation of β-cells, leading to impaired glucose tolerance and decreased levels of serum insulin (Jung et al. 2008), suggesting that autophagy is a protective mechanism in β-cells. Interestingly, a high-fat diet causes profound deterioration of glucose tolerance in Atg7-deficient mice (Ebato et al. 2008). These findings support the hypothesis that autophagy is essential for maintenance of normal β-cell mass and function and that it plays a protective role in diabetes pathogenesis. Dysfunctional autophagy in islets during the aging process may be an important mechanism leading to the development of T2D. Although autophagy decreases with age in the liver (Wohlgemuth et al. 2007) and kidney (Cui et al. 2011), the effects of aging on autophagy in the mammalian pancreatic islets have not yet been elucidated.
In the present study, we used 4-, 14-, and 24-month-old rats (equivalent to 14-, 50-, and 84-year-old humans, respectively), to depict a natural aging model and reflect the pathophysiological characteristics of aging in animals. We evaluated pancreatic β-cell function in vivo and in vitro, and found that old rats had abnormal glucose tolerance, particularly in the case of postprandial hyperglycemia (the values of 2hPG and 2hOGTT AUCg were significantly increased in older rats), although fasting glucose levels were not significantly different in young and older rats, consistent with previous reports (Ihm et al. 2006, 2007; Perfetti et al. 1995). Furthermore, there was significantly less in vitro basal insulin secretion and high-glucose-stimulated insulin secretion in the primary islets of old rats. These results indicate that β-cell function declines with increasing age in rats.
We examined changes in the expression of the autophagy-related proteins LC3/Atg8, beclin-1/Atg6, Atg7, Lamp-2 and SQSTM1/p62, as well as polyubiquitin aggregates, in aging islets. The expansion of the membrane to form early autophagosomes is modulated by Atg7, LC3/Atg8, and Atg9, which control the size of the autophagosomes and the amplitude of autophagy. LC3/Atg8 is crucial for sequestration of large mitochondria and protein aggregates. The intracellular localization of LC3-II punctae and the LC3-II/I ratio have been used to evaluate the extent of autophagy. The results showed that the expression of LC3-II and the LC3II/I ratio decreased gradually with age, possibly as a result of decreased conversion of LC3-I to LC3-II or faster LC3-II degradation. We further detected the expression of Atg7 and Lamp-2 in the islets of rats. Atg7 can activate LC3-I and convert it into LC3-II. The reduced Atg7 expression in the islets of aged rats indicated decreased LC3 conversion. Lamp-2 is required for the proper fusion of lysosomes with autophagosomes during the late stages of autophagy and is a good indicator of the autophagic degradation rate. We found that Lamp-2 expression was significantly reduced in the islets of aged rats, suggesting that autophagic degradation of LC3-II declined with increasing age. The decreased LC3II/I ratio and decreased expression of LC3-II, Atg 7, and Lamp-2 in the islets of old rats indicate that autophagic influx declines in islet cells during the aging process. In addition, we found that beclin-1 was predominantly produced in the islet cell cytoplasm but was hardly detectable in exocrine tissues. Beclin-1 protein expression was most prominent in adult rats, and was higher in 24-month-old rats than in 4-month-old rats. Previous studies have found that beclin-1 levels increase significantly with age in the rat heart (Wohlgemuth et al. 2007) and in skeletal muscle (Wohlgemuth et al. 2009). Beclin-1 plays a central role in autophagy, and promotes crosstalk between apoptosis and autophagy. It also interacts with several cofactors (Atg14L, UVRAG, Bif-1, Rubicon, Ambra1, and survivin) to regulate the lipid kinase Vps-34 and promote the formation of beclin-1–Vps34–Vps15 core complexes, thereby inducing autophagy. In contrast, binding of the beclin-1 BH3 domain to Bcl-2 or Bcl-XL inhibits autophagy (Cao and Klionsky 2007; Kang et al. 2011; Maiuri et al. 2010). Beclin-1 dysfunction has been implicated in many disorders, including cancer and neurodegeneration. It may function through temporally modulated or spatially modulated interactions with its positive and negative regulators in autophagy, rather than a change in its expression.
Many studies have shown that the protein p62 recognizes toxic cellular waste, which is then scavenged by autophagy. Lack of autophagy leads to accumulation of p62-containing aggregates of ubiquitinated proteins, which is detrimental to liver cells as it induces cellular oxidative stress; however, unexpectedly, removal of p62 reverses oxidative stress in autophagy-deficient liver cells (Rusten and Stenmark 2010). Interestingly, autophagy-deficient pancreatic β-cells have inclusion bodies containing large polyubiquitin aggregates and SQSTM1/p62 in their cytoplasm (Ebato et al. 2008; Jung et al. 2008). We found that SQSTM1/p62 accumulated in the islet cells of 24-month-old rats, but was only weakly expressed in the islets of 4- and 14-month-old rats. Accumulation of SQSTM1/p62 is also observed in pancreatic islet cells of Zucker diabetic fatty rats and in insulinoma cells treated with high concentrations of glucose (Kaniuk et al. 2007). We also observed greater levels of SQSTM1/p62 and polyubiquitin aggregates in the islet cells of older rats by Western blotting. Accumulating evidence has indicated that aging tissues exhibit an age-dependent decline in the turnover rate of long-lived proteins, leading to accumulation of damaged proteins, consistent with a defect in autophagy (Marino and Lopez-Otin 2008; Rajawat and Bossis 2008). Increases in the intracellular accumulation of lipofuscin bodies (LBs) and lipid-storing vesicles in human and non-human β-cells with age have been described (Masini et al. 2009a, b). As waste storage organelles, LBs represent the end stage of lysosomal degradation. They are formed through the incorporation of senescent/damaged mitochondria and secretory granules into lysosomes by autophagy (Kurz et al. 2007). Our data suggest that impairment of autophagy in islet cells with age may be an important mechanism leading to the accumulation of misfolded and aggregated proteins in LBs and lipid and polyubiquitin aggregates associated with SQSTM1/p62, which may contribute to decreased β-cell function in the autophagy-impaired islets of old rats and may be related to the increase in the risk of T2D with increasing age.
Mitochondria play a central role in the regulation of aging. They are the primary sites of ROS generation, and enhanced production of ROS and oxidative damage have been found in aging cells, which may be a reason why they are more affected by age than other organelles. If the impaired mitochondria are not removed (due to deficient autophagy), they can generate additional ROS, which further aggravate oxidative damage, forming a vicious circle (Cadenas and Davies 2000). In a model of autophagy deficiency in pancreatic β-cells, impairment of autophagy led to the accumulation of damaged and dysfunctional mitochondria and a corresponding increase in intracellular ROS levels (Hur et al. 2010; Wu et al. 2009). Mice lacking Atg7 in their β-cells exhibited a marked decrease in basal respiration by mitochondria and a significant decrease in mitochondrial oxidative capacity in isolated islets; treatment with the antioxidant N-acetylcysteine (NAC) ameliorated these metabolic defects (Wu et al. 2009). These results demonstrate the potential role of mitochondrial dysfunction and oxidative stress in autophagy-deficient β-cells. 8-OHdG has been used as a marker of oxidative stress-related DNA damage in mitochondria and nuclei (Toyokuni et al. 1997). It is a sensitive biomarker of mitochondrial DNA (mtDNA) oxidative damage as mtDNA is more vulnerable to oxidative damage than nuclear DNA (most of which is protected by histones). We found strong 8-OHdG staining in the islets of old rats, whereas the islets of young rats did not show positive reactions. Our results also reveal swelling of mitochondria in aged rat islets. The increased number of 8-OHdG-positive islet cells and mitochondrial swelling in older rats suggest that declining autophagy with age is associated with increased oxidative stress in rat islets, which contributes to impaired β-cell function.
In conclusion, we clearly demonstrated that insulin secretion by β-cells and autophagic function of islets simultaneously decline with increasing age in rats. Impairment or dysregulation of autophagy with age may cause accumulation of misfolded and aggregated proteins and reduce removal of abnormal mitochondria in β-cells, leading to reduced β-cell function. Apoptotic cell death may not be the only mechanism responsible for the decrease in β-cell function and mass. Prolonged inhibition of autophagy may also contribute to β-cell dysfunction. Our results help to elucidate the role of autophagy in the onset and development of T2D with age and may be useful for exploring new therapies for diabetes prevention and treatment.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China to C.L.L. (nos. 30873412, 81173625), to Y.L. (no. 30801201), from Beijing Nova Program of China to Y.L. (2011116).
Footnotes
Yu Liu, Suozhu Shi and Zhaoyan Gu contributed equally to this work.
Contributor Information
Xiangmei Chen, Email: xmchen301@126.com.
Chunlin Li, Phone: +86-10-66935462, Phone: +86-10-66876345, FAX: +86-10-64720614, Email: lcl301@yahoo.com.cn.
References
- Angelopoulou R, Lavranos G, Manolakou P. ROS in the aging male: model diseases with ROS-related pathophysiology. Reprod Toxicol. 2009;28:167–171. doi: 10.1016/j.reprotox.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Johansen T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy. 2006;2:138–139. doi: 10.4161/auto.2.2.2405. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008;7:3344–3354. doi: 10.4161/cc.7.21.6965. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/S0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17:839–849. doi: 10.1038/cr.2007.78. [DOI] [PubMed] [Google Scholar]
- Cowie CC, Rust KF, Byrd-Holt DD, Eberhardt MS, Flegal KM, Engelgau MM, Saydah SH, Williams DE, Geiss LS, Gregg EW. Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population: National Health And Nutrition Examination Survey 1999–2002. Diabetes Care. 2006;29:1263–1268. doi: 10.2337/dc06-0062. [DOI] [PubMed] [Google Scholar]
- Cui J, Bai XY, Shi S, Cui S, Hong Q, Cai G, Chen X (2012) Age-related changes in the function of autophagy in rat kidneys. Age (Dordr) 34:329-339 [DOI] [PMC free article] [PubMed]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews G, Krippeit-Drews P, Dufer M. Oxidative stress and beta-cell dysfunction. Pflugers Arch. 2010;460:703–718. doi: 10.1007/s00424-010-0862-9. [DOI] [PubMed] [Google Scholar]
- Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8:325–332. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27:495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL, Illert AL, Tanaka Y, Schwarzmann G, Blanz J, Von Figura K, Saftig P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol Biol Cell. 2002;13:3355–3368. doi: 10.1091/mbc.E02-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Du Y, Liu Y, Ma L, Li L, Gong Y, Tian H, Li C (2011) Effect of aging on islet beta-cell function and its mechanisms in Wistar rats. Age (Dordr). 7 Sept 2011. doi:10.1007/s11357-011-9312-7 [DOI] [PMC free article] [PubMed]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur KY, Jung HS, Lee MS. Role of autophagy in beta-cell function and mass. Diabetes Obes Metab. 2010;12(Suppl 2):20–26. doi: 10.1111/j.1463-1326.2010.01278.x. [DOI] [PubMed] [Google Scholar]
- Ihm SH, Matsumoto I, Sawada T, Nakano M, Zhang HJ, Ansite JD, Sutherland DE, Hering BJ. Effect of donor age on function of isolated human islets. Diabetes. 2006;55:1361–1368. doi: 10.2337/db05-1333. [DOI] [PubMed] [Google Scholar]
- Ihm SH, Moon HJ, Kang JG, Park CY, Oh KW, Jeong IK, Oh YS, Park SW. Effect of aging on insulin secretory function and expression of beta cell function-related genes of islets. Diabetes Res Clin Pract. 2007;77(Suppl 1):S150–S154. doi: 10.1016/j.diabres.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Jarvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J Clin Endocrinol Metab. 1999;84:863–868. doi: 10.1210/jc.84.3.863. [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki M, Karim MR. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 2009;452:199–213. doi: 10.1016/S0076-6879(08)03613-6. [DOI] [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniuk NA, Kiraly M, Bates H, Vranic M, Volchuk A, Brumell JH. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes. 2007;56:930–939. doi: 10.2337/db06-1160. [DOI] [PubMed] [Google Scholar]
- Kinasiewicz A, Juszczak M, Pachecka J, Fiedor P. Pancreatic islets isolation using different protocols with in situ flushing and intraductal collagenase injection. Physiol Res. 2004;53:327–333. [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T, Terman A, Brunk UT. Autophagy, ageing and apoptosis: the role of oxidative stress and lysosomal iron. Arch Biochem Biophys. 2007;462:220–230. doi: 10.1016/j.abb.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52:159–164. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Schumann DM, Schulthess F, Oberholzer J, Bosco D, Berney T, Donath MY. Aging correlates with decreased beta-cell proliferative capacity and enhanced sensitivity to apoptosis: a potential role for Fas and pancreatic duodenal homeobox-1. Diabetes. 2006;55:2455–2462. doi: 10.2337/db05-1586. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010;29:515–516. doi: 10.1038/emboj.2009.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Marino G, Lopez-Otin C. Autophagy and aging: new lessons from progeroid mice. Autophagy. 2008;4:807–809. doi: 10.4161/auto.6478. [DOI] [PubMed] [Google Scholar]
- Marsh BJ, Soden C, Alarcon C, Wicksteed BL, Yaekura K, Costin AJ, Morgan GP, Rhodes CJ. Regulated autophagy controls hormone content in secretory-deficient pancreatic endocrine beta-cells. Mol Endocrinol. 2007;21:2255–2269. doi: 10.1210/me.2007-0077. [DOI] [PubMed] [Google Scholar]
- Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F, Marselli L, Masiello P, Marchetti P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia. 2009;52:1083–1086. doi: 10.1007/s00125-009-1347-2. [DOI] [PubMed] [Google Scholar]
- Masini M, Lupi R, Bugliani M, Boggi U, Filipponi F, Masiello P, Marchetti P. A role for autophagy in beta-cell life and death. Islets. 2009;1:157–159. doi: 10.4161/isl.1.2.9372. [DOI] [PubMed] [Google Scholar]
- Newsholme P, Haber EP, Hirabara SM, Rebelato EL, Procopio J, Morgan D, Oliveira-Emilio HC, Carpinelli AR, Curi R. Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol. 2007;583:9–24. doi: 10.1113/jphysiol.2007.135871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109:151–160. doi: 10.1161/CIRCRESAHA.110.237339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfetti R, Rafizadeh CM, Liotta AS, Egan JM. Age-dependent reduction in insulin secretion and insulin mRNA in isolated islets from rats. Am J Physiol. 1995;269:E983–E990. doi: 10.1152/ajpendo.1995.269.6.E983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajawat YS, Bossis I. Autophagy in aging and in neurodegenerative disorders. Hormones (Athens) 2008;7:46–61. doi: 10.14310/horm.2002.1111037. [DOI] [PubMed] [Google Scholar]
- Rajawat YS, Hilioti Z, Bossis I. Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev. 2009;8:199–213. doi: 10.1016/j.arr.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Rusten TE, Stenmark H. p62, an autophagy hero or culprit? Nat Cell Biol. 2010;12:207–209. doi: 10.1038/ncb0310-207. [DOI] [PubMed] [Google Scholar]
- Sakai K, Matsumoto K, Nishikawa T, Suefuji M, Nakamaru K, Hirashima Y, Kawashima J, Shirotani T, Ichinose K, Brownlee M, et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem Biophys Res Commun. 2003;300:216–222. doi: 10.1016/S0006-291X(02)02832-2. [DOI] [PubMed] [Google Scholar]
- Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45:85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- Scheen AJ. Diabetes mellitus in the elderly: insulin resistance and/or impaired insulin secretion? Diabetes Metab. 2005;31(2):5S27–25S34. doi: 10.1016/S1262-3636(05)73649-1. [DOI] [PubMed] [Google Scholar]
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigenaga MK, Gimeno CJ, Ames BN. Urinary 8-hydroxy-2′-deoxyguanosine as a biological marker of in vivo oxidative DNA damage. Proc Natl Acad Sci U S A. 1989;86:9697–9701. doi: 10.1073/pnas.86.24.9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyokuni S, Tanaka T, Hattori Y, Nishiyama Y, Yoshida A, Uchida K, Hiai H, Ochi H, Osawa T. Quantitative immunohistochemical determination of 8-hydroxy-2′-deoxyguanosine by a monoclonal antibody N45.1: its application to ferric nitrilotriacetate-induced renal carcinogenesis model. Lab Invest. 1997;76:365–374. [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, Dunn WA., Jr Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007;10:281–292. doi: 10.1089/rej.2006.0535. [DOI] [PubMed] [Google Scholar]
- Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol. 2009;45:138–148. doi: 10.1016/j.exger.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol. 2010;45:138–148. doi: 10.1016/j.exger.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, Rovira II, Gutkind S, Daniels MP, Komatsu M, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY) 2009;1:425–437. doi: 10.18632/aging.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Lu J, Weng J, Jia W, Ji L, Xiao J, Shan Z, Liu J, Tian H, Ji Q, et al. Prevalence of diabetes among men and women in China. N Engl J Med. 2010;362:1090–1101. doi: 10.1056/NEJMoa0908292. [DOI] [PubMed] [Google Scholar]