

Abstract
The role of epigenetics in the modulation of longevity has not been studied in humans. To this aim, (1) we evaluated the DNA methylation from peripheral leukocytes of 21 female centenarians, their 21 female offspring, 21 offspring of both non-long-lived parents, and 21 young women through ELISA assay, pyrosequencing analysis of Alu sequences, and quantification of methylation in CpG repeats outside CpG islands; (2) we compared the DNA methylation profiles of these populations through Infinium array for genome-wide CpG methylation analysis. We observed an age-related decrease in global DNA methylation and a delay of this process in centenarians’ offspring. Interestingly, literature data suggest a link between the loss of DNA methylation observed during aging and the development of age-associated diseases. Genome-wide methylation analysis evidenced DNA methylation profiles specific for aging and longevity: (1) aging-associated DNA hypermethylation occurs predominantly in genes involved in the development of anatomical structures, organs, and multicellular organisms and in the regulation of transcription; (2) genes involved in nucleotide biosynthesis, metabolism, and control of signal transmission are differently methylated between centenarians’ offspring and offspring of both non-long-lived parents, hypothesizing a role for these genes in human longevity. Our results suggest that a better preservation of DNA methylation status, a slower cell growing/metabolism, and a better control in signal transmission through epigenetic mechanisms may be involved in the process of human longevity. These data fit well with the observations related to the beneficial effects of mild hypothyroidism and insulin-like growth factor I system impairment on the modulation of human lifespan.
Electronic supplementary material
The online version of this article (doi:10.1007/s11357-012-9463-1) contains supplementary material, which is available to authorized users.
Keywords: Epigenetics, Longevity, Centenarians, Centenarians’ offspring, DNA methylation, Aging
Introduction
Epigenetics is destined to change across the lifespan. Loss of global DNA methylation and promoter hypermethylation of several specific genes occur during aging. Epigenetics plays an important role in cellular senescence, human tumorigenesis, and several age-related diseases (Fraga et al. 2007; Bollati et al. 2009; Kim et al. 2010; Choi et al. 2009; Moore et al. 2008; Rakyan et al. 2010; Chambers et al. 2007). Indeed, epigenomic alterations are now increasingly recognized as part of aging and its associated pathologic phenotype (Petronis 2010; Bellizzi et al. 2011). However, the role of epigenetics in the modulation of healthy aging and longevity has not been clearly studied in humans.
Subjects able to reach extreme longevity, such as centenarians, are an extraordinary model to study human longevity (Franceschi and Bonafè 2003; Cevenini et al. 2008). Unfortunately, there are several disadvantages inherent in the study of centenarians: rarity, presence of frailty due to extreme age, and lack of an appropriate control group. In the past, numerous biological parameters measured in centenarians have been compared to a control group of younger subjects in order to identify factors potentially involved in the longevity. This system may be misleading for parameters showing a clear age influence, such as DNA methylation profile (Cevenini et al. 2008). Centenarians’ offspring represent a new approach to identify biological parameters which contribute to human longevity and healthy aging, without the disadvantages of centenarians. They appear to undergo an aging process “better” than that of subjects of the same age, show a lower morbidity and higher survival, and are more numerous than centenarians. In addition, it is possible to compare centenarians’ offspring with a demographically matched control group (subjects matched for age, sex, ethnicity, parent year of birth, but born from non-long-lived parents), thus avoiding cohort effects (Terry et al. 2004; Adams et al. 2008).
The aims of the present study were (1) to characterize and compare the DNA methylation profiles from peripheral leukocytes of female centenarians, their female offspring, female offspring of both non-long-lived parents, and young women and (2) to identify epigenetically modulated genes and pathways potentially involved in the process of aging, healthy aging, and longevity.
Materials and methods
Demographic strategy for enrolment and model description
The subjects enrolled in the present study belonged to a large multicentric project entitled “Does parental longevity impact on the healthy aging of their offspring?” funded by the Italian Ministry of University and Scientific Research. The stringent demographic strategy utilized to select the eligible subjects for this study is illustrated in supplementary Fig. S1. Briefly, 21 female centenarians (born in Northern Italy between the 1900 and 1908) and 21 of their female offspring were recruited, with the constraint that also the other parent was long lived (born in the same birth cohort 1900 and 1908 and died at the age >77 years, 10 years over the average life expectancy at 15 years of age). This selected cohort of centenarians’ offspring was compared with 21 age-matched females (offspring of both non-long-lived parents), with both parents born between 1900 and 1908 but died before the average life expectancy calculated at 15 years of age (67 years if male and 72 years if female) by the Italian mortality tables (see website “Human Mortality Database” of the Max Planck Institute for Demography, Rostock, Germany: http://www.mortality.org/). In addition, we included 21 healthy young females, randomly recruited, with an age ranging between 17 and 34 years, born and living in Northern Italy. The participants' ages were defined by birth certificates or dates of birth as stated on passports or identity cards. Exclusion criteria were the following: presence of cancer at the time of the interview, immunosuppressive (e.g., cyclosporin, methotrexate, glucocorticoids), or anticoagulant therapies.
The study protocol was approved by the Ethical Committee of the Sant’Orsola-Malpighi University Hospital (Bologna, Italy). After obtaining the written informed consent, a standard structured questionnaire was administered to collect information regarding the health status, currently used drugs, clinical history, and lifestyle (Skytthe et al. 2011). This questionnaire was administered by trained staff (nurses and medical doctors). The history of past and current diseases was accurately collected checking the participants medical documentation and the current drug therapy. Venous blood sample was drawn between 7.30 and 9.00 a.m.
DNA extraction and bisulfite treatment of the DNA
Genomic DNA was extracted from peripheral leukocytes in whole blood using the Wizard genomic DNA purification kit (PROMEGA, Madison WI, USA). Samples were processed for DNA isolation as follows: 3 to 5 mL of blood was incubated with a provided cell lysis solution in order to lyse erythrocytes before the leukocytes were isolated by centrifugation (2,000 × g for 10 min at room temperature). The pellet of leukocytes was then suspended in a nuclei lysis solution and digested with proteinase K in sodium dodecyl sulfate buffer at 37 °C for 1 h; then, DNA was extracted by salting out and resuspended in Tris–EDTA buffer. Sodium bisulfite conversion of DNA (500 ng) was performed by the EZ DNA Methylation-Gold Kit (Zymo Research Corporation, Orange, CA) according to the manufacturer's protocol. This assay has a conversion efficiency exceeding 99 %.
Global DNA methylation assay
Global methylation of DNA from extracted peripheral leukocyte DNA was determined using the Methylamp™ Global DNA Methylation Quantification Kit (Epigentek Group Inc., New York, NY, USA) according to the instructions provided by the manufacturer. In this kit, the methylated fraction of DNA is recognized by 5-methylcytosine antibody and quantified through an ELISA-like reaction. The amount of methylated DNA is proportional to the optical density intensity and the degree of DNA methylation can be calculated using the following formula:
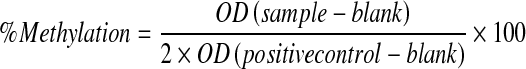 |
where OD is optical density, blank is a buffer without DNA, and positive control is a methylated DNA control provided by the manufacturer.
Repetitive elements PCR and pyrosequencing analyses
Alu PCRs were carried out using 20 ng of bisulfite-treated DNA and 10 pmol forward and reverse primers, one of them being biotinylated. PCR protocols and pyrosequencing conditions, as well as the sequencing primer, were previously described (Bollati et al. 2009). DNA methylation analyses were performed by PyroMark ID instrument (Biotage AB, Uppsala, Sweden) in the PSQ HS 96 System (Biotage), with the Pyro Gold SQA reagent kit (Biotage AB, Uppsala, Sweden) according to the manufacturer’s instructions. Raw data were analyzed using the Q-CpG software v1.0.9 (Biotage AB, Uppsala, Sweden) that calculates the ratio of converted to unconverted cytosine at each CpG, giving the percentage of methylation. For each sample, the methylation value represents the mean between at least two independent PCR and pyrosequencing experiments.
Genome-wide methylation analyses
Genome-wide methylation profile of DNA samples was evaluated through Infinium HumanMethylation27 BeadChip (Illumina, San Diego, CA, USA). This assay determines quantitative measurements of DNA methylation for 27,578 CpG dinucleotides (20,006 CpG localized in CpG islands, 7,572 CpG localized in non-CpG islands) spanning 14,495 genes. The methylation profile was analyzed according to the manufacturer's instructions using Illumina-supplied reagents and conditions. In brief, after bisulfite conversion, 250 ng of DNA was whole-genome amplified (WGA) and enzymatically fragmented. The bisulfite-converted WGA-DNA samples were purified and randomized again prior to hybridization to the HumanMethylation27 BeadChips. During hybridization, the WGA-DNA molecules were annealed to locus-specific DNA oligomers which were linked to individual bead types, one designed against the unmethylated site and one against the methylated site. After hybridization, allele-specific single-base extension provided another level of specificity and incorporated a fluorescent label for detection. The level of methylation was determined at each locus by the intensity of the two possible fluorescent signals, specific for the methylated and unmethylated alleles. DNA methylation values, described as β values, were recorded for each locus in each sample using GenomeStudio software. β value provides a continuous measure of the DNA methylation levels for each CpG site, ranging from 0 in the case of completely unmethylated sites to 1 in completely methylated sites.
Quality control
As described in supplementary Fig. S1, quality control of DNA represented an additional criteria for the enrolment of the study population. Quality control was organized into two steps:
Quality control and quantification of DNA before and after bisulfite conversion. DNA was quantified with NanoDrop (NanoDrop Products Thermo Scientific Wilmington, DE), and quality was assessed by visualization of genomic DNA on 1 % agarose gel electrophoresis. Only DNA samples not fragmented and with a concentration higher than 50 ng/μl were subsequently processed.
Postarray control step. Background-corrected β values (as generated from the GenomeStudio software) and built-in controls were used to evaluate the quality of individual arrays. Samples with low bisulfite conversion (BS) efficiency (BS control intensity values <4,000) were excluded, as well as other outliers that have been detected using box plot of total intensity (unmethylated + methylated values) and histograms of β values. We confirmed that each sample yielded a clear bimodal distribution of methylation β values and excluded samples where this was not the case. Samples were further filtered according to CpG coverage, using GenomeStudio p values of detection of signal above background and the presence of defined β value. The number of detected probes (p value <0.05) was very high for all arrays and varied between 99.3 and 100 %. We computed a matrix demanding at least 99 % coverage per sample and 95 % of global coverage across all samples, resulting in a β value data matrix of dimension 27,564 CpG in 84 samples. Finally, normalization across arrays was performed with GenomeStudio software using a variety of strategies: (a) do-nothing, (b) background normalization, and (c) average normalization. The results were not influenced by the type of normalization. In this manuscript, we reported the results using method (b).
Immunophenotypic characterization
Total white blood cell count was performed by an automated hematology analyzer (Coulter Counter, Beckman, Milan, Italy). Identification of the major lymphocyte subsets (B lymphocytes, T lymphocytes, virgin T lymphocytes, memory T lymphocytes, and NK cells) was performed using the following combination of monoclonal antibodies: CD5/CD19 to identify B and autoreactive B lymphocytes, CD3/CD4/CD8 to identify T lymphocytes subsets, CD28/CD95/CD4 and CD28/CD95/CD8 to identify effector T helper and cytotoxic lymphocytes, and CD3/CD16/CD56 to identify NK cells. Phenotypical analysis of peripheral blood lymphocytes samples was performed on whole blood as previously described (Fagnoni et al. 2000; Nasi et al. 2006). A cytometric approach with fluorochrome-labeled monoclonal antibodies (mAbs, BD Biosciences Pharmingen, San Diego, CA) directly conjugated to fluorescent molecules (fluorescein isothiocyanate, phycoerythrin, phycoerythrin-cy-crhome) staining was used. Phenotypic analyses of cytotoxic and helper subsets were performed placing an electronic gate on CD8+ bright and CD4+ bright cells and evaluating the expression of CD28 vs CD95. A minimum of 30,000 cells per sample was acquired using a FACSCalibur (BD Biosciences) flow cytometer, and data were analyzed with FlowJo (Tree Star Inc., Ashland, OR) software.
Statistical analyses
The locus-by-locus differential methylation analysis was performed using GenomeStudio software. The Illumina Custom error model with false discovery rate correction was applied to data after background normalization. Differences in methylation levels between groups were considered statistically significant when |Diff Score| was >13, corresponding to a q value <0.05. Global methylation, Alu repeats methylation percentage, and β value mean distribution of selected clusters of CpG probes were compared between groups by classical ANOVA and Kruskal–Wallis test, respectively, for parametric and nonparametric data. Newman–Keuls and Dunn tests have been used as posttests, respectively, for parametric and nonparametric data. Bonferroni correction was applied considering the number of statistical tests performed. Hierarchical cluster analysis was performed using Illumina GenomeStudio software. Functional gene ontology (GO) annotation of genes of interest was performed using gene ontology enrichment analysis and visualization tool http://cbl-gorilla.cs.technion.ac.il/ (Eden et al. 2009). Gene functional classification and functional annotation clustering were performed to identify functional gene groups and ontology terms that are significantly overrepresented among genes selected in the Illumina array. p values were derived using hypergeometric tests.
The percentage of CpG sites linked to aging and longevity was calculated and normalized separately for each chromosome using the following formula:
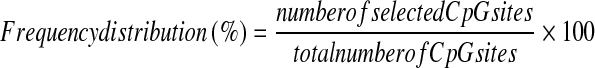 |
The test for overrepresentation of significant probes inside each chromosome has been performed using the hypergeometric cumulative function, considering the total number of probes inside all the chromosomes (NG = 27,564), the total number of significant probes inside all the chromosomes (NS = 217 for longevity, 1,039 for aging), the total number of probes in each chromosome, and the total number of significant probes in each chromosome.
Results
Centenarians’ offspring have a better health status
Our proposed model, described in supplementary Fig. S1 and including female centenarians’ offspring with both long-lived parents (mean age 69.8 + 1.6 years) and female offspring of both non-long-lived parents (72.4 + 1.1 years), seems to be a valid approach to investigate the role of DNA methylation in healthy aging and longevity. Both groups were comparable for age and extremely different in terms of probability to become long lived. Indeed, the prevalence of several diseases (myocardial infarction, cancer, arrhythmia, and hypertension), the proportion of people using medications, and the number of prescribed drugs were significantly lower in centenarians’ offspring than in offspring of both non-long-lived parents (Table 1).
Table 1.
Prevalence of major age-related diseases and pharmacological therapy in centenarians’ offspring (n = 21) and offspring of both non-long-lived parents (n = 21)
| Health assessment | Centenarians’ offspring, n (%) | Offspring of non-long-lived parents, n (%) | Prevalence odds ratio (95 % C.I.) | p value |
|---|---|---|---|---|
| Past diseases | ||||
| Myocardial infarction | 0 (0.0) | 5 (23.8) | 0.43 (0.23–0.63) | 0.048 |
| Cancer | 1 (4.8) | 8 (38.1) | 0.08 (0.01–0.73) | 0.020 |
| Current disease | ||||
| Irregular heart rhythm | 2 (9.5) | 9 (42.9) | 0.14 (0.03–0.76) | 0.032 |
| Hypertension | 4 (19.0) | 18 (85.7) | 0.04 (0.01–0.20) | <0.0001 |
| COPD | 0 (0.0) | 1 (4.8) | 0.49 (0.36–0.67) | 1 |
| Diabetes mellitus | 2 (9.5) | 3 (14.3) | 0.62 (0.09–4.23) | 1 |
| Pharmacological therapy | ||||
| Subjects taking drugs | 15 (71.4) | 21 (100.0) | 0.42 (0.28–0.61) | 0.021 |
| Number of prescribed drugs (mean ± S.E.M.) | 2.74 ± 0.52 | 6.62 ± 0.78 | <0.0001 | |
Medical history: The occurrence of major age-related diseases was collected through a standard structured questionnaire, including past history of myocardial infarction and cancer, and current history of cardiac arrhythmia, high blood pressure, chronic obstructive pulmonary disease (COPD), and diabetes mellitus. Data were expressed as total number of subjects who presented each health condition. Pharmacological therapy: The drug use was determined by two parameters: proportion of subjects taking medications and mean number of prescribed drugs
Examination of the differences between centenarians’ offspring and offspring of non-long-lived parents was performed using chi-square and the independent samples t test analyses
Global DNA and Alu methylation
A significant decrease in global DNA methylation levels was observed with age, as shown by higher methylation percentage in young controls compared to all other groups (Fig. 1a). Interestingly, age-related loss of DNA methylation was less pronounced in centenarians’ offspring, as indicated by the significant higher values of global DNA methylation (p < 0.05) observed in this group compared with offspring of non-long-lived parents.
Fig. 1.
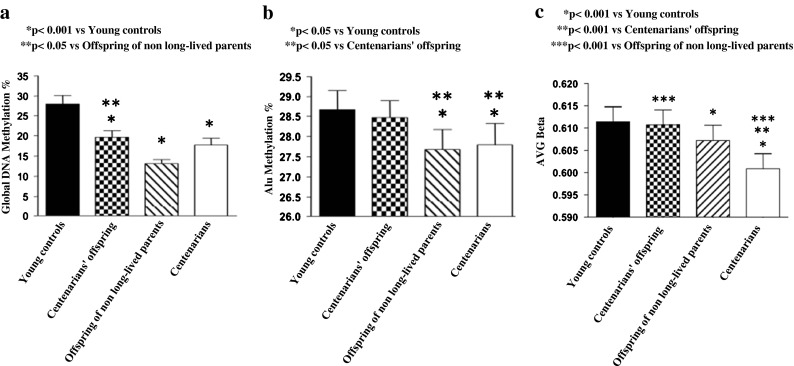
Global DNA methylation (a) and Alu methylation (b) in DNA extracted from peripheral leukocytes of young women (n = 21), female centenarians’ offspring (n = 21), female offspring of both non-long-lived parents (n = 21), and female centenarians (n = 21). Distribution of average (AVG) β methylation values of CpG sites localized in non-CpG islands (c) in enrolled populations, evaluated by genome-wide array
A significant decrease in Alu methylation (Fig. 1b) was observed in centenarians and offspring of both non-long-lived parents (both p < 0.05), while no significant change was detected in centenarians’ offspring, as compared to young controls. Alu methylation was significantly (p < 0.05) higher in centenarians’ offspring compared to offspring of non-long-lived parents.
We performed genome-wide CpG methylation analysis (Infinium HumanMethylation27 BeadChip, Illumina) in DNA from peripheral blood leukocytes of all enrolled subjects. CpG repeats outside CpG islands were moderately to highly methylated (Fig. 1c). These sites, selected according to the criteria of Takai and Jones (2002), are highly enriched in Alu sequences. Mean methylation levels of CpG sites localized in non-CpG islands decreased with age. It is worthy to note that average β methylation value of non-CpG islands was higher in centenarians’ offspring than in offspring of non-long-lived parents (p < 0.001), confirming a better preservation of DNA methylation status in centenarians’ offspring.
Age-dependent methylation profile
Genome-wide methylation analysis, through a locus-by-locus approach, identified CpG sites showing the most significant age-dependent variation in methylation in the four enrolled groups. We identified CpG loci significantly (p < 0.05) hyper- or hypomethylated in all the three groups of old subjects (concurrently in centenarians, centenarians’ offspring, and offspring of non-long-lived parents) vs young controls (Fig. 2a). Average β methylation values of this set of hypermethylated CpG sites (709 loci, corresponding to 607 genes, listed in supplementary Table S1) were significantly higher in centenarians compared to centenarians’ offspring (p < 0.001), offspring of non-long-lived parents (p < 0.001), and young controls (p < 0.001; Fig. 2b, d). A gene ontology analysis of these CpG sites hypermethylated with chronological aging showed enrichment of genes for biological processes and molecular functions involved in the development of anatomical structures, organs, and multicellular organisms, in cell differentiation, and in the regulation of transcription (Table 2). Using the same criteria, we also detected 330 CpG loci (corresponding to 326 genes, listed in supplementary Table S2) showing a significant loss of methylation concurrently in centenarians (p < 0.001), centenarians’ offspring (p < 0.01), and offspring of non-long-lived parents (p < 0.01) compared to young controls (Fig. 2c, e). GO analysis of hypomethylated CpG sites revealed significantly enriched genes for biological processes, including the following categories: defense response to bacterium, regulation of respiratory burst involved in acute inflammatory response, positive regulation of signal transduction/signaling process, and response to bacterium (Table 2). No significant difference has been observed between centenarians’ offspring and offspring of non-long-lived parents in terms of average β values for CpG hyper- and hypomethylated with chronological aging.
Fig. 2.

Aging-associated DNA methylation profile. Venn diagram (a) shows the number of CpG loci significantly hyper- and hypomethylated in centenarians, centenarians’ offspring, and offspring of non-long-lived parents vs young controls. Distribution of average (AVG) β methylation values of CpG sites over the 709 age-hypermethylated (b) and 330 age-hypomethylated (c) loci in enrolled populations. Heat map of age-hypermethylated genes (d) and age-hypomethylated genes (e) in the enrolled populations
Table 2.
Gene ontology analysis of CpG sites hypermethylated and hypomethylated with aging
| GO term | Description | p value |
|---|---|---|
| Hypermethylated | ||
| GO:0048856 | Anatomical structure development | (2.31 × 10−24) |
| GO:0032502 | Developmental process | (1.41 × 10−22) |
| GO:0009653 | Anatomical structure morphogenesis | (7.14 × 10−20) |
| GO:0032501 | Multicellular organismal process | (6.58 × 10−17) |
| GO:0048731 | System development | (9.21 × 10−16) |
| GO:0048869 | Cellular developmental process | (1.78 × 10−13) |
| GO:0007275 | Multicellular organismal development | (6.11 × 10−13) |
| GO:0048513 | Organ development | (1.71 × 10−11) |
| GO:0048598 | Embryonic morphogenesis | (5.46 × 10−11) |
| GO:0003002 | Regionalization | (7.53 × 10−10) |
| GO:0007389 | Pattern specification process | (8.37 × 10−10) |
| GO:0030154 | Cell differentiation | (1.29 × 10−09) |
| GO:0006355 | Regulation of transcription, DNA dependent | (6.93 × 10−09) |
| Hypomethylated | ||
| GO:0042742 | Defense response to bacterium | (4.57 × 10−04) |
| GO:0060264 | Regulation of respiratory burst involved in inflammatory response | (4.90 × 10−04) |
| GO:0009967 | Positive regulation of signal transduction | (6.04 × 10−04) |
| GO:0023056 | Positive regulation of signaling process | (6.94 × 10−04) |
| GO:0009617 | Response to bacterium | (9.87 × 10−04) |
p values were derived using hypergeometric tests
Considering the number of probes for each chromosome, we observed that hyper- and hypomethylated CpG sites associated to aging were not randomly scattered among chromosomes. Hypermethylated loci were mainly located on chromosomes 5 (p < 0.01), 7 (p < 0.05), 8 (p < 0.05), 13 (p < 0.05), and 18 (p < 0.01), while hypomethylated loci were more present on chromosomes 1 and 20 (both p < 0.01; supplementary Fig. S2a)
Healthy aging and long-life methylation profile
In order to identify an epigenetic signature for healthy aging and longevity, a locus-by-locus analysis of CpG sites was performed between centenarians’ offspring and offspring of non-long-lived parents. We selected 150 CpG loci (corresponding to 124 genes, listed in supplementary Table S3) significantly (p < 0.05) hypermethylated in centenarians’ offspring compared to offspring of non-long-lived parents. GO analysis of hypermethylated CpG loci showed enrichment of genes for biological processes, including nucleobase metabolic process, nucleobase, nucleoside, nucleotide, and nucleic acid biosynthetic process (Table 3). Using the same criteria, 67 CpG loci (corresponding to 65 genes, listed in supplementary Table S4) were hypomethylated in centenarians’ offspring compared to offspring of non-long-lived parents. GO analysis of hypomethylated CpG sites revealed enrichment of biological process of “consequence of signal transmission” (Table 3). Hyper- and hypomethylated loci associated to longevity were not randomly scattered among chromosomes, but they were mainly located on chromosomes 19 (both p < 0.05), 21 (p < 0.001, and p < 0.05, respectively) and X (p < 0.001 and p < 10−9, respectively; supplementary Fig. S2b).
Table 3.
Gene ontology analysis of CpG sites hypermethylated and hypomethylated between centenarians’ offspring and old controls
| GO term | Description | p value |
|---|---|---|
| Hypermethylated | ||
| GO:0009112 | Nucleobase metabolic process | (9.35 × 10−4) |
| GO:0034404 | Nucleobase, nucleoside, and nucleotide biosynthetic process | (9.37 × 10−4) |
| GO:0034654 | Nucleobase, nucleoside, nucleotide, and nucleic acid biosynthetic process | (9.80 × 10−4) |
| Hypomethylated | ||
| GO:0023050 | Consequence of signal transmission | (9.40 × 10−4) |
p values were derived using hypergeometric tests
Using more stringent criteria (p < 0.001, β value difference of at least 0.10 between the centenarians’ offspring and offspring of non-long-lived parents), we identified ten and two CpG loci, respectively, hypermethylated and hypomethylated in centenarians’ offspring compared to offspring of non-long-lived parents, corresponding to nine genes (Fig. 3). Six of these genes (SLC38A4, SLC22A18, MGC3207, ECRG4, ATP13A4, AGPAT2) resulted to be hypermethylated and involved in metabolic processes, DUSP22 is a tumor-suppressor gene, while ZNF169 and FLJ32569 remain to be characterized.
Fig. 3.

Average (AVG) β values of 12 CpC loci, corresponding to nine genes, differently methylated between centenarians’ offspring and offspring of non-long-lived parents and selected using highly stringent criteria (p < 0.001 and β value difference of at least 0.10)
We excluded that our results are influenced by a different blood cell distribution between groups. White blood cell count and differential were not different between centenarians’ offspring and offspring of non-long-lived parents (Table S5). In addition, detailed analysis of the main lymphocyte subsets in flow cytometry has shown that there were no significant differences in immunophenotypic characterization of blood cells between both groups (data not shown).
Discussion
While it has been shown that DNA methylation pattern changes with age contributing to the development of several age-related diseases, in literature, there is no evidence on the role of epigenetics in the modulation of human healthy aging and longevity. Our proposed model, including female centenarians’ offspring with both long-lived parents and female offspring of both non-long-lived parents, seems to be a valid approach to answer this question. Both groups were comparable for age and extremely different in terms of probability to become long lived. In addition, a significant lower prevalence of several diseases was detected in centenarians’ offspring than in offspring of non-long-lived parents.
We confirmed an age-dependent decrease in genomic DNA methylation, as evidenced by the methylation analysis of global DNA, Alu sequences, and non-CpG islands. In humans, a loss of genomic DNA methylation has been described to occur in several types of tissue during aging and age-related diseases (Fraga et al. 2007; Bollati et al. 2009; Kim et al. 2010; Choi et al. 2009; Moore et al. 2008). In addition, global genomic hypomethylation of leukocyte DNA has been reported to be an independent risk factor for cancer, atherosclerosis, and cardiovascular disease (Fraga et al. 2007; Kim et al. 2010; Choi et al. 2009; Moore et al. 2008). DNA hypomethylation occurs mostly in transposable DNA repetitive elements, including the Alu and LINE-1 sequences, which play a crucial role in gene regulation and genomic stability. If transcribed, such elements can reinsert into DNA, potentially moving across the genome. Therefore, the decrease in global DNA methylation, observed during aging, could result in increased retrotransposon activity and genomic instability (Wilson et al. 2007; Asada et al. 2006). Hypomethylation of repeated DNA sequences is expected to lead to the transcriptional activation of those sequences that still contain active promoters. This can interfere with cellular transcription activity by disrupting the balance of transcription factors (Wilson et al. 2007). In addition, global DNA hypomethylation seems to increase immunogenicity of self-DNA, contributing to the development of age-associated diseases (Agrawal et al. 2010). Our results showed that this deleterious process is delayed in centenarians’ offspring. Indeed, global DNA methylation and Alu elements' methylation were higher in centenarians’ offspring than in offspring of non-long-lived parents, notwithstanding both groups were comparable for age. These data were fully supported by the evidence of higher average β methylation values of non-CpG islands in centenarians’ offspring than in offspring of non-long-lived parents. Therefore, we cannot exclude that a better preservation of DNA methylation status, leading to an enhanced genomic stability, a more stable transcription system, and a less immunogenic DNA, may delay the onset of age-related diseases and prolong survival in centenarians’ offspring. The apparent better DNA methylation profile of centenarians’ offspring compared to that observed in centenarians may be instead a consequence of a significant difference in age between both groups (about 30 years). In fact, centenarians are not free of loss of global DNA methylation during aging, even if this detrimental process seems to be attenuated in long-lived subjects, at least on the basis of data observed in centenarians’ offspring.
Although genome-wide levels of methylation decrease with age, there is a tendency for DNA methylation to increase mainly in CpG islands localized in the promoter regions of specific genes (Rakyan et al. 2010; Grönniger et al. 2010). Rakyan et al. (2010) recently evaluated genome-scale DNA methylation profiling of whole blood from 93 different healthy females ranging from 49 to 75 years of age, using Illumina HumanMethylation27 BeadChips. The authors reported 213 CpG sites that become more methylated with age and 147 CpG sites that lose methylation with age. A GO analysis of hypermethylated regions showed enrichment of tissue-specific functions, including multicellular organismal development, sequence-specific DNA binding, transcription factor activity, and regulation of transcription. Hypermethylated genes resulted strongly enriched for CpG islands and occurred predominantly at bivalent chromatin domain promoters. This category of promoters is frequently hypermethylated in tumors, pointing to a novel mechanistic link between aberrant hypermethylation in cancer and aging.
In this study, we investigated genome-wide methylation profile using Infinium HumanMethylation27 BeadChip in DNA from peripheral blood of 84 females ranging from 17 to 106 years. We confirmed the impact of aging on DNA methylation, identifying a pattern of 709 CpG loci, exclusively located within CpG islands, that resulted concurrently hypermethylated in all old subjects (centenarians, centenarians’ offspring, and offspring of non-long-lived parents) compared to young controls. Similarly to Rakyan et al. (2010), a GO analysis of these hypermethylated CpG sites showed an enrichment of key developmental genes. In addition, we identified 330 CpG loci hypomethylated with aging. GO analysis of these CpG sites revealed enrichment in categories of genes associated with inflammatory response. This suggests a possible role of epigenetics in the modulation of inflammatory processes in aging (inflammaging; Franceschi et al. 2007; Salvioli et al. 2006). Centenarians’ offspring and offspring of non-long-lived parents had an identical methylation pattern for genes both hyper- and hypomethylated with aging. This last observation was not unexpected, considering that (1) in our experimental design, both centenarians’ offspring and offspring of non-long-lived parents belong to the group of old subjects (see Fig. 2a) that was compared to young subjects and (2) both centenarians’ offspring and offspring of non-long-lived parents were comparable for age.
In order to identify an epigenetic signature for healthy aging and longevity, we compared human genome-wide DNA methylation of centenarians’ offspring with age-matched control subjects, born from both non-long-lived parents. Through a locus-by-locus analysis of CpG sites comparing both groups of subjects, we identified a subset of 150 and 67 CpG sites, respectively, hypermethylated and hypomethylated. These genes may be strictly involved in the process of healthy aging and longevity. GO analysis of hypermethylated CpG loci showed enrichment of genes for few biological processes involved in DNA/RNA synthesis, metabolism, and cellular signaling. In addition, GO analysis of hypomethylated CpG sites revealed significantly enriched biological process of consequence of signal transmission. This category includes steps whereby the downstream processes started by a signal are brought to a conclusion. Although the cause–effect relation is difficult to be assessed in a cross-sectional study, we cannot exclude that a slower cell growing/metabolism and a better control in signal transmission through epigenetic mechanisms may be involved in the process of longevity. This hypothesis is supported by several previous reports:
Insulin-like growth factor I (IGF-I) is a potent growth factor and positive regulator of DNA replication. In animal models, alterations of IGF-I signaling increase the life expectancy (Barbieri et al. 2003). In centenarians, a higher frequency of heterozygous loss of function mutations in the IGF-I receptor gene has been recently described (Suh et al. 2008), suggesting a role of the IGF-I system impairment in the modulation of human lifespan.
A decreased thyroid function might be beneficial in the elderly through its lowering effects on basal metabolic rate and oxidative metabolism with a consequent reduction of DNA damage reactive oxygen species (Peeters 2009). Indeed, high levels of TSH (Gussekloo et al. 2004) and low levels of FT4 (van den Beld et al. 2005) are associated with a better survival in elderly subjects. In addition, centenarians and centenarians’ offspring have higher levels of TSH compared with controls (Atzmon et al. 2009a; Atzmon et al. 2009b). Interestingly, thyroid failure may induce a suppression in several processes involved in nucleotide biosynthesis and DNA replication (Agocha et al. 1997; Ledda-Columbano et al. 2006).
There are some limitations that need to be acknowledged and addressed regarding the present study: (1) We focused our study on DNA methylation profile of leukocytes that represent an easily accessible DNA source widely used in several studies, although DNA methylation is a tissue-specific phenomenon. However, recent papers reported an age-associated hypermethylation of key developmental genes also in skin (Grönniger et al. 2010) and buccal cells (Rakyan et al. 2010), similarly to our data, supporting the hypothesis that these age-related epigenetic changes are not restricted to blood cells compartment. (2) Leukocytes consist of several cell types that may carry different DNA methylation profiles; however, we excluded that our results are influenced by a different cell distribution of blood cells between groups. (3) There is a lack of data on gene/protein expression and (4) the number of subjects analyzed is relatively small.
In conclusion, we confirmed that aging-associated DNA hypermethylation occurs predominantly in genes involved in the development of anatomical structures, organs, and multicellular organisms and in the regulation of transcription. We have also shown that a better preservation of DNA methylation status during life and a specific epigenetic modulation of genes involved in nucleobase, nucleoside, nucleotide, and nucleic acid biosynthetic process and control of signal transmission may contribute to explain the longer lifespan and healthy aging of centenarians’ offspring. This phenomenon may be considered a new aspect of the age remodeling, a continuous adaptation of the body to the deteriorative changes occurring over time. However, it is not clear how relevant these epigenetic changes are in the context of functional changes in gene expression.
Future studies integrating complete analyses of epigenetic processes (DNA methylation and histone posttranscriptional modifications, including methylation, acetylation, ubiquitination, phosphorylation, etc.) with gene expression profile in a wider population and in other tissues are mandatory to confirm the role of epigenetics in the modulation of successful aging and human longevity.
Electronic supplementary material
(PDF 37.6 MB)
Acknowledgments
Financial support to this work has been partially provided by the Italian Ministry of University and Research (MIUR) and by the Istituto Auxologico Italiano. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors would like to thank the Register Office of the city of Milan for its contribution in the collection of data.
References
- Adams ER, Nolan VG, Andersen SL, Perls TT, Terry DF. Centenarian offspring: start healthier and stay healthier. J Am Geriatr Soc. 2008;56:2089–2092. doi: 10.1111/j.1532-5415.2008.01949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agocha A, Lee HW, Eghbali-Webb M. Hypoxia regulates basal and induced DNA synthesis and collagen type I production in human cardiac fibroblasts: effects of transforming growth factor-β 1, thyroid hormone, angiotensin II and basic fibroblast growth factor. J Mol Cell Cardiol. 1997;29:2233–2244. doi: 10.1006/jmcc.1997.0462. [DOI] [PubMed] [Google Scholar]
- Agrawal A, Tay J, Yang GE, Agrawal S, Gupta S. Age-associated epigenetic modifications in human DNA increase its immunogenicity. Aging. 2010;2:93–100. doi: 10.18632/aging.100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada K, Kotake Y, Asada R, Saunders D, Broyles RH, Towner RA, Fukui H, Floyd RA. LINE-1 hypomethylation in a choline-deficiency-induced liver cancer in rats: dependence on feeding period. J Biomed Biotechnol. 2006;1:17142. doi: 10.1155/JBB/2006/17142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzmon G, Barzilai N, Hollowell JG, Surks MI, Gabriely I. Extreme longevity is associated with increased serum thyrotropin. J Clin Endocrinol Metab. 2009;94:1251–1254. doi: 10.1210/jc.2008-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzmon G, Barzilai N, Surks MI, Gabriely I. Genetic predisposition to elevated serum thyrotropin is associated with exceptional longevity. J Clin Endocrinol Metab. 2009;94:4768–4775. doi: 10.1210/jc.2009-0808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri M, Bonafè M, Franceschi C, Paolisso G. Insulin/IGF-I-signaling pathway: an evolutionarily conserved mechanism of longevity from yeast to humans. Am J Physiol Endocrinol Metab. 2003;285:1064–1071. doi: 10.1152/ajpendo.00296.2003. [DOI] [PubMed] [Google Scholar]
- Bellizzi D, D’Aquila P, Montesanto A, Corsonello A, Mari V, Mazzei B, Lattanzio F, Passarino G (2011) Global DNA methylation in old subjects is correlated with frailty. Age. doi:10.1007/s11357-011-9216-6 [DOI] [PMC free article] [PubMed]
- Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130:234–239. doi: 10.1016/j.mad.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevenini E, Invidia L, Lescai F, Salvioli S, Tieri P, Castellani G, Franceschi C. Human models of aging and longevity. Expert Opin Biol Ther. 2008;8:1393–405. doi: 10.1517/14712598.8.9.1393. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, James SR, Link PA, McCann SE, Hong CC, Davis W, Nesline MK, Ambrosone CB, Karpf AR. Association between global DNA hypomethylation in leukocytes and risk of breast cancer. Carcinogenesis. 2009;30:1889–1897. doi: 10.1093/carcin/bgp143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinforma. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagnoni FF, Vescovini R, Passeri G, Bologna G, Pedrazzoni M, Lavagetto G, Casti A, Franceschi C, Passeri M, Sansoni P. Shortage of circulating naive CD8+ T cells provides new insights on immunodeficiency in aging. Blood. 2000;95:2860–2868. [PubMed] [Google Scholar]
- Fraga MF, Agrelo R, Estell M. Cross-talk between aging and cancer: the epigenetic language. Ann NY Acad Sci. 2007;1100:60–74. doi: 10.1196/annals.1395.005. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Bonafè M. Centenarians as a model for healthy aging. Biochem Soc Trans. 2003;31:457–461. doi: 10.1042/BST0310457. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Grönniger E, Weber B, Heil O, Peters N, Stäb F, Wenck H, Korn B, Winnefeld M, Lyko F. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genetics. 2010;6:e1000971. doi: 10.1371/journal.pgen.1000971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussekloo J, van Exel E, de Craen AJ, Meinders AE, Frölich M. Thyroid status, disability and cognitive function, and survival in old age. JAMA. 2004;292:2591–2599. doi: 10.1001/jama.292.21.2591. [DOI] [PubMed] [Google Scholar]
- Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW. DNA methylation as a biomarker for cardiovascular disease risk. PLoS One. 2010;5:e9692. doi: 10.1371/journal.pone.0009692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledda-Columbano GM, Molotzu F, Pibiri M, Cossu C, Perra A, Columbano A. Thyroid hormone induces cyclin D1 nuclear translocation and DNA synthesis in adult rat cardiomyocytes. FASEB J. 2006;20:87–94. doi: 10.1096/fj.05-4202com. [DOI] [PubMed] [Google Scholar]
- Moore LE, Pfeiffer RM, Poscablo C, Real FX, Kogevinas M, Silverman D, García-Closas R, Chanock S, Tardón A, Serra C, Carrato A, Dosemeci M, García-Closas M, Esteller M, Fraga M, Rothman N, Malats N. Genomic DNA hypomethylation as a biomarker for bladder cancer susceptibility in the Spanish Bladder Cancer Study: a case-control study. Lancet Oncol. 2008;9:359–366. doi: 10.1016/S1470-2045(08)70038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasi M, Troiano L, Lugli E, Pinti M, Ferraresi R, Monterastelli E, Mussi C, Salvioli G, Franceschi C, Cossarizza A. Thymic output and functionality of the IL-7/IL-7 receptor system in centenarians: implications for the neolymphogenesis at the limit of human life. Aging Cell. 2006;5:167–175. doi: 10.1111/j.1474-9726.2006.00204.x. [DOI] [PubMed] [Google Scholar]
- Peeters RP. Thyroid function and longevity: new insights into an Old Dilemma. J Clin Endocrinol Metab. 2009;94:4658–4660. doi: 10.1210/jc.2009-2198. [DOI] [PubMed] [Google Scholar]
- Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, Leslie RD, Deloukas P, Spector TD. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvioli S, Capri M, Valensin S, Tieri P, Monti D, Ottaviani E, Franceschi C. Inflamm-aging, cytokines and aging: state of the art, new hypotheses on the role of mitochondria and new perspectives from systems biology. Curr Pharm Des. 2006;12:3161–3171. doi: 10.2174/138161206777947470. [DOI] [PubMed] [Google Scholar]
- Skytthe A, Valensin S, Jeune B, Cevenini E, Balard F, Beekman M, Bezrukov V, Blanche H, Bolund L, Broczek K, Carru C, Christensen K, Christiansen L, Collerton JC, Cotichini R, de Craen AJ, Dato S, Davies K, De Benedictis G, Deiana L, Flachsbart F, Gampe J, Gilbault C, Gonos ES, Haimes E, Hervonen A, Hurme MA, Janiszewska D, Jylhä M, Kirkwood TB, Kristensen P, Laiho P, Leon A, Marchisio A, Masciulli R, Nebel A, Passarino G, Pelicci G, Peltonen L, Perola M, Poulain M, Rea IM, Remacle J, Robine JM, Schreiber S, Scurti M, Sevini F, Sikora E, Skouteri A, Slagboom PE, Spazzafumo L, Stazi MA, Toccaceli V, Toussaint O, Törnwall O, Vaupel JW, Voutetakis K, Franceschi C, GEHA consortium Design, recruitment, logistics, and data management of the GEHA (Genetics of Healthy Ageing) project. Exp Gerontol. 2011;46:934–945. doi: 10.1016/j.exger.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh Y, Atzmon G, Cho MO, Hwang D, Liu B, Leahy DJ, Barzilai N, Cohen P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. PNAS. 2008;105:3438–3442. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. PNAS. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry DF, Wilcox MA, McCormick MA, Pennington JY, Schoenhofen EA, Andersen SL, Perls TT. Lower all-cause, cardiovascular, and cancer mortality in centenarians offspring. J Am Geriatr Soc. 2004;52:2074–2076. doi: 10.1111/j.1532-5415.2004.52561.x. [DOI] [PubMed] [Google Scholar]
- Van den Beld AW, Visser TJ, Feelders RA, Grobbee DE, Lamberts SW. Thyroid hormone concentrations, disease, physical function, and mortality in elderly men. J Clin Endocrinol Metab. 2005;90:6403–6409. doi: 10.1210/jc.2005-0872. [DOI] [PubMed] [Google Scholar]
- Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775:138–162. doi: 10.1016/j.bbcan.2006.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 37.6 MB)