

Abstract
Thirty five years ago mesotrypsin was first isolated from the human pancreas. It was described as a minor trypsin isoform with the remarkable property of near total resistance to biological trypsin inhibitors. Another unusual feature of mesotrypsin was discovered later, when it was found that mesotrypsin has defective affinity toward many protein substrates of other trypsins. As the younger sibling of the two major trypsins secreted by the pancreas, cationic and the anionic trypsin, it has been speculated to represent an evolutionary waste with no apparent function. We know now that mesotrypsin is functionally very different from the other trypsins, with novel substrate specificity that hints at distinct physiological functions. Recently, evidence has begun to emerge implicating mesotrypsin in direct involvement in cancer progression. This review will explore the biochemical characteristics of mesotrypsin and structural insights into its specificity, function, and inhibition.
Keywords: Trypsin, mesotrypsin, serine protease, protease inhibitors, protein crystallography, substrate specificity, cancer progression
Introduction
Proteases are encoded by roughly 3% of all genes in organisms ranging from bacteria to plants to humans [1]. They are universally distributed throughout the human body and play essential roles in many important physiological and pathological functions. One of the most abundant mechanistic classes of proteases is the serine protease group, named for the key nucleophilic serine involved in catalysis. Trypsins are a group of serine proteases that are produced and secreted as zymogens by the pancreas and activated by enteropeptidase cleavage in the duodenum, where they act as major digestive enzymes [2].
Due to the potential for proteolytic tissue damage, elaborate mechanisms have evolved to regulate proteases at multiple levels. One mechanism involves complexation of proteases with endogenous protein protease inhibitors. Regulation of the protease/inhibitor balance is crucial, and the upset of this balance is in evidence in numerous pathological conditions, including forms of cancer, emphysema, cystic fibrosis, chronic obstructive pulmonary disease, acute respiratory distress syndrome, asthma, cirrhosis, thrombosis, dementia, pancreatitis, and psoriasis [3-8]. One major group of endogenous inhibitors of trypsins and other serine proteases are the ‘canonical’ inhibitors, named for a protease-binding loop of highly characteristic backbone conformation [9-12]. A canonical inhibitor fulfills the paradoxical function of binding to trypsin or another serine protease in a substrate-like manner, and yet acting as an inhibitor rather than a substrate [11,13]. A particularly impressive example is the interaction of the canonical inhibitor bovine pancreatic trypsin inhibitor (BPTI) with bovine trypsin, where at neutral pH, K i ≈ 5 × 10-14 M [14,15], and k cat ≈ 8 × 10-10 s-1 [16,17].
Human mesotrypsin, encoded by the PRSS3 gene, has been described as a defective human trypsin due to its compromised ability to cleave protein substrates, and its physiological role has been a mystery [18-20]. While mesotrypsin has long been known to be resistant to inhibition by protein protease inhibitors [18,19,21,22], in 2003 it was discovered that mesotrypsin possesses an extraordinary catalytic capability for hydrolyzing the reactive sites of canonical trypsin inhibitors SPINK1 and soybean trypsin inhibitor [19]. In the past five years, evidence has continued to mount suggesting that mesotrypsin may have evolved very specifically to carry out the unique task of proteolyzing canonical inhibitors as substrates [23-25]. Here, we will describe the structural and enzymatic features of mesotrypsin that account for this unusual catalytic capability. We will also explore emerging evidence for its role in cancer, and consider the potential for development of potent and selective mesotrypsin inhibitors.
Human trypsin genes and proteins
Two major human trypsinogens, trypsinogen 1 and trypsinogen 2, were first purified in 1969 [26]. Trypsinogen 1 is the most abundant, accounting for ~13% of proteins in human pancreatic juice, while trypsinogen 2 accounts for ~6% of proteins in human pancreatic juice [27]. A third, minor trypsinogen isoform, originally referred to as “zymogen X”, was isolated by Rinderknecht et al. in 1979 [27]. It occurs in very low concentrations and probably accounts for <0.5% of proteins in human pancreatic juice. Human trypsinogens I, II and “zymogen X” were renamed on the basis of their relative isoelectric points as cationic, anionic and mesotrypsinogen, respectively [18,27]. The three human trypsinogens are encoded by different genes; those for cationic trypsinogen (PRSS1) and anionic trypsinogen (PRSS2) are located at proximal loci on chromosome 7q35, while the gene encoding mesotrypsinogen (PRSS3) is found on chromosome 9p13 [28].
Differentially-spliced forms of mesotrypsinogen, transcribed from alternative promoters, are expressed in different tissues [29-31]. Trypsinogen 4, which may utilize an unconventional CUG translation initiation codon [30], is highly expressed in brain tissue [32,33], and expressed at lower levels in many tissues and some tumors [29,34-39]. Trypsinogen 5, the most recently identified isoform, appears to have more restricted expression limited primarily to brain, intestine, uterus, and keratinocytes [29]. The multiple zymogen forms differ only at the N-terminus encoded by exon 1, such that processing of any of the isoforms by removal of the prodomain results in active mesotrypsin of identical amino acid sequence [31]. Trypsinogens 4 and 5 lack recognizable signal sequences, but a feature-based algorithm for predicting non-classical and leaderless secreted proteins [40] scores these zymogens as likely candidates for nonclassical secretion. There is some evidence for processing of the prodomain of trypsinogen 4 and deposition of activated mesotrypsin in the extracellular neuronal matrix of the brain [32], and likewise for activation of trypsinogens 4 and 5 by enteropeptidase in the granular layer of the epidermis [29]. Although active mesotrypsin protein has not been directly detected in tumors, mesotrypsin activity has been implicated in tumor progression by studies using PRSS3 silencing or mesotrypsin inhibition in cancer models, as described below, under “Role of mesotrypsin in cancer”.
Sequence and structural differences between mesotrypsin and other human trypsins
The three trypsins belong to the chymotrypsin superfamily of serine endopeptidases that are characterized by the catalytic triad His-57, Asp-102 and Ser-195 (chymotrypsinogen numbering) [41]. They are all stabilized by Ca2+ as a ligand, and strictly cleave peptide bonds after Arg or Lys optimally at pH around 8.0 [18]. Mesotrypsin shows high sequence homology with the major digestive trypsins (Figure 1). Cationic trypsin (Uniprot: P07477) and anionic trypsin (Uniprot: P07478) are 96% identical, while mesotrypsin (Uniprot: P35030) shares 87.8% and 88.7% identity with cationic and anionic trypsin, respectively. Mesotrypsin exhibits unique sequence and structural features that contribute to distinct specificity and functional properties, most notably Arg-193, which is a highly conserved glycine residue in most other serine proteases.
Figure 1.
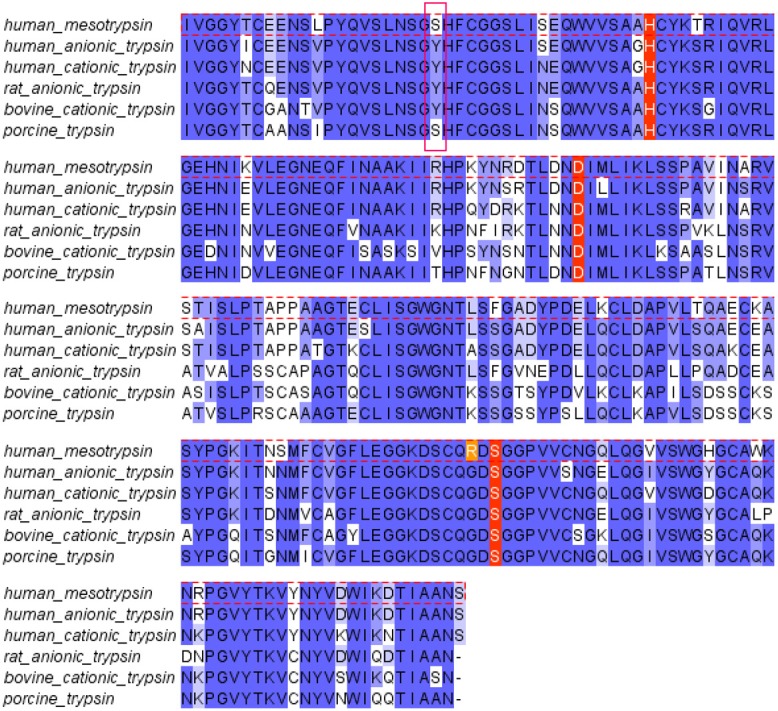
Sequence alignment of human trypsins. Human mesotrypsin displays a high degree of conservation with other mammalian trypsins. Two important differences are the substitution of Arg for the conserved Gly-193 (highlighted in orange) and the substitution of Tyr-39 with Ser (pink box).
Structural studies of mesotrypsin (Table 1) have revealed that Arg-193 contributes to an atypical clustering of positive electrostatic surface potential on the primed side of the substrate binding cleft (which interacts with substrate residues C-terminal to the cleavage site), in the vicinity of the S2′ subsite [22]. Co-crystal structures with several polypeptide trypsin inhibitors, along with mutagenesis studies, have demonstrated steric and in some cases electrostatic repulsion between Arg-193 and the P2′ residue of a bound substrate or inhibitor (Figure 2) [25,42,43]. The structures reveal that Arg-193 must undergo considerable conformational rearrangements to allow binding of substrates or inhibitors with bulky P2′ residues, while analysis of the kinetics of proteolysis show that this reduces affinity toward peptide inhibitors or substrates by several orders of magnitude compared to other trypsins [23,25,42,43]. As a result, mesotrypsin is uniquely defiant to inhibition by many polypeptide serine protease inhibitors (Table 2) [18,19,21,22], and is also inefficient in the cleavage of many tryptic sites within protein substrates, for example in the activation of other pancreatic zymogens [18,19]. Mutation of mesotrypsin Arg-193 to Gly, as is found at this position in most other serine proteases, has been reported to restore sensitivity to inhibition by canonical trypsin inhibitors [19].
Table 1.
Crystal structures of mesotrypsin complexes
| PDB ID | Enzyme | Inhibitor | Resolution (Å) | Reference |
|---|---|---|---|---|
| 1H4W | mesotrypsin | benzamidine | 1.70 | Katona et al. [22] |
| 1R9P | mesotrypsin-S195A | BPTI | 1.40 | Salameh et al. [43] |
| 3L33 | mesotrypsin-S195A | APPI | 2.48 | Salameh et al. [25] |
| 3L3T | mesotrypsin-S195A | APPI-R15K | 2.38 | Salameh et al. [25] |
| 3P92 | mesotrypsin-S195A | BPTI-K15R/R17G | 1.60 | Salameh et al. [42] |
| 3P95 | mesotrypsin-S195A | BPTI-K15R/R17D | 1.30 | Salameh et al. [42] |
| 4DG4 | mesotrypsin-S39Y/S195A | BPTI | 1.40 | Salameh et al. [24] |
Figure 2.

Unique structural features of the mesotrypsin active site. Illustration of major structural differences between cationic trypsin and mesotrypsin. A. In the cationic trypsin complex with BPTI (PDB: 1TRN), the a highly conserved Gly-193 forms an H-bond with His-40 and Tyr-39 forms an H-bond with the main chain amide nitrogen of the P4′ residue of BPTI. B. By contrast, the mesotrypsin complex with BPTI (PDB: 2R9P) features fewer intermolecular H-bonds, as well as the potential for steric and/or electrostatic repulsion by Arg-193 of an inhibitor or substrate possessing a bulky side chain at the P2′ position. In both panels, the Arg-17 P2′ side chain of BPTI has been omitted for clarity.
Table 2.
Comparison of trypsin and mesotrypsin inhibition by canonical serine protease inhibitors
| Enzyme | Inhibitor | K i (M) | Reference |
|---|---|---|---|
| bovine trypsin | bovine pancreatic trypsin inhibitor (BPTI) | 6 × 10-14 | [14,15] |
| bovine trypsin | soybean trypsin inhibitor (SBTI) | 1 × 10-11 | [68] |
| porcine trypsin | SPINK1 (human) | 3 × 10-11 | [15] |
| bovine trypsin | Bowman-Birk inhibitor (soybean) | 8 × 10-10 | [69] |
| bovine trypsin | Curcurbita maxima trypsin inhibitor I | 2 × 10-12 | [70] |
| bovine trypsin | Schistocerca gregaria trypsin inhibitor | 3 × 10-12 | [71] |
| bovine trypsin | sunflower trypsin inhibitor | 1 × 10-10 | [72] |
| porcine trypsin | bovine pancreatic trypsin inhibitor (BPTI) | 1 × 10-11 | [73] |
| porcine trypsin | APPI (human) | 1 × 10-10 | [73] |
| human cationic trypsin | bovine pancreatic trypsin inhibitor (BPTI) | 2 × 10-11 | [43] |
| human cationic trypsin | APPI (human) | 1.8 × 10-10 | [23] |
|
| |||
| human mesotrypsin | bovine pancreatic trypsin inhibitor (BPTI) | 1.4 × 10-5 | [25,43] |
| human mesotrypsin | soybean trypsin inhibitor (SBTI) | 4.2 × 10-7 | [22] |
| human mesotrypsin | SPINK1 (human) | 1.5 × 10-6 | [19] |
| human mesotrypsin | APPI (human) | 1.4 × 10-7 | [23,43] |
Another relatively conserved residue near the trypsin active site, Tyr-39, is substituted by Ser-39 in mesotrypsin (Figure 1). This particular residue was investigated recently to analyze its role in interactions with polypeptide trypsin inhibitors [24]. Tyr-39, but not Ser-39, forms a hydrogen bond with the main chain amide nitrogen of the P4′ residue of a bound protease inhibitor (Figure 2). The presence of Ser-39 relative to Tyr-39 results in a 4- to 13-fold poorer binding affinity with the protease inhibitors examined. Thus, we concluded that the presence of Ser-39 in mesotrypsin, and corresponding absence of a single H-bond to the inhibitor backbone, makes a small but significant functional contribution to the resistance of mesotrypsin to inhibition [24]. Further studies may explore whether additional residue substitutions found in mesotrypsin contribute further to its unique inhibitor resistance and specificity.
Substrates and physiological function of mesotrypsin
Serine proteases in general carry out vital responsibilities relating to all aspects of human physiology, including roles in food digestion, reproduction, blood coagulation, signal transduction, apoptosis, and the immune response [44]. The identification of genes for many serine proteases of yet-unknown function suggests that additional roles remain to be uncovered [1]. Given its low concentration in the digestive juice and its apparent ectopic expression across a range of diverse tissues, the physiological function of mesotrypsin has remained enigmatic, but recent biochemical studies offer suggestions and clues. Compared to known physiological protein substrates of other trypsins, mesotrypsin is a poor enzyme. It fails to activate pancreatic zymogens, and also shows reduced capacity to degrade trypsinogens [19]. It is also substantially compromised in its ability to cleave protease activated receptors (PARs) [45-47]. Intriguingly, however, mesotrypsin displays enhanced catalytic activity, relative to other trypsins, toward one class of protein substrates: the canonical inhibitors of serine proteases [19,23,25,43].
Canonical inhibitors ordinarily function as extremely tight-binding but proteolysis resistant substrate mimics for their cognate proteases, via a standard mechanism in which the peptide bond targeted for cleavage is thermodynamically stable [11,13,48,49]. Due to intramolecular stabilization that maintains the inhibitory binding loop in a canonical conformation, perfectly prearranged for insertion into the protease active site [9,10,12], affinity is typically 5-7 orders of magnitude tighter than the comparable interaction with an unstructured peptide substrate [50,51]. Rather than being cleaved and released as would be a normal substrate, the canonical inhibitor lingers in the protease active site, with the reverse (peptide religation) reaction outpacing the forward (cleavage) reaction [11,48].
Mesotrypsin breaks this pattern—the effect of Arg-193, Ser-39, and perhaps additional residues unique to mesotrypsin results in a reduction in inhibitor affinity, from the picomolar range typical of protease/canonical inhibitor interactions to the micromolar range more typical of protease/substrate interactions (Tables 2 and 3). Unlike most other proteases, mesotrypsin appears to recognize its substrates in part via “conformational specificity”, such that presentation of a peptide substrate in the context of a stabilized canonical loop is essential for efficient substrate recognition [23]. Interestingly, the sequence and structural features of mesotrypsin that confer this binding specificity also dramatically enhance the rate with which mesotrypsin cleaves canonical inhibitors, bringing the kinetic constants K m, k cat, and the specificity constant k cat/K m in line with those of other proteases for their specific substrates (Table 3). The structural explanation for this gain of function appears to involve the weakening of favorable interactions, and introduction of unfavorable interactions, between mesotrypsin and the primed-side residues of the canonical binding loop [24,25,43]. This results in expulsion of these residues from the active site upon inhibitor cleavage, disfavoring religation of the cleaved inhibitor.
Table 3.
Comparison of kinetic constants for proteolysis of specific substrates by mesotrypsin and other serine proteases
| Enzyme | Substrate | K m (M) | k cat (s-1) | k cat/K m (M-1s-1) | Reference |
|---|---|---|---|---|---|
| mesotrypsin | APPI | 1.4 × 10-7 | 0.042 | 3.0 × 105 | [23] |
| mesotrypsin | SBTI | 1.5 × 10-6 | ~0.16† | ~1.0 × 105 † | [19] |
| cationic trypsin | chymotrypsinogen A | 2.1 × 10-5 | 2.3 | 1.1 × 105 | [74] |
| enteropeptidase | cationic trypsinogen | 1.4 × 10-6 | 35 | 2.5 × 107 | [75] |
| t-PA | Lys-plasminogen | 7.6 × 10-6 | 0.2 | 2.9 × 104 | [76] |
| u-PA | Lys-plasminogen | 5.4 × 10-6 | 1.7 | 3.1 × 105 | [77] |
Values are approximations from data shown in reference [19], based on an apparent initial rate of hydrolysis of ~0.04 s-1 in a reaction containing 500 nM SBTI, a concentration 3-fold below below the K m as estimated by the reported K i.
Given these observations, we have asked whether some of the canonical serine protease inhibitors might be in fact physiological substrates of mesotrypsin. In the digestive system, one obvious potential use of mesotrypsin’s unique digestive capability could be the inactivation and breakdown of protease inhibitors in the diet. Canonical inhibitors are widely distributed across the plant and animal kingdoms and are especially enriched in many plant food sources such as legumes, grains, and vegetables [11,52]. The Sahin-Tóth laboratory first demonstrated the rapid hydrolysis of the reactive site bond of the canonical soybean trypsin inhibitor (SBTI) [19]; from their published data we have estimated the kinetic constants for this cleavage to resemble those of other protease/ substrate interactions (Table 3).
In tissues and organs outside of the digestive system, mesotrypsin may be involved in the inactivation or clearance of some of the dozens of endogenous human canonical protease inhibitors. In search of potential mesotrypsin substrates produced in prostate cancer cells, we conducted an affinity-based proteomic screen to identify secreted trypsin inhibitors cleaved by mesotrypsin [23]. We found that the amyloid precursor protein (APP), which contains a protease inhibitor domain belonging to the I2 or Kunitz-BPTI family and is secreted as a physiological inhibitor of factor XIa (sAPP/protease nexin 2), is rapidly and selectively cleaved by mesotrypsin within the Kunitz protease inhibitor domain (APPI). Cleavage of APPI by mesotrypsin compromises its inhibition of other serine proteases, including cationic trypsin and factor XIa, by two orders of magnitude [23]. Considering that APP is a ubiquitously expressed transmembrane protein and is coexpressed with mesotrypsin in a number of tissues, it likely represents the first endogenous physiological substrate of mesotrypsin to be identified. Processing by mesotrypsin may regulate the protease inhibitory function of sAPP/protease nexin 2 in vivo, and may also modulate other activities of APP that involve the Kunitz domain. Further studies are needed to determine whether other endogenous human protease inhibitors may also be physiological substrates of mesotrypsin.
Role of mesotrypsin in cancer
While normal expression of the PRSS3 gene encoding mesotrypsin is highest in pancreas and brain with very limited expression elsewhere [29,53-55], we and others have found PRSS3 to be transcriptionally upregulated with cancer progression in epithelial cancers including lung, colon, breast, pancreas, and prostate [35-38,56]. The first report to offer mechanistic insight into the potential role of mesotrypsin in cancer progression was published in 2009 [37]. In this study, Hockla et. al. identified PRSS3 upregulation with malignant progression in a breast cancer cell line progression series. Whereas knockdown of PRSS3 by RNA interference approaches correlated closely with suppression of malignant growth in 3D cultures, treatment of cells with recombinant purified mesotrypsin enhanced the malignant growth phenotype [37]. Employing a proteomic screen of cleavage products from cell culture conditioned media, we identified the cell surface glycoprotein CD109 as a potential mesotrypsin substrate involved in driving malignancy; however it is not yet known whether the shedding of CD109 is directly catalyzed or indirectly mediated by mesotrypsin [37].
Another study, by Jiang et al., described the involvement of PRSS3 in the progression of pancreatic cancer [38]. They found that PRSS3 expression correlated with metastasis and poor survival in pancreatic cancer patients. Using several cell culture and mouse xenograft models of pancreatic cancer, they found that over-expression of PRSS3 promoted invasion and proliferation of pancreatic cancer cells in vitro, as well as metastasis in vivo, while suppression of PRSS3 expression reduced cell invasion and delayed progression to metastasis [38]. The effects of mesotrypsin appeared to be mediated at least in part by upregulation of VEGF expression via the PAR1-mediated ERK pathway [38].
Most recently, the involvement of mesotrypsin in prostate cancer progression has been established by the Radisky group [36]. After observing that PRSS3 expression was upregulated in metastatic prostate tumors, and that its expression in primary tumors was associated with recurrence, we used a mouse orthotopic model with bioluminescent imaging to show that PRSS3/mesotrypsin expression is critical for prostate cancer metastasis. Silencing of PRSS3 inhibited anchorage-independent growth of prostate cancer cells in soft agar assays, and suppressed invasiveness in Matrigel trans-well assays and three-dimensional (3D) cell culture models. In addition, treatment with recombinant mesotrypsin directly promoted an invasive cellular phenotype in prostate cancer cells, a specific effect that required the proteolytic activity of mesotrypsin; neither cationic trypsin nor a mesotrypsin mutant lacking activity could similarly drive this invasive phenotype [36].
Engineering polypeptide inhibitors for mesotrypsin
Protein therapeutics represent a growing segment of the drug discovery field, accounting for a quarter of recent new drug approvals [57]. Several polypeptide serine protease inhibitors, some already clinically approved [58,59] or under study as investigational drugs, show high solubility, low toxicity, limited antigenicity, stability to digestion, and in some cases even retention of activity after oral administration [60-64], characteristics indicative of high potential as protein therapeutics. Since mesotrypsin has been implicated in progression of several different cancers as discussed above in “Role of mesotrypsin in cancer”, we hypothesize that potent and selective inhibitors of mesotrypsin could potentially offer novel therapeutics. However, targeting mesotrypsin presents special challenges: mesotrypsin is resistant to inhibition by many polypeptide serine protease inhibitors, and possesses enhanced catalytic capability for hydrolyzing some inhibitors as highly specific substrates. A further challenge in the development of therapeutic mesotrypsin inhibitors is likely to be the identification of sufficiently selective inhibitors, since mesotrypsin belongs to a large family of closely structurally related trypsin-like proteases. However, by taking advantage of the unique active site features of mesotrypsin, it may be possible to develop novel and selective, potentially therapeutic, inhibitors.
Natural polypeptide inhibitors of the Kunitz-BPTI family present feasible leads for the development of a therapeutic mesotrypsin inhibitor, since members of this family include several established and investigational drugs targeting other serine proteases [59,60,63-67]. The Kunitz domain scaffold is in general highly stable; however, mesotrypsin possesses highly enhanced catalytic activity toward some Kunitz domain inhibitors, which may represent physiological substrates rather than inhibitors as discussed above in “Substrates and physiological function of mesotrypsin” [23]. Therefore, it will be critical that we select or engineer a scaffold possessing adequate resistance to mesotrypsin degradation. The stability of Kunitz inhibitors toward mesotrypsin varies dramatically; for example we have shown that bovine pancreatic trypsin inhibitor (BPTI) is 300 times more stable to mesotrypsin proteolysis than is APPI [25]. BPTI and APPI share the characteristic Kunitz domain fold, in which the binding loop makes the majority of close contacts with the inhibited enzyme. This loop is supported by a compact scaffold with a three dimensional structure maintained by hydrophobic packing and three disulfide bonds. BPTI and APPI differ only at the P1 and P2′ residues within the binding loop, and share 45% amino acid identity throughout the scaffold. Mesotrypsin affinity toward the inhibitors is nearly exclusively modulated by the binding loop sequence, as mutation at the P1 and P2′ positions of BPTI creates an inhibitor with affinity essentially equal to that of APPI, and vice versa [25]. Surprisingly, these mutations confer minimal effects on cleavage rates; thus, the greater stability of BPTI to mesotrypsin hydrolysis is largely attributable to the scaffold, rather than to the binding loop sequence [25]. As a result, a scaffold can be selected for optimal proteolytic stability, and then the binding loop modified to confer optimal affinity and enhance selectivity.
In optimizing affinity, we have found the P1 residue to be of particular importance, since Arg at this position (versus Lys) favors tighter binding by a factor of six [25]. The amino acid nature of the P2′ residue is also extremely important, as bulky and charged residues strongly disfavor binding, whereas acidic residues facilitate cleavage [42]. We have recently engineered a polypeptide inhibitor based upon the BPTI scaffold and incorporating mutations at the P1 position (Lys-15 to Arg) and at the P2′ position (Arg-17 to Gly) [42]. This new inhibitor, designated BPTI-K15R/R17G, is a relatively stable, high affinity mesotrypsin inhibitor with an equilibrium binding constant K i of 5.9 nM, a >2000-fold improvement in affinity over native BPTI. This engineered inhibitor demonstrated high efficacy for inhibiting mesotrypsin in assays of breast cancer cell malignant growth and pancreatic cancer cell invasion [42]. It also suppressed prostate cancer cell invasion to a similar extent as did PRSS3 gene silencing [36]. Our work to date suggests that inhibition of mesotrypsin activity may provide a novel modality for treatment of cancer, although further improvements in inhibitor selectivity will be important before clinical potential can be realized.
Concluding remarks
Mesotrypsin is a very unusual enzyme, possessing active site steric and electrostatic features unique among human serine proteases. These distinctive active site features are responsible for the resistance of mesotrypsin to many polypeptide trypsin inhibitors, and are also responsible for the unique substrate specificity of mesotrypsin toward protein protease inhibitors. Based on the observation that other trypsins are unable to recapitulate the activity of mesotrypsin in driving the invasive phenotype of prostate cancer cells [36], we speculate that these same physical and chemical features are critical in the ability of mesotrypsin to drive cancer metastasis [36,38]. Importantly, these same distinctive features may provide an opportunity to develop new inhibitors, complementary in shape and charge to the mesotrypsin active site, which will selectively target mesotrypsin for therapeutic purposes.
Acknowledgements
Support was provided by US National Cancer Institute (CA154387) and US Department of Defense (PC094054) to ESR.
Disclosure of conflict of interest
None.
References
- 1.Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40:D343–350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rinderknecht H. Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig Dis Sci. 1986;31:314–321. doi: 10.1007/BF01318124. [DOI] [PubMed] [Google Scholar]
- 3.Bitoun E, Chavanas S, Irvine AD, Lonie L, Bodemer C, Paradisi M, Hamel-Teillac D, Ansai S, Mitsuhashi Y, Taieb A, de Prost Y, Zambruno G, Harper JI, Hovnanian A. Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol. 2002;118:352–361. doi: 10.1046/j.1523-1747.2002.01603.x. [DOI] [PubMed] [Google Scholar]
- 4.Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency--a model for conformational diseases. N Engl J Med. 2002;346:45–53. doi: 10.1056/NEJMra010772. [DOI] [PubMed] [Google Scholar]
- 5.Kiraly O, Boulling A, Witt H, Le Marechal C, Chen JM, Rosendahl J, Battaggia C, Wartmann T, Sahin-Toth M, Ferec C. Signal peptide variants that impair secretion of pancreatic secretory trypsin inhibitor (SPINK1) cause autosomal dominant hereditary pancreatitis. Hum Mutat. 2007;28:469–476. doi: 10.1002/humu.20471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lomas DA, Belorgey D, Mallya M, Onda M, Kinghorn KJ, Sharp LK, Phillips RL, Page R, Crowther DC, Miranda E. Polymerisation underlies alpha1-antitrypsin deficiency, dementia and other serpinopathies. Front Biosci. 2004;9:2873–2891. doi: 10.2741/1444. [DOI] [PubMed] [Google Scholar]
- 7.Lomas DA, Mahadeva R. Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest. 2002;110:1585–1590. doi: 10.1172/JCI16782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Z, Yang P. Role of imbalance between neutrophil elastase and alpha 1-antitrypsin in cancer development and progression. Lancet Oncol. 2004;5:182–190. doi: 10.1016/S1470-2045(04)01414-7. [DOI] [PubMed] [Google Scholar]
- 9.Bode W, Huber R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur J Biochem. 1992;204:433–451. doi: 10.1111/j.1432-1033.1992.tb16654.x. [DOI] [PubMed] [Google Scholar]
- 10.Hubbard SJ, Campbell SF, Thornton JM. Molecular recognition. Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J Mol Biol. 1991;220:507–530. doi: 10.1016/0022-2836(91)90027-4. [DOI] [PubMed] [Google Scholar]
- 11.Krowarsch D, Cierpicki T, Jelen F, Otlewski J. Canonical protein inhibitors of serine proteases. Cell Mol Life Sci. 2003;60:2427–2444. doi: 10.1007/s00018-003-3120-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radisky ES, Koshland DE Jr. A clogged gutter mechanism for protease inhibitors. Proc Natl Acad Sci U S A. 2002;99:10316–10321. doi: 10.1073/pnas.112332899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laskowski M Jr, Kato I. Protein inhibitors of proteinases. Annu Rev Biochem. 1980;49:593–626. doi: 10.1146/annurev.bi.49.070180.003113. [DOI] [PubMed] [Google Scholar]
- 14.Castro MJ, Anderson S. Alanine point-mutations in the reactive region of bovine pancreatic trypsin inhibitor: effects on the kinetics and thermodynamics of binding to beta-trypsin and alpha-chymotrypsin. Biochemistry. 1996;35:11435–11446. doi: 10.1021/bi960515w. [DOI] [PubMed] [Google Scholar]
- 15.Maywald F, Boldicke T, Gross G, Frank R, Blocker H, Meyerhans A, Schwellnus K, Ebbers J, Bruns W, Reinhardt G, Schnabel E, Schroder W, Fritz H, Collins J. Human pancreatic secretory inhibitor (PSTI) produced in active form and secreted from Escherichia coli . Gene. 1988;68:357–369. doi: 10.1016/0378-1119(88)90038-8. [DOI] [PubMed] [Google Scholar]
- 16.Quast U, Engel J, Steffen E, Tschesche H, Kupfer S. Kinetics of the interaction of alpha-chymotrypsin with trypsin kallikrein inhibitor (Kunitz) in which the reactive-site peptide bond Lys-15--Ala-16 is split. Eur J Biochem. 1978;86:353–360. doi: 10.1111/j.1432-1033.1978.tb12317.x. [DOI] [PubMed] [Google Scholar]
- 17.Quast U, Engel J, Steffen E, Tschesche H, Kupfer S. Stopped-flow kinetics of the resynthesis of the reactive site peptile bond in kallikrein inhibitor (Kunitz) by beta-trypsin. Biochemistry. 1978;17:1675–1682. doi: 10.1021/bi00602a015. [DOI] [PubMed] [Google Scholar]
- 18.Rinderknecht H, Renner IG, Abramson SB, Carmack C. Mesotrypsin: a new inhibitor-resistant protease from a zymogen in human pancreatic tissue and fluid. Gastroenterology. 1984;86:681–692. [PubMed] [Google Scholar]
- 19.Szmola R, Kukor Z, Sahin-Toth M. Human mesotrypsin is a unique digestive protease specialized for the degradation of trypsin inhibitors. J Biol Chem. 2003;278:48580–48589. doi: 10.1074/jbc.M310301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahin-Toth M. Human mesotrypsin defies natural trypsin inhibitors: from passive resistance to active destruction. Protein Pept Lett. 2005;12:457–464. doi: 10.2174/0929866054395356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nyaruhucha CN, Kito M, Fukuoka SI. Identification and expression of the cDNA-encoding human mesotrypsin(ogen), an isoform of trypsin with inhibitor resistance. J Biol Chem. 1997;272:10573–10578. doi: 10.1074/jbc.272.16.10573. [DOI] [PubMed] [Google Scholar]
- 22.Katona G, Berglund GI, Hajdu J, Graf L, Szilagyi L. Crystal structure reveals basis for the inhibitor resistance of human brain trypsin. J Mol Biol. 2002;315:1209–1218. doi: 10.1006/jmbi.2001.5305. [DOI] [PubMed] [Google Scholar]
- 23.Salameh MA, Robinson JL, Navaneetham D, Sinha D, Madden BJ, Walsh PN, Radisky ES. The amyloid precursor protein/protease nexin 2 Kunitz inhibitor domain is a highly specific substrate of mesotrypsin. J Biol Chem. 2010;285:1939–1949. doi: 10.1074/jbc.M109.057216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salameh MA, Soares AS, Alloy A, Radisky ES. Presence versus absence of hydrogen bond donor Tyr-39 influences interactions of cationic trypsin and mesotrypsin with protein protease inhibitors. Protein Sci. 2012;21:1103–12. doi: 10.1002/pro.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salameh MA, Soares AS, Navaneetham D, Sinha D, Walsh PN, Radisky ES. Determinants of affinity and proteolytic stability in interactions of Kunitz family protease inhibitors with mesotrypsin. J Biol Chem. 2010;285:36884–36896. doi: 10.1074/jbc.M110.171348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Figarella C, Clemente F, Guy O. On zymogens of human pancreatic juice. FEBS Lett. 1969;3:351–353. doi: 10.1016/0014-5793(69)80176-6. [DOI] [PubMed] [Google Scholar]
- 27.Rinderknecht H, Renner IG, Carmack C. Trypsinogen variants in pancreatic juice of healthy volunteers, chronic alcoholics, and patients with pancreatitis and cancer of the pancreas. Gut. 1979;20:886–891. doi: 10.1136/gut.20.10.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen JM, Ferec C. Genes, cloned cDNAs, and proteins of human trypsinogens and pancreatitis-associated cationic trypsinogen mutations. Pancreas. 2000;21:57–62. doi: 10.1097/00006676-200007000-00052. [DOI] [PubMed] [Google Scholar]
- 29.Nakanishi J, Yamamoto M, Koyama J, Sato J, Hibino T. Keratinocytes synthesize enteropeptidase and multiple forms of trypsinogen during terminal differentiation. J Invest Dermatol. 2010;130:944–952. doi: 10.1038/jid.2009.364. [DOI] [PubMed] [Google Scholar]
- 30.Nemeth AL, Medveczky P, Toth J, Siklodi E, Schlett K, Patthy A, Palkovits M, Ovadi J, Tokesi N, Nemeth P, Szilagyi L, Graf L. Unconventional translation initiation of human trypsinogen 4 at a CUG codon with an N-terminal leucine. A possible means to regulate gene expression. Febs J. 2007;274:1610–1620. doi: 10.1111/j.1742-4658.2007.05708.x. [DOI] [PubMed] [Google Scholar]
- 31.Rowen L, Williams E, Glusman G, Linardopoulou E, Friedman C, Ahearn ME, Seto J, Boysen C, Qin S, Wang K, Kaur A, Bloom S, Hood L, Trask BJ. Interchromosomal segmental duplications explain the unusual structure of PRSS3, the gene for an inhibitor-resistant trypsinogen. Mol Biol Evol. 2005;22:1712–1720. doi: 10.1093/molbev/msi166. [DOI] [PubMed] [Google Scholar]
- 32.Toth J, Siklodi E, Medveczky P, Gallatz K, Nemeth P, Szilagyi L, Graf L, Palkovits M. Regional distribution of human trypsinogen 4 in human brain at mRNA and protein level. Neurochem Res. 2007;32:1423–1433. doi: 10.1007/s11064-007-9327-8. [DOI] [PubMed] [Google Scholar]
- 33.Wiegand U, Corbach S, Minn A, Kang J, Muller-Hill B. Cloning of the cDNA encoding human brain trypsinogen and characterization of its product. Gene. 1993;136:167–175. doi: 10.1016/0378-1119(93)90460-k. [DOI] [PubMed] [Google Scholar]
- 34.Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem. 2004;279:13532–13539. doi: 10.1074/jbc.M312090200. [DOI] [PubMed] [Google Scholar]
- 35.Diederichs S, Bulk E, Steffen B, Ji P, Tickenbrock L, Lang K, Zanker KS, Metzger R, Schneider PM, Gerke V, Thomas M, Berdel WE, Serve H, Muller-Tidow C. S100 family members and trypsinogens are predictors of distant metastasis and survival in early-stage non-small cell lung cancer. Cancer Res. 2004;64:5564–5569. doi: 10.1158/0008-5472.CAN-04-2004. [DOI] [PubMed] [Google Scholar]
- 36.Hockla A, Miller E, Salameh MA, Copland JA, Radisky DC, Radisky ES. PRSS3/Mesotrypsin is a therapeutic target for metastatic prostate cancer. Mol Cancer Res. 2012;10:1555–1566. doi: 10.1158/1541-7786.MCR-12-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hockla A, Radisky DC, Radisky ES. Mesotrypsin promotes malignant growth of breast cancer cells through shedding of CD109. Breast Cancer Res Treat. 2010;124:27–38. doi: 10.1007/s10549-009-0699-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang G, Cao F, Ren G, Gao D, Bhakta V, Zhang Y, Cao H, Dong Z, Zang W, Zhang S, Wong HH, Hiley C, Crnogorac-Jurcevic T, Lemoine NR, Wang Y. PRSS3 promotes tumour growth and metastasis of human pancreatic cancer. Gut. 2010;59:1535–1544. doi: 10.1136/gut.2009.200105. [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi T, Shuman MA, Craik CS. Reverse biochemistry: use of macromolecular protease inhibitors to dissect complex biological processes and identify a membrane-type serine protease in epithelial cancer and normal tissue. Proc Natl Acad Sci U S A. 1999;96:11054–11061. doi: 10.1073/pnas.96.20.11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17:349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 41.Higaki JN, Gibson BW, Craik CS. Evolution of catalysis in the serine proteases. Cold Spring Harb Symp Quant Biol. 1987;52:615–621. doi: 10.1101/sqb.1987.052.01.070. [DOI] [PubMed] [Google Scholar]
- 42.Salameh MA, Soares AS, Hockla A, Radisky DC, Radisky ES. The P(2)′ residue is a key determinant of mesotrypsin specificity: engineering a high-affinity inhibitor with anticancer activity. Biochem J. 2011;440:95–105. doi: 10.1042/BJ20110788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salameh MA, Soares AS, Hockla A, Radisky ES. Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J Biol Chem. 2008;283:4115–4123. doi: 10.1074/jbc.M708268200. [DOI] [PubMed] [Google Scholar]
- 44.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 45.Grishina Z, Ostrowska E, Halangk W, Sahin-Toth M, Reiser G. Activity of recombinant trypsin isoforms on human proteinase-activated receptors (PAR): mesotrypsin cannot activate epithelial PAR-1, -2, but weakly activates brain PAR-1. Br J Pharmacol. 2005;146:990–999. doi: 10.1038/sj.bjp.0706410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knecht W, Cottrell GS, Amadesi S, Mohlin J, Skaregarde A, Gedda K, Peterson A, Chapman K, Hollenberg MD, Vergnolle N, Bunnett NW. Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J Biol Chem. 2007;282:26089–26100. doi: 10.1074/jbc.M703840200. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Luo W, Wartmann T, Halangk W, Sahin-Toth M, Reiser G. Mesotrypsin, a brain trypsin, activates selectively proteinase-activated receptor-1, but not proteinase-activated receptor-2, in rat astrocytes. J Neurochem. 2006;99:759–769. doi: 10.1111/j.1471-4159.2006.04105.x. [DOI] [PubMed] [Google Scholar]
- 48.Estell DA, Wilson KA, Laskowski M Jr. Thermodynamics and kinetics of the hydrolysis of the reactive-site peptide bond in pancreatic trypsin inhibitor (Kunitz) by Dermasterias imbricata trypsin 1. Biochemistry. 1980;19:131–137. doi: 10.1021/bi00542a020. [DOI] [PubMed] [Google Scholar]
- 49.Buczek O, Krowarsch D, Otlewski J. Thermodynamics of single peptide bond cleavage in bovine pancreatic trypsin inhibitor (BPTI) Protein Sci. 2002;11:924–932. doi: 10.1110/ps.4460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Radisky ES, King DS, Kwan G, Koshland DE Jr. The role of the protein core in the inhibitory power of the classic serine protease inhibitor, chymotrypsin inhibitor 2. Biochemistry. 2003;42:6484–6492. doi: 10.1021/bi034275d. [DOI] [PubMed] [Google Scholar]
- 51.Tan NH, Kaiser ET. Synthesis and characterization of a pancreatic trypsin inhibitor homologue and a model inhibitor. Biochemistry. 1977;16:1531–1541. doi: 10.1021/bi00627a001. [DOI] [PubMed] [Google Scholar]
- 52.Rawlings ND, Tolle DP, Barrett AJ. Evolutionary families of peptidase inhibitors. Biochem J. 2004;378:705–716. doi: 10.1042/BJ20031825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ge X, Yamamoto S, Tsutsumi S, Midorikawa Y, Ihara S, Wang SM, Aburatani H. Interpreting expression profiles of cancers by genome-wide survey of breadth of expression in normal tissues. Genomics. 2005;86:127–141. doi: 10.1016/j.ygeno.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 54.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, Lancet D, Shmueli O. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- 56.Yang L, Zhang L, Wu Q, Boyd DD. Unbiased screening for transcriptional targets of ZKSCAN3 identifies integrin beta 4 and vascular endothelial growth factor as downstream targets. J Biol Chem. 2008;283:35295–35304. doi: 10.1074/jbc.M806965200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szymkowski DE. Creating the next generation of protein therapeutics through rational drug design. Curr Opin Drug Discov Devel. 2005;8:590–600. [PubMed] [Google Scholar]
- 58.Leader B, Baca QJ, Golan DE. Protein therapeutics:a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 59.Cicardi M, Levy RJ, McNeil DL, Li HH, Sheffer AL, Campion M, Horn PT, Pullman WE. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523–531. doi: 10.1056/NEJMoa0905079. [DOI] [PubMed] [Google Scholar]
- 60.Fritz H, Wunderer G. Biochemistry and applications of aprotinin, the kallikrein inhibitor from bovine organs. Arzneimittelforschung. 1983;33:479–494. [PubMed] [Google Scholar]
- 61.Kennedy AR. Chemopreventive agents: protease inhibitors. Pharmacol Ther. 1998;78:167–209. doi: 10.1016/s0163-7258(98)00010-2. [DOI] [PubMed] [Google Scholar]
- 62.Kennedy AR. The Bowman-Birk inhibitor from soybeans as an anticarcinogenic agent. Am J Clin Nutr. 1998;68:1406S–1412S. doi: 10.1093/ajcn/68.6.1406S. [DOI] [PubMed] [Google Scholar]
- 63.Kobayashi H, Suzuki M, Hirashima Y, Terao T. The protease inhibitor bikunin, a novel anti-metastatic agent. Biol Chem. 2003;384:749–754. doi: 10.1515/BC.2003.083. [DOI] [PubMed] [Google Scholar]
- 64.Kobayashi H, Yagyu T, Inagaki K, Kondo T, Suzuki M, Kanayama N, Terao T. Therapeutic efficacy of once-daily oral administration of a Kunitz-type protease inhibitor, bikunin, in a mouse model and in human cancer. Cancer. 2004;100:869–877. doi: 10.1002/cncr.20034. [DOI] [PubMed] [Google Scholar]
- 65.Devy L, Rabbani SA, Stochl M, Ruskowski M, Mackie I, Naa L, Toews M, van Gool R, Chen J, Ley A, Ladner RC, Dransfield DT, Henderikx P. PEGylated DX-1000: pharmacokinetics and antineoplastic activity of a specific plasmin inhibitor. Neoplasia. 2007;9:927–937. doi: 10.1593/neo.07544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li W, Wang BE, Moran P, Lipari T, Ganesan R, Corpuz R, Ludlam MJ, Gogineni A, Koeppen H, Bunting S, Gao WQ, Kirchhofer D. Pegylated kunitz domain inhibitor suppresses hepsin-mediated invasive tumor growth and metastasis. Cancer Res. 2009;69:8395–8402. doi: 10.1158/0008-5472.CAN-09-1995. [DOI] [PubMed] [Google Scholar]
- 67.Williams A, Baird LG. DX-88 and HAE: a developmental perspective. Transfus Apher Sci. 2003;29:255–258. doi: 10.1016/S1473-0502(03)00170-8. [DOI] [PubMed] [Google Scholar]
- 68.Laskowski M Jr, Finkenstadt WR. Study of protein-protein and of protein-ligand interactions by potentiometric methods. Methods Enzymol. 1972;26:193–277. doi: 10.1016/s0076-6879(72)26012-8. [DOI] [PubMed] [Google Scholar]
- 69.Ascenzi P, Amiconi G, Bolognesi M, Menegatti E, Guarneri M. Binding of the soybean Bowman-Birk proteinase inhibitor and of its chymotrypsin and trypsin inhibiting fragments to bovine alpha-chymotrypsin and bovine betatrypsin. A thermodynamic study. J Mol Recognit. 1990;3:192–196. doi: 10.1002/jmr.300030504. [DOI] [PubMed] [Google Scholar]
- 70.Otlewski J, Zbyryt T. Single peptide bond hydrolysis/resynthesis in squash inhibitors of serine proteinases. 1. Kinetics and thermodynamics of the interaction between squash inhibitors and bovine beta-trypsin. Biochemistry. 1994;33:200–207. doi: 10.1021/bi00167a026. [DOI] [PubMed] [Google Scholar]
- 71.Patthy A, Amir S, Malik Z, Bodi A, Kardos J, Asboth B, Graf L. Remarkable phylum selectivity of a Schistocerca gregaria trypsin inhibitor: the possible role of enzyme-inhibitor flexibility. Arch Biochem Biophys. 2002;398:179–187. doi: 10.1006/abbi.2001.2686. [DOI] [PubMed] [Google Scholar]
- 72.Luckett S, Garcia RS, Barker JJ, Konarev AV, Shewry PR, Clarke AR, Brady RL. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J Mol Biol. 1999;290:525–533. doi: 10.1006/jmbi.1999.2891. [DOI] [PubMed] [Google Scholar]
- 73.Kitaguchi N, Takahashi Y, Oishi K, Shiojiri S, Tokushima Y, Utsunomiya T, Ito H. Enzyme specificity of proteinase inhibitor region in amyloid precursor protein of Alzheimer’s disease: different properties compared with protease nexin I. Biochim Biophys Acta. 1990;1038:105–113. doi: 10.1016/0167-4838(90)90017-a. [DOI] [PubMed] [Google Scholar]
- 74.Sahin-Toth M. Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J Biol Chem. 2000;275:22750–22755. doi: 10.1074/jbc.M002943200. [DOI] [PubMed] [Google Scholar]
- 75.Nemoda Z, Sahin-Toth M. The tetra-aspartate motif in the activation peptide of human cationic trypsinogen is essential for autoactivation control but not for enteropeptidase recognition. J Biol Chem. 2005;280:29645–29652. doi: 10.1074/jbc.M505661200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Madison EL, Coombs GS, Corey DR. Substrate specificity of tissue type plasminogen activator. Characterization of the fibrin independent specificity of t-PA for plasminogen. J Biol Chem. 1995;270:7558–7562. doi: 10.1074/jbc.270.13.7558. [DOI] [PubMed] [Google Scholar]
- 77.Lijnen HR, Van Hoef B, Collen D. Influence of cyanogen-bromide-digested fibrinogen on the kinetics of plasminogen activation by urokinase. Eur J Biochem. 1984;144:541–544. doi: 10.1111/j.1432-1033.1984.tb08499.x. [DOI] [PubMed] [Google Scholar]