

Abstract
Background
To investigate the association of DNA nucleotide excision repair (NER) defects with neurological degeneration, cachexia and cancer, we performed autopsies on 4 adult xeroderma pigmentosum (XP) patients with different clinical features and defects in NER complementation groups XP-A, XP-C or XP-D.
Results
The XP-A (XP12BE) and XP-D (XP18BE) patients exhibited progressive neurological deterioration with sensorineural hearing loss. The clinical spectrum encompassed severe cachexia in the XP-A (XP12BE) patient, numerous skin cancers in the XP-A and two XP-C (XP24BE and XP1BE) patients and only few skin cancers in the XP-D patient. Two XP-C patients developed internal neoplasms including glioblastoma in XP24BE and uterine adenocarcinoma in XP1BE. At autopsy, the brains of the 44 yr XP-A and the 45 yr XP-D patients were profoundly atrophic and characterized microscopically by diffuse neuronal loss, myelin pallor and gliosis. Unlike the XP-A patient, the XP-D patient had a thickened calvarium, and the brain showed vacuolization of the neuropil in the cerebrum, cerebellum and brainstem, and patchy Purkinje cell loss. Axonal neuropathy and chronic denervation atrophy of the skeletal muscles were observed in the XP-A patient, but not in the XP-D patient.
Conclusions
These clinical manifestations and autopsy findings indicate advanced involvement of the central and peripheral nervous system. Despite similar defects in DNA repair, different clinicopathological phenotypes are seen in the four cases, and therefore distinct patterns of neurodegeneration characterize XP-D, XP-A and XP-C patients.
Keywords: DNA damage, DNA repair, Neurodegeneration, Glioblastoma, Cachexia
Background
Xeroderma pigmentosum (XP) is a rare, inherited disorder of DNA repair characterized by sun sensitivity of exposed tissues (skin, eye, oral mucous membranes) and a greater than 10,000-fold increased risk of cutaneous, ocular, and tongue neoplasms [1-4]. Some XP patients also have neurologic manifestations [3-11]. There are seven DNA nucleotide excision repair (NER)-deficient complementation groups designated XP-A through XP-G and an XP variant form with defective trans-lesion DNA polymerase eta [12,13]. Recently we reported on 106 XP patients seen at NIH over the past 4 decades [1]. Progressive neurologic degeneration was present in 24% (25/106), primarily in XP-D (n=16) and XP-A (n=6) complementation groups. Their core clinical features included loss of intellectual function, abnormal speech, mutism, areflexia, ataxia, peripheral neuropathy, and loss of the ability to walk. Progressive sensorineural hearing loss was a strong indicator of neurological degeneration [14]. The postmortem findings of XP, including involvement of the nervous system, have been described in the literature [6,7,10,15-34]. Most of these patients were in complementation group XP-A. There were no post-mortem reports on XP-D patients.
Here we report the post-mortem findings in 4 XP cases with different clinical features (Figure 1 and Table 1). The XP-A and XP-D patients showed severe progressive neurologic deterioration. The XP-A and XP-C patients presented with hundreds of skin cancers. The two XP-C patients died of internal neoplasms including glioblastoma in XP24BE and cervical adenocarcinoma with tumor metastasis to multiple organs and systems in XP1BE.
Figure 1.

XP patients studied. A and B: Case 1 XP-A patient (A) at age 17y with numerous freckle-like pigmented lesions on sun exposed skin and (B) at 37y with marked cachexia and thinning of subcutaneous tissues of face and chest. She had more that 100 surgical procedures on her face for removal of skin lesions. C: Case 2 XP-D patient at age 40y. She had been well protected from sun exposure since early childhood and had only few pigmented lesions and skin cancers. D: Case 3 XP-C patient, at age 29y with multiple freckle-like pigmented lesion on sun exposed skin and cheilitis. The patient underwent many surgical procedures for removal of skin cancers on her face. E and F: Case 4 XP-C patient, at age 28y (E) with multiple pigmented lesions, telangiectasia, cheilitis and corneal clouding. Multiple surgical procedures were performed on her face for removal of skin cancers and at age 48y (F) following exenteration of both orbits for treatment of ocular squamous cell carcinomas.
Table 1.
Clinical and autopsy abnormalities in XP-A (XP12BE) and XP-D (XP18BE) patients compared to two XP-C patients
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| |
XP-A (XP12BE) (GM05509)* |
XP-D (XP18BE) (CRL1275)**(XPKABE) |
XP-C (XP24BE) (GM11638)* |
XP-C (XP1BE) (GM10881)* |
| |
late onset juvenile form XP |
XP with neurological degeneration |
no neurological disease |
subclinical neurodegeneration |
| Age at death/Gender/DOB |
d44 yr/F b1965 |
d45 yr/F b1964 |
d35 yr/F b1972 |
d49 yr/F b1944 |
| XP complementation group |
XP-A |
XP-D |
XP-C |
XP-C |
| Mutations (see text for details) |
compound heterozygote |
compound heterozygote |
compound heterozygote |
homozygous |
|
CLINICAL FINDINGS |
|
|
|
|
| Height |
145.5 cm (<3 %ile) |
165 cm (50–75 %ile) |
153.3 cm - 34 yr (10 %ile) |
160 cm (25–50 %ile) |
| Weight |
32 kg (<3 %ile) |
60.6 kg (50 %ile) |
56.7 kg - 35 yr (50 %ile) |
76.6 kg (50–75 %ile) |
| Cachexia? |
yes |
no |
no |
no |
| Acute skin sunburning on minimal exposure? |
yes |
yes |
no |
no |
| Freckle-like skin lesions on sun exposed skin? |
yes |
yes |
yes |
yes |
| Skin cancer?1 |
>200 BCC and 1 SCC |
7 SCC and 2 BCC |
>200 (190-BCC,3 SCC and ~50 MIS) |
>200 (>35 BCC, >37 SCC, 2 soft tissue sarcoma, 28 MIS and 6 Mel) |
| Microcephaly? |
yes (2%ile) |
no |
no |
no |
| Hearing |
progressive high frequency sensorineural hearing loss |
progressive high frequency sensorineural hearing loss |
normal |
subclinical high frequency sensorineural hearing loss at 48 yr |
| Deep tendon reflexes |
absent |
absent (1995) |
normal |
normal at 37 yr, ankle hyporeflexia at 43 yr |
| Able to walk? |
no |
no |
yes |
yes |
| Able to talk? |
no |
no |
yes |
yes |
| Able to care for self? |
no |
no |
yes |
yes |
| Difficulty swallowing? |
yes - G-tube - age 37 yr |
yes - G-tube - age 44 yr |
normal |
normal |
| Primary ovarian failure? |
no |
unknown |
yes |
yes |
| Anterior eye abnormalities |
bilateral pinguecula, exposure keratopathy2 |
bilateral pinguecula, exposure keratopathy2 |
corneal scar, pterygium |
blateral orbital exenterations for infiltrative SCC of globe3 |
| Eye - retinal degeneration |
optic nerve atrophy2 |
no2 |
no |
unknown |
| Imaging of brain |
diffuse cerebral and cerebellar atrophy (41 yr −2006) |
minimal cortical atrophy (19 yr- 1983) |
left frontal lobe tumor |
slight cerebral cortical atrophy and ventricular enlargement (44 yr) |
|
AUTOPSY FINDINGS |
|
|
|
|
| Thick calvarium? |
no |
yes; cortical sclerosis, no tumor seen |
no |
no |
| Brain weight [normal 1240 g (1050–1550)] |
660g (~ 6mo) |
760g (~1 yr) |
1330g [normal] |
1300g [normal] |
| Brain atrophy? |
yes - diffuse |
yes - diffuse |
no atrophy - tumor |
no, except optic nerves secondary to orbital exenteration |
| Dilated brain ventricles? |
yes |
yes |
asymmetric due to tumor |
no |
| Thin corpus callusum? |
yes |
yes |
no remarkable features |
no |
| Histological neuronal loss? |
yes - hippocampus, pons, medulla, midbrain, thinned cortex, small cerebellum |
yes – outer cortex (neuronal loss and vacuolization resembling status spongiosis), hippocampus (CA2 and CA3 regions), basal ganglia, cerebellum |
no remarkable features |
no |
| Histological gliosis? |
yes - midbrain, pons, medulla, basal ganglia, thalamus, hippocampus |
yes - cortex, hippocampus |
no remarkable features |
yes - molecular layer of cerebellum |
| Histological myelin pallor? |
yes - temporal lobe, frontal lobe, cerebellum |
yes - basal ganglia, cerebellum |
no remarkable features |
no remarkable features |
| Cerebellum |
Atrophy, loss of Purkinje cells with axonal swelling, Bergmann astrocytosis |
atrophy, patchy Purkinje cell loss |
no remarkable features |
moderate to marked Purkinje cell loss with Bergmann astrocyte proliferation |
| Dorsal root ganglia |
no remarkable features |
no remarkable features |
no remarkable features |
severe neuronal loss |
| Histology of peripheral nerves |
minimal focal perivascular inflammation in the adjacent connective tissue |
no pathologic changes |
femoral nerve - unremarkable |
median nerve mild focal interstital fibrosis, sural nerve - no pathologic diagnosis |
| EM of peripheral nerves |
axonal neuropathy |
not done |
not done |
not done |
| Eye pathology? |
neovascularization of cornea, optic atrophy2 |
neovascularization of cornea, cataract2 |
not done |
sockets of orbits lined with skin |
| Histology of muscles |
myofiber type -grouping |
no pathologic changes |
unremarkable |
angulated fibers of skeletal muscles |
| Histology of Pharynx |
inflammation and fibrosis |
chronic inflammation |
normal |
normal |
| Esophagus |
no pathologic changes |
T-lymphocyte infiltration of Auerbach's plexus |
no pathologic changes |
no pathologic changes |
| Larynx |
no pathologic changes |
ulceration with chronic and acute inflammation |
pink mucosa |
delicate pink mucosa |
| Lungs |
normal |
bronchopneumonia |
bilateral pneumonia |
pulmonary emboli |
| Thyroid |
normal |
normal |
cystic nodule filled with pink, amprphous material, consistent with goiter |
follicular adenomas |
| Ovaries |
no pathologic changes |
no pathologic changes |
small - microscopic fibrosis, no follicles |
covered by tumor |
| Uterus |
leiomyomas |
adenomyosis |
small - calcified nodules 1 cm, leiomyomas |
covered by tumor |
| Breasts |
fibrocystic changes |
fibrocystic changes |
no masses |
no masses |
| Cause of death | XP-related neurologic degeneration | XP-related neurologic degeneration | Glioblastoma WHO grade IV. Tumor cells positive for GFAP and IDH1, negative for EGFR. P53 positive in <5% tumor cells. | metastasis of well differentiated mucinous adenocarcinoma of uterine endocervix |
1BCC - basal cell carcinoma, SCC -squamous cell carcinoma, MIS - melanoma in situ 2From Ramkumar et al. (reference 26) 3From Gaasterland et al. (reference 48) *Coriell Institute for Medical Research cell culture identification number **American Type Culture Collection cell culture identification number.
Results
Case histories
Case 1 (XP12BE - XP-A)
The patient was a 44 year-old female with XP who had been followed at NIH since diagnosis at age 4 years [4,8,9,14,26,35] (Figure 1A and B and Table 1). She experienced the first acute sun-sensitivity reaction at age 3 months with erythema and swelling after minimal sun exposure. Over time she developed freckle-like pigmentation on the sun exposed skin of her face, neck and dorsal upper extremities. Her first pathologically documented skin malignancy was a sclerosing basal cell carcinoma of the cheek at age 8 years. Subsequently, the patient developed more than two hundred skin cancers, which were mostly basal cell carcinomas (BCC) with 1 squamous cell carcinoma (SCC). At age 19 years, she was treated with oral 13-cis retinoic acid for skin cancer prevention [36]. She responded well with reduction in new skin cancer frequency from 43 BCC in two years before treatment to 3 new BCC after two years of treatment. Eighteen months after the discontinuation of 13-cis retinoic acid, she developed 18 new skin cancers. In the following years, she practiced increased protection from the sun with improved protective clothing and sunblocking agents. She experienced progressive neurologic decline, which further contributed to reduced outdoor exposure. Over time, fewer skin cancers developed and her overall XP-related skin pigmentation faded with only few lesions present.
While her early developmental milestones were within normal range, neurological signs were noted at age 7 years, with loss of deep tendon reflexes. At age 8 years, an audiogram revealed bilateral hearing loss at 8,000 Hertz, which progressed over the years [9,14]. Learning difficulties and an IQ of 79 were noted at age 15 yr. Computerized tomography (CT) of the brain at age 20 showed no focal abnormalities and no appreciable atrophy (Figure 2A). At age 21 years, truncal ataxia became more prominent and tongue movement appeared uncoordinated. At the age of 22, she began to lose pain sensation, the Romberg test became positive and she exhibited child-like behavior [9]. By age 37 years, she was immobile and required assistance to walk. Because she was unable to swallow solid food without choking a gastrostomy tube (G-tube) was placed for nutritional support. Despite this therapeutic measure, she continued to become markedly cachectic. At age 41 years, there was evidence of prominent brain atrophy on imaging studies (Figure 2B). Neurological deterioration progressed inexorably and she became completely unresponsive and expired at the age of 44. An unrestricted autopsy was performed. Compound heterozygous mutations were detected in the XPA DNA repair gene (Table 1) with G>T intron 3 splice acceptor, and G>C splice donor of exon 4 [37,38]. The post-UV DNA repair rate (unscheduled DNA synthesis (UDS)) of the cultured skin fibroblasts was about 1% of normal [4,35,39,40]. Post-UV survival of cultured skin fibroblasts was markedly reduced [41].
Figure 2.

Case 1. XP-A A: Two consecutive Computed Tomography (CT) images of the brain obtained at age 20y. There are no focal brain abnormalities and no appreciable brain atrophy. B: Two axial T1-weighted Magnetic Resonance Images (MRI) of the brain in the same patient obtained at age 41y. There is evidence of prominent brain atrophy manifested by enlargement of the ventricles and widening of the intracranial subarachnoid spaces. C: Gross atrophy of the right brain hemisphere (right, comparable image to 2B) in comparison to the normal brain at the same age and gender (left). Loss of brain substance appears to be greater in the white matter. D: Thinning of cortex of the right temporal lobe (H&E, x 40). E: Luxol fast blue stain of the right temporal lobe showing patchy loss of myelination.
Case 2 (XP18BE- XP-D)
This 45-year-old female patient with XP had a history of progressive neurologic deficits [14,26] (Figure 1C and Table 1). She presented to the NIH at age 5 years with hearing difficulty and developmental delay. In addition, she had marked photosensitivity with blistering on minimal sun exposure. At age 8 years, SCC was removed from her nose. Subsequently, she practiced extensive early sun protection and only developed 9 skin cancers during her lifetime (2 BCC and 7 SCC). At age 13 years, she was first noted to have high frequency sensorineural hearing loss and at age 30 years, a repeat audiologic evaluation revealed profound sensorineural hearing loss [14]. At age 31 she developed ataxia with a broad-based gait, eventually requiring confinement to a wheelchair at age 36. By age 39 years, she was unable to care for herself and needed to read lips and write notes in order to communicate. She developed progressive dysphagia and at age 44 years a gastrostomy tube was placed for nutritional support that proved successful for weight maintenance. Her final admission was for pneumonia and sepsis. She expired 3 days later and an unrestricted autopsy was performed. Laboratory testing demonstrated XP-D complementation group (Table 1) with compound heterozygous mutations in the XPD (ERCC2) gene: R616P and R683W. Cultured skin fibroblasts (XPKABE) showed post-UV UDS of 25-50% of normal but markedly reduced post-UV survival [41].
Case 3 (XP24BE- XP-C)
Case 3 was a 35-year-old female diagnosed with XP at age 6 years (Figures 1D and 3 and Table 1). During her disease course, she developed almost 200 skin cancers including BCC, SCC, 11 melanomas in situ (Figure 3G and H), and one invasive melanoma (0.65 mm). At age 11 years, she participated in a study with oral 13-cis retinoic acid (isotretinoin) for skin cancer chemoprevention with a good response [36]. She developed premature menopause before age 30 years.
Figure 3.

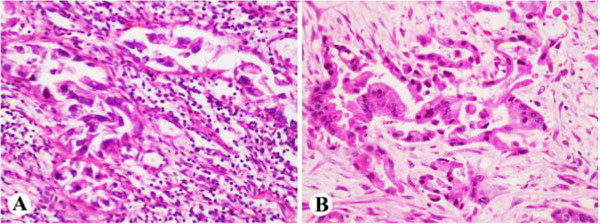
Case 3. XP-C A-B: MRI images of brain tumor. A: Axial post contrast T1-weighted MRI image of the brain obtained at age 29y showing a space occupying lesion in the left frontal lobe. The lesion demonstrates decrease signal intensity and does not enhance. The lack of enhancement after contrast indicates low histologic grade of this tumor. B: Axial post contrast T1-weighted MRI image of the brain in the same patient obtained five years later. There is evidence of tumor recurrence after surgery. The tumor demonstrates abnormal increased enhancement on the post contrast scan. This feature is indicative of malignant transformation. C: Histologic features of the primary tumor (as shown in A), a low grade astrocytoma (inset shows slightly atypical cells). D: Histologic features of the recurrent tumor, a glioblastoma. The tumor is characterized by increased cellularity, and pseudo-palisading necrosis (arrow), multinucleated tumor cell is shown in inset. E: Immunohistochemistry shows that the tumor cells are positive for IDH1 (X200). F: Pigmented lesion showing irregular size, shape and color. G: Histology of F showing melanoma in situ (x400).
At age 29 years, she was found to have an asymptomatic left frontal lobe, non-enhancing brain mass lesion (Figure 3A). The patient elected follow-up with serial MRIs. However, two years later, the lesion enlarged and became symptomatic. She underwent craniotomy with subtotal resection of the mass. Pathologic examination revealed a low grade astrocytoma (Figure 3C and D). She was treated postoperatively with external beam radiation therapy with a total dose of 5,940 cGy. As with other XP patients receiving radiotherapy [42], she had a normal skin response. The patient was stable until age of 34 years, when she developed expressive aphasia and fatigue. Imaging revealed recurrent tumor with abnormal increase of enhancement on the post-contrast scan (Figure 3B), a feature considered indicative of malignant transformation. Resection showed that the tumor had progressed to glioblastoma (Figure 3E and F). Worsening aphasia and right-sided hemiparesis complicated her postoperative course. She began temozolomide therapy. She did not have any unusual side effects to standard doses of temozolomide which was well tolerated. The patient’s condition continued to deteriorate, and she expired at home. A partially restricted autopsy was performed. Laboratory testing showed compound heterozygous mutations in the XPC gene (Table 1): A155X and 83bp insertion of intron 5.1 with stop codon 34 codons downstream [43].
Case 4 (XP1BE- XP-C)
Case 4 was a 49-year-old Caucasian female with XP, who had been followed at the NIH since age 24 years [4,10,14,44,45] (Figure 1E and F and Table 1). She had more than 200 biopsy-proven BCC and SCC and developed approximately 38 separate primary melanomas, half of which were in-situ lesions [46]. A number of these lesions responded to intra-lesional alpha interferon [47]. Bilateral orbital exenterations were performed for recurrent squamous cell carcinomas of the cornea and globe at age 32 and 36 years, followed by 5,000 rads of irradiation with a normal response [48]. In addition, she was found to have subclinical, asymptomatic neurologic disease, including right peroneal nerve weakness and subclinical hearing loss, without mental deterioration [10,14,44]. She had multiple urinary tract infections, chronic glomerulonephritis following left nephrectomy at age 40 years for staghorn calculi, premature menopause secondary to hypogonadotropic hypogonadism, and fatty liver. Just prior to death, she experienced shortness of breath and developed a superaventricular tachycardia. An unrestricted autopsy was performed [10]. She had mutations in the XPC gene (Table 1): homozygous deletion of AA Exon 8 del AA 1396–7, fs188 - stop 5 codons downstream [49]. Cultured skin fibroblasts had about 10% of normal post-UV UDS [4,35,39]. Post-UV skin fibroblast cell survival was reduced [41].
Pathological examinations
Case 1 (XP12BE- XP-A)
Neuropathology
Gross description: The brain weighed 660 g (normal for ~ 6 month old infant) and was bisected fresh (Figure 2C and Table 1). The leptomeninges were smooth and transparent. There was no venous congestion or evidence of acute subarachnoid hemorrhage. Generalized brain atrophy with marked dilatation of lateral ventricles and thinning of the corpus callosum was noted. The gyri/sulci were grossly within normal limits. Examination of the basal cerebral vasculature revealed no dilatation, thrombosis, atheroma, or other abnormalities. Coronal sectioning of the right cerebral hemispheres confirmed diffuse atrophy (Figure 2C) with marked ventricular dilatation. While the cortical ribbon was thin throughout the hemispheres, the loss of brain substance seemed greater in the subjacent white matter. Deep gray structures including the mamillary body, thalami, subthalamic nuclei, basal ganglia, and the hippocampus were grossly unremarkable. Serial sectioning of the right brainstem and cerebellum revealed no grossly significant abnormalities. The spinal leptomeninges were thin and translucent and the subarachnoid space was free of exudates. The spinal arteries and veins were unremarkable. Significant eye findings included exposure keratopathy and bilateral optic atrophy as previously described [26].
Microscopic description of sections from the midbrain, pons, medulla and hippocampus revealed neuronal loss and gliosis. In addition increased capillary density was seen in the thalamus. Sections from the right frontal and temporal cortex (Figure 2D) showed neuronal loss and gliosis accompanied by microglial activation. In addition, patchy white matter rarefaction was seen and highlighted by a LFB stain (Figure 2E). There were no neuritic plaques, tangles or neuronal inclusions. Sections from the right cerebellum demonstrated overall atrophy of the folia. Focal loss of Purkinje cells with increased numbers of torpedoes (Purkinje cell axonal swellings) and associated Bergmann astrocytosis was seen at the junction of the granular and molecular layers. The granular layer was mostly preserved with only mild rarefaction. A section from the dura was unremarkable. Electron microscopy of the radial nerve showed increased interstitial collagen with reduced density of large and myelinated fibers, axonal atrophy and occasional regeneration clusters consistent with axonal neuropathy (Figure 4A). Rare thinly myelinated fibers were seen. Denervation atrophy (esterase positive angular atrophic fibers, pyknotic nuclear clumps) of the skeletal muscles was present. Fiber type grouping, however, was not a feature. Temporal bone histology revealed marked atrophy of the cochlear sensory epithelium and neurons [14].
Figure 4.
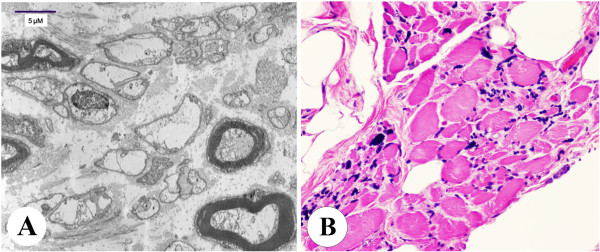
Case 1. XP-A Axonal neuropathy and denervation atrophy of skeletal muscle. A: Electron microscopy showing mild to moderate reduction in the number of large myelinated axons accompanied by increased collagen and axonal degeneration and axonal atrophy consistent with axonal neuropathy. B: Skeletal muscle showing marked variation in fiber size, with rounded and angulated atrophic fibers, pyknotic nuclear clumps, fibrosis with fat replacement.
Other findings: The indicated cause of death was XP-related neurologic degeneration. Hemangioma of the lip, seborrheic keratosis, uterine leiomyoma and fibrocystic change in bilateral breasts were also identified (Table 1).
Case 2 (XP18BE- XP-D)
Neuropathology
Gross description of the skull was notable for a substantially thickened calvarium, measuring 2.3 cm (frontal) (Figure 5A), 1.7 cm (occipital) and 1.0 cm (temporal). There were two boney excrescences measuring 1.4x1.2x0.4 cm and 1.2x1.0x0.6 cm in the right middle fossa of the skull base. The brain weighed 760 g (normal for ~ 1 yr old child) and showed diffuse widening of the sulci consistent with global atrophy (Table 1). The cerebral gyri/sulci were unremarkable and the hemispheres were symmetric. Coronal sections of the hemispheres confirmed the diffuse cerebral atrophy with dilated ventricles and a thin corpus callosum, but no focal lesions were seen (Figure 5B). Cerebellar hemispheres and vermis also appeared atrophic. Examination of peripheral nerves including both brachial plexuses, and radial, median, ulnar, femoral, sural and vagal nerves was unremarkable.
Figure 5.

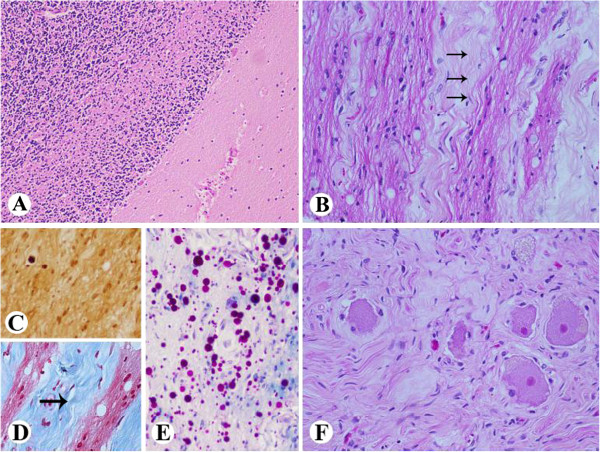
Case 2 XP-D Neuropathologic changes. A: Thick calvarium (2.3 cm, frontal). B: Global cerebral atrophy with dilated ventricles and thinned corpus callosum. C-D: Thinning of the cortical ribbon with vacuolization of the neuropil, neuronal loss and gliosis in the right frontal (C, x100) and parietal cortex (D, x200). E-F: Patchy loss of Purkinje cells in the right cerebellum with Bergman gliosis and axonal torpedos (E, x40; F, x200). G: Patchy myelin pallor in the right cerebellum (x100). H: GFAP highlights the reactive astrocytes in parietal lobe (x200).
Microscopic description: Sections from the right frontal and parietal cortex showed thinning of the cortical ribbon with extensive neuronal loss widespread vacuolization of the neuropil (Figure 5C and 5D). Vacuolization was most prominent in the deep cortical layers (resembling the pattern of coarse vacuolation termed status spongiosis); neuronal loss and gliosis were accompanying features. Neurofibrillary tangles, plaques or other inclusions or storage products were not identified. Sections from the hippocampus demonstrated atrophy, gliosis, and pyramidal cell loss and vacuolization of the neuropil. Hippocampal neuronal loss most prominently involved the CA2 and CA3 sectors. Sections from the right basal ganglia and mid brain-pontine junction revealed extensive neuronal loss and gliosis, Figure 6 (A-C). Sections from the right cerebellum showed focal loss of Purkinje cells and accompanying Bergmann gliosis (Figure 5E and 5F) with patchy myelin pallor of the cerebellar white matter (Figure 5G). A section from the grossly described boney excrescence in the right middle fossa showed cortical sclerosis with no evidence of tumor. Immunohistochemistry and special stains for GFAP and LFB were performed on sections from the cortex, hippocampus and cerebellum. The GFAP stain highlighted reactive astrocytes (Figure 5H).
Figure 6.

Case 2 XP-D. Hippocampal sections showed neuronal loss involving CA2/CA3 sectors (A, X200); Neuropil disorganization seen in basal ganglia (B, X200) and midbrain-pontine areas (C, X200).
Histology of the peripheral nerves (Figure 7A and 7B), pharyngeal muscle and esophageal muscle were unremarkable. Examination of the esophagus showed infiltration of the ganglia of Auerbach’s plexus by CD3 positive T-lymphocytes (Figure 7C and 7D).
Figure 7.

Case 2 XP-D. Neuropathologic changes. A and B, No specific abnormality was appreciated in peripheral nerve stained with H&E (A, x100) and LFB/PAS (B, x100). C and D, Periganglial lymphocytic infiltration of Auerbach’s plexus in the esophagus (C, x40; D, x 200).
Other findings: The immediate cause of death was aspiration pneumonia. Focal patchy consolidation, bronchial mucus plugging and a cluster of plant (vegetable) cells with alveolar and interstitial infiltration by neutrophils and lymphocytes were seen in both lungs. A small (0.2 cm), hemangioma was present on the patient’s back. Laryngeal ulceration, bilateral breast fibrocystic changes, mild renal glomerulosclerosis, subserosal uterine adenomyosis, and cholelithisasis were also present (Table 1). There was no ocular neoplasm identified. The eye findings were reported previously [26], and consisted of bilateral pinguecula, corneal pannus, exposure keratopathy, retinal gliosis and bilateral peripheral retinal pigmentary degeneration.
Case 3 (XP24BE- XP-C)
Neuropathology
Gross findings: There were several metal plates on the frontal bone indicating prior surgery. The cranial vault appeared of normal thickness. The brain weighed 1330 g fresh (normal for adult) (Table 1). The leptomeninges over the left frontal region were slightly distorted with multiple sutures; the leptomeninges over the right hemisphere were unremarkable. The entire brain appeared edematous and the left cerebral hemisphere was enlarged, and with midline shift, but no obvious herniations were identified. Examination of the arteries of the circle of Willis and their major branches revealed no evidence of atherosclerosis, thrombus formation or malformations. The superficial veins and cranial nerves were unremarkable. Coronal sectioning of the cerebral hemispheres revealed an ill-defined tumor mass infiltrating the left frontal and temporal lobes, crossing the corpus callosum and invading the right cerebral hemisphere. The tumor was tan/white, soft with grey gelatinous areas, resembling tumor necrosis. Serial transverse sectioning of the brain stem and cerebellum revealed no abnormalities. The spinal cord was grossly unremarkable.
Microscopic findings: The prior primary tumor as shown in Figure 3A was a low grade astrocytoma (Figure 3C) with only slightly pleomorphic tumor cells (Figure 3C inset) that stained positive with GFAP. The proliferative index of the tumor cells was low as indicated by the MIB-1 staining. At autopsy, the diffusely infiltrative astrocytic tumor showed high- grade features characterized by frequent mitotic figures, microvascular proliferation, and pseudopalisading necrosis (Figure 3D). Scattered multinucleated tumor cells were seen (Figure 3D inset). The tumor cells immunolabeled with GFAP and IDH1 (Figure 3E), and were negative for EGFR. MIB-1 stain indicated a high proliferative rate and p53 stain showed focal positivity in less than 5% of tumor cells. The sections from the remaining brain and spinal cord did not show significant pathologic changes.
Other findings included bilateral pneumonia with bacterial and fungal infection, which was the immediate cause of her death (Table 1). The ovaries were small with microscopic fibrosis with no follicles observed. A cutaneous melanoma in situ (Figure 3F and 3G) and a basal cell carcinoma were identified a few weeks before the patient expired. Several pigmented skin lesions were sampled at autopsy, including lentiginous melanocytic proliferation, atypical melanocytic proliferation, severely atypical melanocytic proliferation and seborrheic keratosis. No other skin cancers were identified.
Case 4 (XP1BE- XP-C)
Neuropathology
Gross description: The brain weighed 1300 gm (normal for an adult) (Table 1) and was bisected fresh. The dura, cerebral hemispheres, and infratentorial structures were externally unremarkable. The optic nerves and chiasm were atrophic. There was patchy non-occlusive atherosclerosis involving the basilar artery and circle of Willis. Sequential sections through the hemispheres, brain stem, and cerebellum revealed no focal lesions. The spinal arteries and veins were unremarkable. Peripheral nerves including median, sural nerves and celiac plexus revealed no gross abnormalities.
Microscopic description: The cerebellum showed focal loss of Purkinje cells (Figure 8A) with accompanying Bergmann astrocyte proliferation, and gliosis of the molecular layer. The hemispheric cortical grey matter and subcortical white matter were unremarkable. The pituitary was unremarkable. Sections of optic nerves and geniculate nuclei revealed degenerative changes (Figure 8B and 8C) including marked axonal loss with gliosis and resultant fibrosis of the fibro-vascular bundles and trans-synaptic degeneration in the lateral geniculate body with gliosis, consistent with the patient’s 10-year history of bilateral orbital exenterations [48]. Sections of spinal cord revealed no pathologic changes; however, the dorsal root ganglia showed severe neuronal loss with sclerotic foci occupying previous neuronal sites (Figure 8D) as reported previously [10]. Peripheral ganglia (celiac) were unremarkable.
Figure 8.
Case 4 XP-C Neuropathologic changes. A: The cerebellum shows focal loss of Purkinje cells (x100). B-E: Sections of optic nerves and geniculate nuclei reveal degenerative changes, including axonal loss, gliosis, and fibrosis (arrows) in keeping with the patient’s 10 year history of bilateral orbital exenterations (B. H&E x200; C. immunostain for Bielschowsky, x200 D. Masson stain, x200, E. LFB/PAS stain, x200). F. Dorsal root ganglia show severe neuronal loss and accompanying fibrosis (H&E, x200).
Multiple pulmonary emboli were the immediate cause of her death. The underlying cause of the pulmonary emboli was a clinically unsuspected well differentiated mucinous adenocarcinoma of endocervical origin (Figure 9A), which directly invaded the surrounding pelvic organs including ovaries and sigmoid colon, causing diffuse abdominal carcinomatosis that involved the diaphragm (Figure 9B), omentum, appendix, large and small bowel, and metastasized to multiple splenic hilar lymph nodes, spleen and lungs.
Figure 9.
Case 4 XP-C Carcinomatosis. Well differentiated mucinous adenocarcinoma of endocervical origin (A, x200), caused extensive abdominal carcinomatosis and involved diaphragm (B, x200).
Other findings included obesity, glomerulonephritis, multiple simple renal parenchymal cysts, steatohepatitis, nodular regenerative hyperplasia of liver, right ventricular dilatation and left ventricular hypertrophy, and thyroid follicular adenomas. In addition, there was mild to moderate atherosclerosis of the left anterior descending artery, and moderate to severe atherosclerosis of the right coronary artery.
Discussion
Neurodegeneration in XP patients
The present study represents the first detailed description of the postmortem neuropathologic findings in an XP-D patient. These features have been systematically compared to an XP-A and two XP-C patients (Table 1). Of note, all four patients were included in a large series of XP patients followed at the NIH for many years permitting close observation of disease progression and the effects of treatment [1,14]. Major clinical neurologic abnormalities in XP patients include sensorineural hearing loss, peripheral neuropathy, microcephaly and dementia. The XP-A and XP-D patients were diagnosed with XP at age 4 and 5 years, respectively, and developed high frequency sensorineural hearing loss at a young age. Their neurologic status showed progressive decline with loss of ability to walk and talk leading to inability to care for themselves and death at age 44 and 45 years of age (Table 1).
The core neuropathologic findings of this study are profound global cerebral and pancerebellar atrophy in the XP-A and XP-D cases. The most striking gross finding was generalized brain atrophy (660 and 760 g) with brain size similar to that of a 6 to 12 month old infant [50]. Consistent with what has been described previously in XP-A patients [4-6,11], we found brain atrophy involving all lobes. There was diffuse neurodegeneration with neuronal loss and gliosis of both hemispheres, similar changes in the cerebellum, basal ganglia, and corticospinal tracts. Retinal gliosis was identified in both cases [26]. In addition, the XP-D patient had a thickened calvarium, vacuolization of the deep cortical neuropil, patchy Purkinje cell loss, and infiltration of the ganglia of esophageal Auerbach’s plexus by T-lymphocytes. Intriguingly, calvarial thickening which has been described in XP-A patients from Finland [5] was not observed in the XP-A patient.
Both XP-A and XP-D patients developed dysphagia, which poses difficult challenges for clinical management. In addition to CNS dysfunction, we carefully evaluated the peripheral neuromuscular system, and found axonal neuropathy and chronic denervation atrophy of skeletal muscle including those that related to swallowing in the XP-A patient. The peripheral neuropathy has been previously reported and involves both motor and sensory nerves. The underlying pathogenic mechanism is thought to be a neuronopathy [7,51]. In contrast, significant peripheral neuronopathy was not identified in the XP-D patient where section of the esophagus showed T-cell infiltrates of Auerbach’s plexus. This finding suggests an immune-mediated mechanism may be associated with acute autonomic and sensory neuropathy leading to impairment of both superficial and deep sensation in the XP-D patient.
Both the XP-A and the XP-D patients had severe progressive neurodegeneration. However, while the swallowing difficulty led to the placement of a G-tube for feeding of both patients, only the XP-A patient developed cachexia. The XP-D patient retained her weight, which suggests that cachexia is not directly related to the extent of neurodegeneration or the inability to swallow.
A patient with XP/Cockayne syndrome complex was described with a defect in the XP-G gene (XP20BE) [27]. He died at age 6 yr and had marked growth retardation with short stature and severe cachexia. However the autopsy findings of XP20BE were different from the XP-A and XP-D cases. His brain appeared proportionally small and not atrophic with a grossly normal cortical ribbon and an unusual tigroid pattern of demyelination. In addition, the patient had cataracts, and retinal pigmentary atrophy typical of Cockayne syndrome.
With respect to the two XP-C cases, one case showed unexpected carcinomatosis with endocervical adenocarcinoma. The other patient developed a glioblastoma, which arose from a pre-existing low-grade, diffuse astrocytoma. This observation is furthered supported by the IDH1 labeling of the tumor cells. It has been reported that as high as 90% of diffuse and anaplastic gliomas and secondary glioblastomas (arising from a pre-existing low-grade astrocytoma) are IDH1-positive [52]. The generalized neurodegenerative changes seen in the XP-A and XP-D cases were not observed in the XP-C cases although the older patient had focal loss of Purkinje cells and, neuronal loss in the dorsal root ganglion.
There was a clinical correlation between the skin abnormalities and the presence of progressive neurological degeneration. In a longitudinal study of 73 XP patients with audiograms examined at the NIH, all 17 patients who developed sensorineural hearing loss and progressive neurological degeneration had a history of acute burning on minimal sun exposure, including the XP-A and XP-D patients in this report [14]. This burning history was not present in the XP-C patients and they did not show progressive neurological degeneration. Further, cultured skin cells from XP patients with a history of burning on minimal sun exposure and neurological degeneration were hypersensitive to killing by ultraviolet radiation [41,53].
Cancer in XP patients
Our previous data have shown that 65% (69/106) of XP patients developed sunlight-induced skin cancers. Non-melanoma skin cancer was increased 10,000-fold and melanoma was increased 2,000-fold in patients under age 20 [1,2]. The XP-A, and XP-C patients developed hundreds of skin cancers (Table 1). The XP-A patient and XP-C patient XP24BE were treated with oral 13-cis-retinoic acid for chemoprevention of skin cancer [36]. They both had a good response, with no further skin malignancies identified at autopsy. The XP-D patient was sun protected from early childhood and developed only 9 skin cancers.
The XP-C patients developed internal cancers with a glioma in XP24BE and a cervical adenocarcinoma in XP1BE. XP patients have been estimated to have an 11-fold increase in internal neoplasms and a 50-fold increase in cancers of the central nervous system [2]. The reported tumors include Schwannoma in a 73 y/o [1], spinal cord astrocytoma in a 24 y/o, gastric cancer in a 50 y/o and lung cancer in a 37 y/o smoker [42]. Some carcinogenic components of cigarette smoke bind to DNA forming adducts that are repaired by the NER system. It is thought that the lung cancers in the young XP patients are related to mutations resulting from this unrepaired damage. The underlying mechanism of induction of the CNS neoplasms is not known.
DNA repair defects in XP patients
Defects in the repair-enzyme genes XPA through XPG are known to cause XP [12,13,54]. Cells from XP patients with mutations in the XPC DNA repair gene have defective global genome DNA repair and normal transcription coupled repair. In contrast, cells from patients with mutations in the CSA or CSB genes have defects only in transcription coupled repair. These patients have clinical Cockayne syndrome with severe progressive neurological degeneration with short stature and retinal degeneration but without an increase in cancer [12,54]. However, cells from XP patients with mutations in the XPA and XPD genes have defective global genome repair and in addition, defective transcription coupled DNA repair. This additional transcription coupled DNA repair defect is associated with progressive neurological degeneration in humans (see for example [5]) and in mouse model systems [55]. Patients with trichothiodystrophy have mutations in the XPD gene in association with developmental abnormalities (including absent myelin in the brain) as well as sulfur deficient brittle hair, cataracts, bone abnormalities and increased susceptibility to infections, but no increase in cancer [12,54,56]. The level of DNA repair does not correlate with the clinical phenotype. Thus, while cells from XP-A patients with severe neurological degeneration generally have <2% of normal post-UV unscheduled DNA synthesis (UDS) in cultured skin fibroblasts, cells from XP-D patients, who also have severe neurological degeneration, have 25-50% UDS and cells from Cockayne syndrome patients have normal UDS. Further, cells from XP-C patients with no neurological abnormalities have 10% UDS [4,35,39,41,56]. Thus the relationship of mutations in the DNA repair genes to clinical phenotypes is complex and the precise mechanisms of these differences are not understood.
XP patients have defective repair of UV induced DNA damage to skin and eyes. Unrepaired DNA damage can lead to increased cell death and to a high frequency of somatic mutations in the surviving, dividing cells. This process is believed to underlie the skin sun sensitivity and high frequency of sunlight-induced cancers in XP patients. Skin tumors from XP patients frequently have evidence of UV type mutations [57,58]. There is a good correlation between post-UV killing and neurological degeneration in cultured cells from XP patients. Thus, skin fibroblasts from XP-A and XP-D patients with severe neurological degeneration are more sensitive to post-UV killing than are cells from XP-C patients who have no neurological abnormalities [41]. However, UV radiation does not reach the brain because of shielding from the scalp and bones of the skull. It has been hypothesized that oxidative DNA damage generated through metabolism or other sources might produce DNA damage that is not repaired in the non-dividing cells of the nervous system leading to progressive neuronal death [8]. Most types of oxidative DNA damage are repaired by base excision repair, a process that is not defective in XP cells. Indeed, both XP-C patients received therapeutic doses of x-radiation (which produces oxidative damage) for treatment (brain tumor in XP24BE and orbital tumors in XP1BE and spinal cord astrocytoma in another XP-C patient [42]) (Table 1) with a normal response. However, certain types of products of oxidative DNA damage, such as cyclopurine deoxynucleosides, are repaired by the NER system. Impaired repair of this type of oxidative DNA damage has been proposed as a mechanism of the progressive neurodegeneration seen in these XP patients [59,60].
Conclusions
In conclusion, while all 4 XP patients had defects in NER genes, they had markedly different phenotypes with different clinical consequences. Three of the patients had hundreds of skin cancers early in life, and oral therapy with retinoids and increased sun protection proved effective in 2 of them. None of these XP patients died from their skin cancers. Two died from internal neoplasms (glioblastoma and endocervical adenocarcinoma) and the other two adult patients died from progressive neurodegeneration. Understanding the complex relationships of these DNA repair defects to these causes of death are a challenge for future investigations.
Methods
Neuropathological examination
The patients were examined at the NIH under protocols approved by the Institutional Review Board of the National Cancer Institute. They gave their permission for use of identifying images. The families of the patients have given their consent for performance of the autopsies. The brain, peripheral nerves, muscles were fixed with 10% buffered formalin and embedded in paraffin. Histological examinations were performed on 5 μm thick sections using hematoxylin–eosin (HE). Selected sections were evaluated with special stains (luxol fast blue (LFB), Bielschowsky, Masson’s trichrome) and immunostains (glial fibrillary acidic protein (GFAP), synaptophysin, NeuN, neurofilament proteins, KP-1, beta-amyloid, Isocitrate dehydrogenase 1 (IDH1), p53, EGFR, MIB-1). For immunohistochemical analyses, 5 μm thick sections were serially cut in selected brain regions and brain tumors. The control subjects had no neurological abnormalities and showed no morphological changes in the brain. Slides were incubated in Tris with 3% goat serum for 15 min and then incubated for 1–2 h at room temperature with primary antibody. Detection was carried out using an automated slide stainer (DAKO; Autostainer) with a horseradish peroxidase/3,3′-diaminobenzidine polymer-based detection system (DAKO; Envision+). No staining was observed in the sections incubated in the absence of either antibody.
Abbreviations
BCC: Basal cell carcinomas; NER: Nucleotide excision repair; NIH: National Institutes of Health; SCC: squamous cell carcinoma; UDS: unscheduled DNA synthesis; UV: ultraviolet radiation; XP: xeroderma pigmentosum
Competing interests
The authors declare no competing interests.
Authors’ contributions
The autopsies were performed by J-PL, Y-CL, QL, PPA, KM, SH, DLL, DEK and JCO; The slides and images were interpreted by MMQ, MA, C-HL, MT, EJR, and JCO; The radiological images were interpreted by NP; The patients were cared for and clinical data collected by DT, JJD, HAF and KHK; DNA repair and mutations were determined by SK; The study was conceived and designed by MMQ, JCO, JJD and KHK. The manuscript was drafted and revised critically for intellectual content by J-PL, Y-CL, MMQ, EJR and KHK. All authors read and approved the final manuscript.
Contributor Information
Jin-Ping Lai, Email: Jinping.LaiXX@cshs.org.
Yen-Chun Liu, Email: prairieliuyc@gmail.com.
Meghna Alimchandani, Email: meghna.alimchandani@nih.gov.
Qingyan Liu, Email: qingyan212@gmail.com.
Phyu Phyu Aung, Email: aungpatho@gmail.com.
Kant Matsuda, Email: kant_matsuda@hotmail.com.
Chyi-Chia R Lee, Email: leechy@mail.nih.gov.
Maria Tsokos, Email: mtsokos@bidmc.harvard.edu.
Stephen Hewitt, Email: hewitts@mail.nih.gov.
Elisabeth J Rushing, Email: elisabeth.rushing@gmail.com.
Deborah Tamura, Email: tamurad@mail.nih.gov.
David L Levens, Email: levensd@mail.nih.gov.
John J DiGiovanna, Email: jdigiova@mail.nih.gov.
Howard A Fine, Email: ward.Fine@nyumc.org.
Nicholas Patronas, Email: NPatronas@cc.nih.gov.
Sikandar G Khan, Email: skhan@helix.nih.gov.
David E Kleiner, Email: kleinerd@mail.nih.gov.
J Carl Oberholtzer, Email: oberholtzerc@mail.nih.gov.
Martha M Quezado, Email: quezadom@mail.nih.gov.
Kenneth H Kraemer, Email: kraemerk@nih.gov.
Acknowledgment
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (NIH). We thank Drs. Leomar Ballester and Avi Rosenberg for their assistance with preparation of the manuscript.
References
- Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki S, Emmert S, Pike KM, Raziuddin A, Plona TM, DiGiovanna JJ, Tucker MA, Kraemer KH. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48:168–176. doi: 10.1136/jmg.2010.083022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–1021. doi: 10.1001/archderm.1994.01690080084012. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. doi: 10.1001/archderm.1987.01660260111026. [DOI] [PubMed] [Google Scholar]
- Robbins JH, Kraemer KH, Lutzner MA, Festoff BW, Coon HG. Xeroderma pigmentosum. An inherited disease with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann Intern Med. 1974;80:221–248. doi: 10.7326/0003-4819-80-2-221. [DOI] [PubMed] [Google Scholar]
- Anttinen A, Koulu L, Nikoskelainen E, Portin R, Kurki T, Erkinjuntti M, Jaspers NG, Raams A, Green MH, Lehmann AR, Wing JF, Arlett CF, Marttila RJ. Neurological symptoms and natural course of xeroderma pigmentosum. Brain. 2008;131:1979–1989. doi: 10.1093/brain/awn126. [DOI] [PubMed] [Google Scholar]
- Itoh M, Hayashi M, Shioda K, Minagawa M, Isa F, Tamagawa K, Morimatsu Y, Oda M. Neurodegeneration in hereditary nucleotide repair disorders. Brain Dev. 1999;21:326–333. doi: 10.1016/S0387-7604(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Kanda T, Oda M, Yonezawa M, Tamagawa K, Isa F, Hanakago R, Tsukagoshi H. Peripheral neuropathy in xeroderma pigmentosum. Brain. 1990;113:1025–1044. doi: 10.1093/brain/113.4.1025. [DOI] [PubMed] [Google Scholar]
- Robbins JH. Xeroderma pigmentosum. Defective DNA repair causes skin cancer and neurodegeneration. JAMA. 1988;260:384–388. doi: 10.1001/jama.1988.03410030100038. [DOI] [PubMed] [Google Scholar]
- Robbins JH, Brumback RA, Mendiones M, Barrett SF, Carl JR, Cho S, Denckla MB, Ganges MB, Gerber LH, Guthrie RA. Neurological disease in xeroderma pigmentosum. Documentation of a late onset type of the juvenile onset form. Brain. 1991;114:1335–1361. doi: 10.1093/brain/114.3.1335. [DOI] [PubMed] [Google Scholar]
- Robbins JH, Kraemer KH, Merchant SN, Brumback RA. Adult-onset xeroderma pigmentosum neurological disease–observations in an autopsy case. Clin Neuropathol. 2002;21:18–23. [PubMed] [Google Scholar]
- Ueda T, Kanda F, Aoyama N, Fujii M, Nishigori C, Toda T. Neuroimaging features of xeroderma pigmentosum group A. Brain Behav. 2012;2:1–5. doi: 10.1002/brb3.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- Totonchy MB, Tamura D, Pantell MS, Zalewski C, Bradford PT, Merchant SN, Nadol J, Khan SG, Schiffmann R, Pierson TM, Wiggs E, Griffith AJ, DiGiovanna JJ, Kraemer KH, Brewer CC. Auditory analysis of xeroderma pigmentosum 1971–2012: hearing function, sun sensitivity and DNA repair predict neurological degeneration. Brain. 2013;136:194–208. doi: 10.1093/brain/aws317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sanctis C, Cacchione A. L'idiozia xerodermica. Rivista Sperimentale di Freniatria e Medicina Legale delle Alienazioni Mentali. 1932;56:269–292. [Google Scholar]
- Hayashi M. Roles of oxidative stress in xeroderma pigmentosum. Adv Exp Med Biol. 2008;637:120–127. doi: 10.1007/978-0-387-09599-8_13. [DOI] [PubMed] [Google Scholar]
- Hayashi M. Oxidative stress in developmental brain disorders. Neuropathology. 2009;29:1–8. doi: 10.1111/j.1440-1789.2008.00888.x. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Araki S, Kohyama J, Shioda K, Fukatsu R. Oxidative nucleotide damage and superoxide dismutase expression in the brains of xeroderma pigmentosum group A and Cockayne syndrome. Brain Dev. 2005;27:34–38. doi: 10.1016/j.braindev.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Araki S, Kohyama J, Shioda K, Fukatsu R, Tamagawa K. Brainstem and basal ganglia lesions in xeroderma pigmentosum group A. J Neuropathol Exp Neurol. 2004;63:1048–1057. doi: 10.1093/jnen/63.10.1048. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Itoh M, Araki S, Kumada S, Shioda K, Tamagawa K, Mizutani T, Morimatsu Y, Minagawa M, Oda M. Oxidative stress and disturbed glutamate transport in hereditary nucleotide repair disorders. J Neuropathol Exp Neurol. 2001;60:350–356. doi: 10.1093/jnen/60.4.350. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Miwa-Saito N. Tanuma N. Neuropathology: Kubota M Brain vascular changes in Cockayne syndrome; 2011. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Ohto T. Shioda K. Brain Dev: Fukatsu R Lesions of cortical GABAergic interneurons and acetylcholine neurons in xeroderma pigmentosum group A; 2011. [DOI] [PubMed] [Google Scholar]
- Kenyon GS, Booth JB, Prasher DK. Rudge P Neuro-otological abnormalities in xeroderma pigmentosum with particular reference to deafness. Brain. 1985;108(Pt 3):771–784. doi: 10.1093/brain/108.3.771. [DOI] [PubMed] [Google Scholar]
- Kohji T, Hayashi M, Shioda K, Minagawa M, Morimatsu Y, Tamagawa K, Oda M. Cerebellar neurodegeneration in human hereditary DNA repair disorders. Neurosci Lett. 1998;243:133–136. doi: 10.1016/S0304-3940(98)00109-8. [DOI] [PubMed] [Google Scholar]
- Okayasu I, Satoh Y, Irimajiri T, Mitoh Y. Fetal case of xeroderma pigmentosum–first report of an autopsy case. Acta Pathol Jpn. 1978;28:459–464. doi: 10.1111/j.1440-1827.1978.tb01269.x. [DOI] [PubMed] [Google Scholar]
- Ramkumar HL, Brooks BP, Cao X, Tamura D, DiGiovanna JJ, Kraemer KH, Chan CC. Ophthalmic manifestations and histopathology of xeroderma pigmentosum: two clinicopathological cases and a review of the literature. Surv Ophthalmol. 2011;56:348–361. doi: 10.1016/j.survophthal.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55:1442–1449. doi: 10.1212/WNL.55.10.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed WB, Landing B, Sugarman G, Cleaver JE, Melnyk J. Xeroderma pigmentosum. Clinical and laboratory investigation of its basic defect. JAMA. 1969;207:2073–2079. doi: 10.1001/jama.207.11.2073. [DOI] [PubMed] [Google Scholar]
- Reed WB, MARY SB, NICKEL WR. Xeroderma pigmentosum with neurological complications: The deSanctis-Cacchione syndrome. Arch Dermatol. 1965;91:224–226. doi: 10.1001/archderm.1965.01600090032005. [DOI] [PubMed] [Google Scholar]
- Reed WB, Sugarman GI, Mathis RA. DeSanctis-Cacchione syndrome. A case report with autopsy findings. Arch Dermatol. 1977;113:1561–1563. doi: 10.1001/archderm.1977.01640110081013. [DOI] [PubMed] [Google Scholar]
- Roytta M, Anttinen A. Xeroderma pigmentosum with neurological abnormalities. A clinical and neuropathological study. Acta Neurol Scand. 1986;73:191–199. doi: 10.1111/j.1600-0447.1986.tb10586.x. [DOI] [PubMed] [Google Scholar]
- Takagi M. Xeroderma pigmentosum–report of two autopsy cases (author's transl) Kokubyo Gakkai Zasshi. 1979;46:389–396. [PubMed] [Google Scholar]
- Wang Y, Tan XH, DiGiovanna JJ, Lee CC, Stern JB, Raffeld M, Jaffe ES, Kraemer KH. Genetic diversity in melanoma metastases from a patient with xeroderma pigmentosum. J Invest Dermatol. 2010;130:1188–1191. doi: 10.1038/jid.2009.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano K. Xeroderma pigmentosum with disorders of the central nervous system; a histopathological study. Folia Psychiatr Neurol Jpn. 1950;4:143–175. [PubMed] [Google Scholar]
- Kraemer KH, Coon HG, Petinga RA, Barrett SF, Rahe AE, Robbins JH. Genetic heterogeneity in xeroderma pigmentosum: complementation groups and their relationship to DNA repair rates. Proc Natl Acad Sci U S A. 1975;72:59–63. doi: 10.1073/pnas.72.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer KH, DiGiovanna JJ, Moshell AN, Tarone RE, Peck GL. Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N Engl J Med. 1988;318:1633–1637. doi: 10.1056/NEJM198806233182501. [DOI] [PubMed] [Google Scholar]
- States JC, Myrand SP. Splice site mutations in a xeroderma pigmentosum group A patient with delayed onset of neurological disease. Mutat Res. 1996;363:171–177. doi: 10.1016/0921-8777(96)00004-3. [DOI] [PubMed] [Google Scholar]
- Terunuma A, Ye J, Emmert S, Khan SG, Kraemer KH, Vogel JC. Ultraviolet light selection assay to optimize oligonucleotide correction of mutations in endogenous xeroderma pigmentosum genes. Gene Ther. 2004;11:1729–1734. doi: 10.1038/sj.gt.3302344. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, De Weerd-Kastelein EA, Robbins JH, Keijzer W, Barrett SF, Petinga RA, Bootsma D. Five complementation groups in xeroderma pigmentosum. Mutat Res. 1975;33:327–340. doi: 10.1016/0027-5107(75)90208-0. [DOI] [PubMed] [Google Scholar]
- Petinga RA, Andrews AD, Tarone RE, Robbins JH. Typical xeroderma pigmentosum complementation group A fibroblasts have detectable ultraviolet light-induced unscheduled DNA synthesis. Biochim Biophys Acta. 1977;479:400–410. doi: 10.1016/0005-2787(77)90033-8. [DOI] [PubMed] [Google Scholar]
- Andrews AD, Barrett SF, Robbins JH. Xeroderma pigmentosum neurological abnormalities correlate with colony-forming ability after ultraviolet radiation. Proc Natl Acad Sci U S A. 1978;75:1984–1988. doi: 10.1073/pnas.75.4.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna JJ, Patronas N, Katz D, Abangan D, Kraemer KH. Xeroderma pigmentosum: spinal cord astrocytoma with 9-year survival after radiation and isotretinoin therapy. J Cutan Med Surg. 1998;2:153–158. doi: 10.1177/120347549800200308. [DOI] [PubMed] [Google Scholar]
- Khan SG, Oh KS, Shahlavi T, Ueda T, Busch DB, Inui H, Emmert S, Imoto K, Muniz-Medina V, Baker CC, DiGiovanna JJ, Schmidt D, Khadavi A, Metin A, Gozukara E, Slor H, Sarasin A, Kraemer KH. Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis. 2006;27:84–94. doi: 10.1093/carcin/bgi204. [DOI] [PubMed] [Google Scholar]
- Robbins JH, Brumback RA, Moshell AN. Clinically asymptomatic xeroderma pigmentosum neurological disease in an adult: evidence for a neurodegeneration in later life caused by defective DNA repair. Eur Neurol. 1993;33:188–190. doi: 10.1159/000116932. [DOI] [PubMed] [Google Scholar]
- Robbins JH, Polinsky RJ, Moshell AN. Evidence that lack of deoxyribonucleic acid repair causes death of neurons in xeroderma pigmentosum. Ann Neurol. 1983;13:682–684. doi: 10.1002/ana.410130621. [DOI] [PubMed] [Google Scholar]
- Stern JB, Peck GL, Haupt HM, Hollingsworth HC, Beckerman T. Malignant melanoma in xeroderma pigmentosum: search for a precursor lesion. J Am Acad Dermatol. 1993;28:591–594. doi: 10.1016/0190-9622(93)70079-9. [DOI] [PubMed] [Google Scholar]
- Turner ML, Moshell AN, Corbett DW, Stern JB, Roth MJ, DiGiovanna J, Horn TD, Kraemer KH. Clearing of melanoma in situ with intralesional interferon alfa in a patient with xeroderma pigmentosum. Arch Dermatol. 1994;130:1491–1494. doi: 10.1001/archderm.1994.01690120027003. [DOI] [PubMed] [Google Scholar]
- Gaasterland DE, Rodrigues MM, Moshell AN. Ocular involvement in xeroderma pigmentosum. Ophthalmology. 1982;89:980–986. doi: 10.1016/s0161-6420(82)34692-8. [DOI] [PubMed] [Google Scholar]
- Li L, Bales ES, Peterson CA, Legerski RJ. Characterization of molecular defects in xeroderma pigmentosum group C. Nat Genet. 1993;5:413–417. doi: 10.1038/ng1293-413. [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Quantitative growth and development of human brain. Arch Dis Child. 1973;48:757–767. doi: 10.1136/adc.48.10.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara N, Kumamoto T, Takasawa H, Tsubaki T, Origuchi Y. The peripheral neuropathy in De Sanctis-Cacchione syndrome. Histological, ultrastructural, and morphometric studies. Acta Neuropathol (Berl) 1982;56:194–200. doi: 10.1007/BF00690635. [DOI] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JH, Moshell AN. DNA repair processes protect human beings from premature solar skin damage: evidence from studies on xeroderma pigmentosum. J Invest Dermatol. 1979;73:102–107. doi: 10.1111/1523-1747.ep12532789. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience. 2007;145:1388–1396. doi: 10.1016/j.neuroscience.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, Van DP I, De Waard MC, Haasdijk ED, Brandt R, Vermeij M, Rijksen Y, Maas A, Van SH, Hoeijmakers JH. Van der Horst GT Age-related neuronal degeneration: complementary roles of nucleotide excision repair and transcription-coupled repair in preventing neuropathology. PLoS Genet. 2011;7:1002405. doi: 10.1371/journal.pgen.1002405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghri S, Tamura D, Kraemer KH, DiGiovanna JJ. Trichothiodystrophy: a systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J Med Genet. 2008;45:609–621. doi: 10.1136/jmg.2008.058743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya-Grosjean L, Sarasin A. The role of UV induced lesions in skin carcinogenesis: an overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat Res. 2005;571:43–56. doi: 10.1016/j.mrfmmm.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Wang Y, DiGiovanna JJ, Stern JB, Hornyak TJ, Raffeld M, Khan SG, Oh KS, Hollander MC, Dennis PA, Kraemer KH. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci U S A. 2009;106:6279–6284. doi: 10.1073/pnas.0812401106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ. The case for 8,5'-cyclopurine-2'-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience. 2007;145:1407–1417. doi: 10.1016/j.neuroscience.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waard MC, van dP I, Zuiderveen BN, Comley LH, Haasdijk ED, Rijksen Y, Ridwan Y, Zondag G, Hoeijmakers JH, Elgersma Y, Gillingwater TH, Jaarsma D. Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol. 2010;120:461–475. doi: 10.1007/s00401-010-0715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]