

Abstract
Protein A affinity chromatography is the standard purification process for the capture of therapeutic antibodies. The individual IgG-binding domains of protein A (E, D, A, B, C) have highly homologous amino acid sequences. From a previous report, it has been assumed that the C domain has superior resistance to alkaline conditions compared to the other domains. We investigated several properties of the C domain as an IgG-Fc capture ligand. Based on cleavage site analysis of a recombinant protein A using a protein sequencer, the C domain was found to be the only domain to have neither of the potential alkaline cleavage sites. Circular dichroism (CD) analysis also indicated that the C domain has good physicochemical stability. Additionally, we evaluated the amino acid substitutions at the Gly-29 position of the C domain, as the Z domain (an artificial B domain) acquired alkaline resistance through a G29A mutation. The G29A mutation proved to increase the alkaline resistance of the C domain, based on BIACORE analysis, although the improvement was significantly smaller than that observed for the B domain. Interestingly, a number of other amino acid mutations at the same position increased alkaline resistance more than did the G29A mutation. This result supports the notion that even a single mutation on the originally alkali-stable C domain would improve its alkaline stability. An engineered protein A based on this C domain is expected to show remarkable performance as an affinity ligand for immunoglobulin.
Keywords: protein engineering, IgG-binding domain, therapeutic antibody, affinity ligand, physicochemical stability
Introduction
Protein A is a popular affinity ligand for the purification of monoclonal antibodies (Mabs). Protein A is a cell wall-associated protein expressed by the Gram-positive bacterium Staphylococcus aureus.1 Protein A consists of a tandem repeat of five highly homologous IgG-Fc binding domains, designated E, D, A, B, and C, and a cell-wall anchoring region, designated XM, from the N terminus.2
Due to its high purification efficiency, protein A affinity chromatography has become the standard process for the capture of Mabs from a cell culture supernatant in the industrial manufacturing of therapeutic Mabs.1,3 However, particular care has to be taken to minimize contamination when producing Mabs for therapeutic use as the column is commonly re-used. A cleaning-in-place (CIP) step is often integrated into the purification process cycle in order to remove contaminants such as proteins, lipids, nucleic acids and microbes.1,4 However, some of the more stringent requirements for the CIP step remain to be addressed. Sodium hydroxide (NaOH) appears to be the most effective cleaning agent for use in the CIP step. NaOH is able to remove tightly bound impurities and inactivate microorganisms at low cost.4 Thus, it is advantageous to use matrices that can tolerate high pH levels.
Among the various proteinous ligands, protein A is relatively resistant to alkaline conditions but cannot resist highly alkaline conditions (>0.1M NaOH).1 Therefore, protein engineering methods were investigated toward increasing the alkaline stability of its IgG-Fc binding domains. The Z domain is the most commonly used artificial protein A and has superior chemical stability to its native constructs.1,5 The Z domain is an engineered analog of the B domain originally developed for use in the affinity purification step of fusion protein production. The Z domain contains two amino acid substitutions relative to the B domain (A1V and G29A). The G29A mutation is the major contributor to its improved chemical stability due to modification of the alkali-susceptible Asn–Gly (at residues 28–29) sequence. The amino acid substitution to an asparagine residue is another popular modification to improve the chemical stability of proteins.1 Asparagine is known to be susceptible to high pH levels through deamidation or backbone cleavage.6–8 As these chemical reactions are dependent on hydroxide ions, the reaction rates are accelerated by increases in pH level. The asparagine modifications are highly dependent on protein sequence and conformation, and these modifications have been thoroughly investigated in the case of protein A.9,10
On the other hand, the C domain is already thought to have superior resistance to alkaline conditions compared to the other domains. One of the reasons for the high chemical stability of the C domain is speculated to be the presence of Thr-23 in place of the Asn-23 observed in the other domains. Experimental data obtained using the N23T mutant of the Z domain are reported in the above-mentioned study on asparagine modifications.9 To date, only a few experiments have been performed on the individual and native IgG-binding domains of protein A, although much effort has gone into studies on the protein engineering of protein A. In view of the considerable industrial significance of protein A as an affinity ligand for therapeutic antibodies, this study was conducted to examine the potential for increasing the alkaline stability of the C domain. First, we identified the alkaline cleavage sites in a recombinant protein A using a protein sequencer. Second, we investigated the thermostability of the B and C domains by Circular dichroism (CD) analysis. In addition, we investigated the potential applicability of the C domain, in which a number of mutations were introduced, as a scaffold protein. Finally, we evaluated various amino acid substitutions at the Gly-29 position of the C domain, as the Z domain is known to acquire alkaline resistance via a G29A mutation.
Results
Alkaline cleavage site analysis
In order to identify the cleavage sites of protein A under alkaline conditions, protein sequencing analysis was carried out after the exposure of protein A to a high pH solution (0.5M NaOH). The recombinant protein A (XM region deleted) was used to trace the position of the cleavage sites in the individual IgG-binding domains (E, D, A, B, and C) in a single experiment. In addition, we investigated the protease cleavage sites in protein A by exposure to CHO cell culture supernatant. CHO cells are universally used in the industrial manufacture of therapeutic antibodies.11,12 Therefore, resistance to proteases in CHO cell culture supernatant is also important feature for affinity ligands used for the capture of IgG.13
The recombinant protein A was incubated in an alkaline solution (0.5M NaOH) or CHO cell culture supernatant. SDS-PAGE was then performed on samples of the incubated mixtures to isolate fragments cleaved by the hydroxide ions under the alkaline conditions or in the presence of the proteases.
Figure 1 shows the SDS-PAGE profiles of protein A after the cleavage treatments. The recombinant protein A showed a well-purified single band pattern (approximately 30 kDa) before exposure to 0.05M NaOH or CHO cell supernatant, as shown in the analyte lane in Figure 1(A). In addition, the protein A was observed to be stable at 25°C for a long period. On the other hand, in the alkali treatment lane in Figure 1(A), the main band of protein A showed a slightly higher molecular mass after alkali treatment. Additionally, some bands were detected in the molecular weight range of 10 ∼ 25 kDa, which is lower than the molecular weight of protein A. The SDS-profiles of protein A after protease treatment are shown in Figure 1(B). Here, also, only the band derived from protein A was visible because the concentration of proteases included in the CHO supernatant was significantly lower than that of protein A. Although no shift in the main band of protein A was observed, as was the case after alkali treatment, some bands showing a lower molecular weight than that of protein A were detected. The isolated protein fragment bands of interest were analyzed by N-terminal protein sequencing. Cleavage was confirmed at the Gln–Ser sequence in the D domain (residues 13–14 and 43–44) under alkaline conditions and at the Gly–Glu sequence in the E and D domains (residues 44–45 in the E domain and 49–50 in the D domain) by protease treatment.
Figure 1.
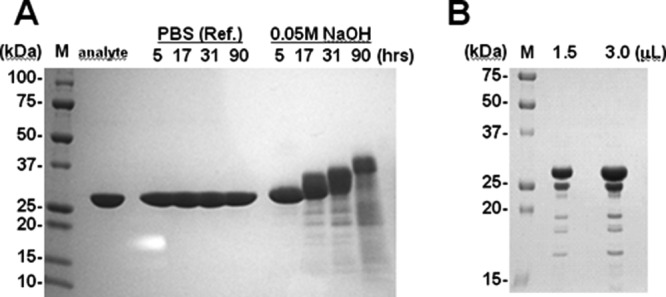
SDS-PAGE profiles of protein A after alkali (A) and CHO-protease (B) treatment. An analyte of protein A was used as the test substance. PBS (Ref.) and 0.05M NaOH were used for treatment, and the treatment period (in hours) is indicated. Loaded sample volumes of protein A after CHO-protease treatment are indicated in panel (B). The left lane of each panel is the molecular weight marker (M), and the molecular weight of each band is indicated. Each of the loaded samples is indicated at the top of each panel.
Figure 2 shows sequence alignment of the five IgG-binding domains and their cleavage sites. We added possible cleavage sites based on this experiment and alignment as the three-dimensional structure (topology) of each IgG-binding domain is thought to be basically the same. The C domain is the only one to have a Val–Ser sequence at residues 40–41 (residues 43–44 in the D domain).
Figure 2.
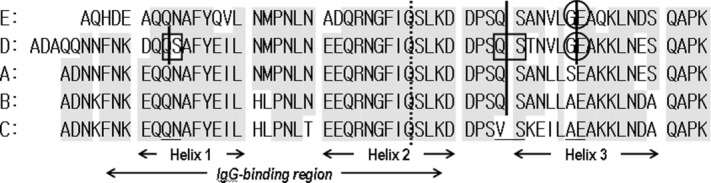
Analysis of the sensitivities of each IgG-binding domain to alkaline conditions and CHO protease. Sequence alignment of the IgG-binding domains of Protein A is shown and conserved residues are shaded. Cleavage sites under alkaline conditions, detected using a protein sequencer, are enclosed in squares. Cleavage sites when immersed in CHO supernatant, detected with a protein sequencer, are enclosed in circles. Vertical lines indicate possible cleavage sites based on this experiment.
Thermostability analysis by CD spectroscopy
The 58-residue (56 residues in the E domain and 61 residues in the D domain) IgG-binding domains of protein A form a three-helix bundle structure.14–16 The C domain had a far-UV CD spectrum with negative ellipticity at 208 and 222 nm, which is typical of alpha-helical proteins. The ellipticity at 222 nm allows us to follow the structural state of the C domain relative to increases in temperature simply by looking at the change in ellipticity. The CD spectra of the C domain were recorded at every five degrees from 25 to 85°C, as shown in Figure 3. As a reference, the CD spectra of the B domain, the standard domain among the protein A domains, were also recorded in the same manner. There appear to be few differences between the CD spectra obtained in these experiments and those from previous reports.9,17 As a result, the mid-point temperature of the thermal unfolding curve (Tm) was found to be 67.5 (±2)°C for the B domain, as shown in Figure 3. The Tm for the C domain is predicted to be approximately 72.5°C, although completely unfolded data points (more than 85°C) were not measured because of limitations with regard to the thermocirculator used. The C domain appears to have relatively higher conformational stability than does the B domain.
Figure 3.
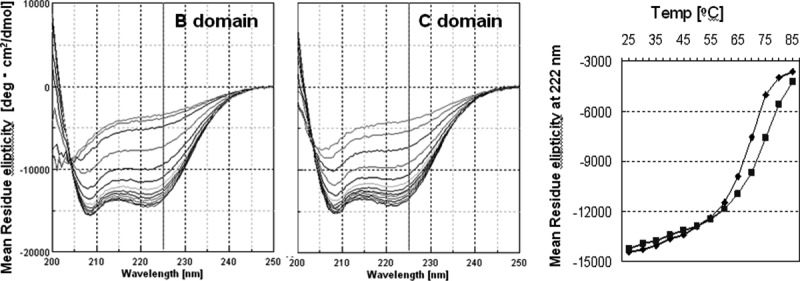
CD analysis of the thermostability of the B and C domains of protein A. Right spectra are superpositions of each CD spectra from 25 to 85°C of the B (left) or C (right) domains. The graph on the right shows plots of the CD spectra (at 222 nm) of the B (closed diamonds) or C (closed squares) domains.
BIACORE analysis of alkaline stability in relation to IgG-binding capacity
We carried out a BIACORE analysis to evaluate the residual IgG-binding capacity after alkali treatment. The BIACORE system is based on a surface plasmon resonance (SPR) method that allows measurement of biospecific interactions in real-time as changes in mass concentrations on a sensor surface. We monitored changes in the binding capacity of each purified recombinant protein for human IgG following exposure to NaOH. In this analysis, the protein concentrations before and after alkali treatment were adjusted to remain the same, while the concentration of a protein showing IgG-binding activity should change. As curve fitting using protein concentration as a variable is difficult, the concentration was considered to be constant in this curve fitting analysis. In this case, the concentration change in a protein showing IgG-binding activity is reflected in the parameter Rmax, which is the maximum binding capacity. Therefore, alkaline stability was evaluated as the value of Rmax after alkali exposure relative to that before alkali exposure [residual IgG-binding capacity (%)].
Protein samples comprising a single domain, the C domain (C-Wild), B domain (B-Wild), Z domain (B-A1V/G29A, Z-Wild), a G29A mutant of the C domain (C-G29A), and the same mutant of the B domain (B-G29A), were used. The substitution of Gly to Ala at residue 29 is the only acknowledged mutation to improve chemical stability.5,16 We evaluated the change over time of the alkaline stability of each protein as the residual IgG-binding capacity after exposure to 1.0M NaOH at room temperature, as shown in Figure 4. As alkali-exposure time progressed, the C domain appeared to show higher residual IgG-binding capacity than of that did the B domain in both the wild-type and G29A mutant. A G29A mutation should improve the alkaline stability of the B or C domain, thus it is reasonable that B-G29A showed almost the same residual IgG-binding capacity after alkali exposure as that of Z-Wild. C-wild showed a similar curve to Z-wild for residual IgG-binding capacity after alkali exposure over time. We performed a similar experiment using exposure to 0.5M NaOH at 30°C for 20 h. The change induced by the alkaline treatment can be used as a control of repeatability at a definite temperature and short time period, providing the facilities are maintained at a neutral pH. We confirmed that the residual IgG-binding capacity after long alkali-exposure of the C domain was significantly higher than that of the B domain, as shown in Figure 5. The data for C domain incubated for 25 h was added, and showed a residual IgG-binding capacity of about 30%. These data were used as controls for the following experiments performed under optimized alkali treatment conditions.
Figure 4.
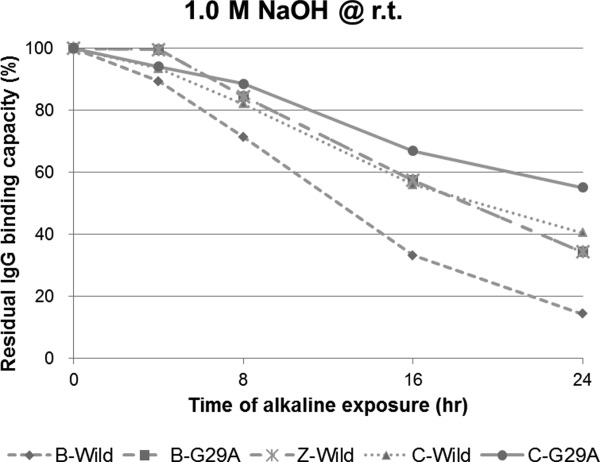
Comparison of the alkaline stability among “well-known” single domains of protein A. Residual binding capacity is ratio of the value after alkali treatment (1.0 M NaOH, 4, 8, 16, and 24 h) to the value before alkali treatment.
Figure 5.
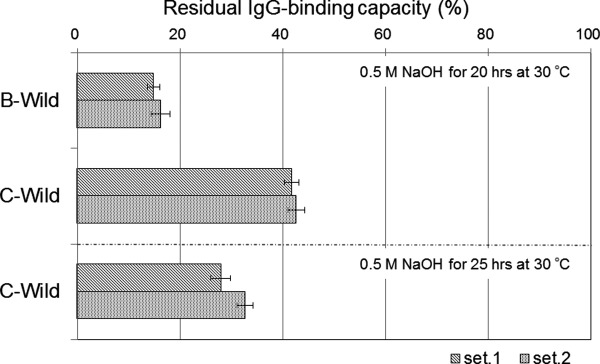
Comparison of the alkaline stability between B domain and C domain of protein A. Residual binding capacity is ratio of the value after alkali treatment (1.0M NaOH, 25 h, at 30°C) to the value before alkali treatment. Standard deviations of the mean (N = 3) in each set (set1, set2) are shown as error bars.
Mutational screening study using the BIACORE system
Although the G29A mutation has been widely used in a large number of reports, a surprisingly limited amount of information exists on other amino acid substitutions at the same position. We also evaluated various amino acid substitutions at the Gly-29 position of the C domain by BIACORE analysis, as the Z domain is known to acquire alkaline resistance via a G29A mutation. We prepared thirteen variants in which amino acid substitutions were introduced at the Gly-29 position of the C domain. Valine (V), leucine (L), isoleucine (I), methionine (M), serine (S), threonine (T), asparagine (D), gluthamine (E), histigine (H), arginine (R), phenylalanine (F), tyrosine (Y), and tryptophan (W) were selected as amino acid substitutions in place of the Al-Gly substitution at residue 29. Asparagine (N) and glutamine (Q) were not selected due to their high susceptibility to deamidation. Proline (P) was not selected in order to avoid destabilization of the α-helix structure.21 Cystein (C) and lysine (K) were also not selected as these amino acids are useful in the immobilization of the affinity ligand.22 Gly-29 is located near the IgG-Fc binding site of protein A so it is not suitable as a point of immobilization.
Following the preparation of the C domain Gly-29 mutants (single domain), the residual binding capacity after the alkali treatment (0.5M NaOH, 30°C, 25 h) of each mutant was evaluated using BIACORE. The affinity constants KA (M−1, ka/kd) of all the mutants were in the order of 107 M−1 (Supporting Information Table S3), which is similar to that of the single domain of protein A.17,19 Figure 6 shows a comparison of the residual IgG-binding capacities among the C domain Gly-29 mutants. This assay shows with reproducible results that the alkaline stability of C-G29A is higher than that of the C-Wild domain. It should be noted that some other amino acid substitutions to Gly-29 improve the alkaline stability more effectively than does G29A, with G29E, G29R, G29S, G29I, and G29L mutations all showing similar or larger increases in the alkaline stability of the C domain than that observed for G29A, while the results for G29Y, G29M, and G29W showed distinctively larger increases in the alkaline stability of the C domain than that observed for G29A.
Figure 6.
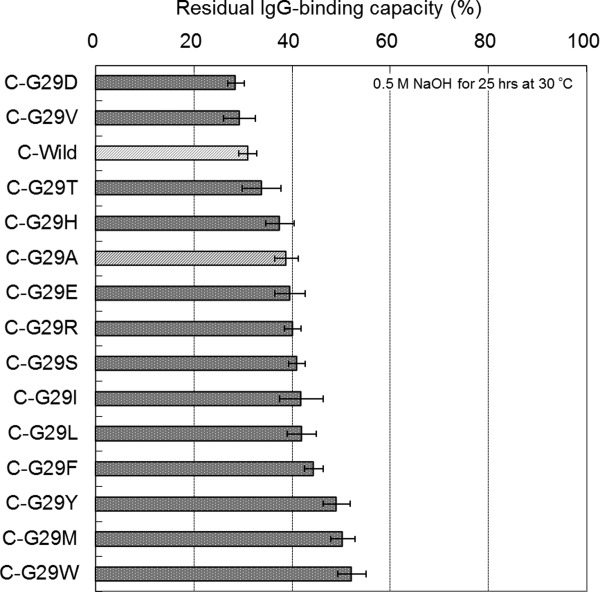
Comparison of the alkaline stability among the C domain mutants of protein A. Residual binding capacity is the ratio of the value after alkali treatment (0.5M NaOH, 25 h) to the value before alkali treatment. Standard deviations of the mean (N = 3) are shown as error bars.
In order to confirm rough trend for the effects of other amino acid substitutions at the Gly-29 position of the other domains of protein A, a number of B domain Gly-29 mutants (G29V, G29R, and G29W) were also prepared. The residual binding capacity after alkali treatment of each mutant was evaluated using BIACORE as described above, except that the incubation time of alkali treatment was changed from 25 to 20 h to optimize performance for the comparison of B domain mutants. Figure 7 shows a comparison of the residual binding capacities among B domain Gly-29 mutants. The G29V mutation showed a smaller increase in alkaline stability than that of G29A. Meanwhile, the G29W mutation showed a larger increase in alkaline stability than that of G29A.
Figure 7.
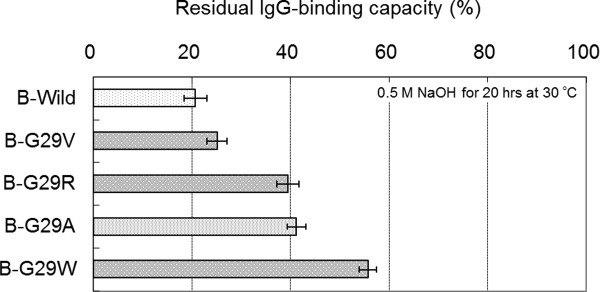
Comparison of the alkaline stability among the B domain mutants of protein A. Residual binding capacity is the ratio of the value after alkali treatment (0.5M NaOH, 20 h) to the value before alkali treatment. Standard deviations of the mean (N = 3) are shown as error bars.
Discussion
Properties of the C domain as an IgG-Fc capture ligand
It has been assumed that the C domain has superior resistance to alkaline conditions compared to the other domains. Thr-23, an amino acid found only in the C domain, is easily predicted to have a beneficial effect on alkaline stability, as experimental data showed that the introduction of a N23T mutation in the Z domain led to increased alkaline stability.9 However, only a few experiments have been performed on the C domain of protein A. This study was first conducted to examine the potential alkaline stability of the C domain. As a result of the cleavage site analysis of protein A, an unknown and inherent advantage was unexpectedly found. Val-40, one of the unique residues of the C domain, appears to have a positive effect on its alkaline stability. Previously, the only predictable feature in the sequence of the C domain was thought be Thr-23, as experimental data showed that the introduction of a N23T mutation in the Z domain led to increased alkaline stability.9 However, no cleavage sites were detected around residue 23 in any of the domains in this experiment. Val-40 might be another important residue characterizing the high alkaline stability of the C domain. The C domain also lacks a protease-susceptible Gly–Glu sequence. This feature is significant in that resistance to proteases is also required for industrial IgG-capture media used with CHO cell culture supernatants.4 It is conceivable that the C domain is, therefore, suitable as a scaffold for an affinity ligand for industrial IgG-capture media.
Studies on the stability of protein A have mainly focused on the “engineered” domains of protein A, particularly the Z domain mutants.9 Hence, few studies have centered on the stability of the domains, particularly the “native” domains, of protein A. CD analysis indicated that the C domain has relatively higher conformational stability than does the B domain. For example, high thermostability should play a role in high-temperature cleaning for the CIP process in the manufacture of Mabs. In a previous report, a computer simulation comparing the native fold with the mirror image fold indicated a unique feature in the native fold of the C domain18 The simulation pointed to the possibility that a substitution of Thr-23 in the C domain (numbered Thr-24 in that report) for Asn-23 in the other domains is responsible for differences in the energetic features between the C and other domains. Thr-23 may be responsible for the improved thermostability of the C domain as it constitutes the N-cap of helix II.
BIACORE analysis provided new insight into the residual IgG-binding capacity of the native domains (B and C domain) of protein A after alkali treatment. As shown in Figure 5, the alkaline stability of the C domain was significantly higher than that of the B domain. The C domain appears to have almost the same alkaline stability as the Z domain, which gains its improved alkaline stability via a G29A mutation, which is not inconsistent similar alkaline stabilities demonstrated by the mutant B-G29A and Z domains. Hence, it is reasonable to speculate that the alkaline stability of the C-G29A mutant is higher than that of the Z domain. As for alkaline stability in relation to the IgG-binding capacity of the domains, the significant positive effect of Thr-23 in the C domain, reported previously,9 was confirmed in our study (Supporting Information, Figure S1).
Remarkable alkaline stability of C domain having only one amino acid substitution
BIACORE analysis allows the efficient evaluation of improvements in the alkaline stability of a protein by mutation. The methodology developed using BIACORE enables us to evaluate many mutations simultaneously, allowing us to perform systematic mutational screening studies. We, therefore, focused on other amino acid substitutions at the Gly-29 position of the C domain. An amino acid substitution of Gly-29 had been made in order to avoid the Asn–Gly sequence, which is the most sensitive alkali cleavage site.5 The mutation to Asn-28 has been avoided in the past as the asparagine side chain of Asn-28 plays a role in interactions with the IgG-Fc region.19,20 In contrast, Gly-29 is not involved in the interactions with IgG-Fc,5 although it is conserved in all protein A domains, as shown in Figure 2. Nilson, who produced the Z domain, reported that the reason for selecting alanine as the amino acid substitution candidate was that he wished to make only minimal changes to the structure so that a mutation to an even larger side chain was not considered.5 Although this sounds reasonable, we decided that other amino acid substitutions to Gly-29 were still worth confirming experimentally.
A number of other amino acid mutations at the same position increased the alkaline resistance more than did the G29A mutation, as shown in Figure 6. The relative relationship in the effects of the mutations in improving the alkaline stability of the B domain was almost the same as that for the C domain, as shown in Figure 7. Previously, there have been reports that the rate of deamidation is affected by the size of the amino acid side-chain on the C-terminal side of the Asn residue at neutral to alkaline pH levels.23 Certainly, in our results, larger side-chain amino acid substitutions tended to afford greater improvements in the alkaline stability of the C domain. Of course, it is hard to explain everything based on this trend as the secondary and tertiary structures in proteins also play an important role in the rate of deamidation.10 For example, the substitution of Val for Ala leads to a decrease in the alkaline stability although the size of the Val side-chain is larger than that of Ala. This substitution may cause destabilization of the α-helix due to its β-branched side-chain.24 In terms of the effectiveness of Gly-29 substitution, the substitutions made in the C domain were not as effective as those in the B domain. It is conceivable, therefore, that the inherent high alkaline stability of the C domain allows relatively little improvement via the mutations in comparison to the B domain.
Additionally, the acidic stabilities of the representative variant a number of C domain Gly-29 mutants were confirmed (Supporting Information Figure S2). It is also important to maintain the acidic tolerance of these mutants with wild-type Protein A domains, because acidic buffers are normally used to elute bound IgG from the Protein A affinity column.1,3 Basically, mutations to Gly-29 appear to have little effect on acidic tolerance.
Since the mutational effects on the alkaline stability of proteins are closely connected with related factors, our mutational screening study was useful as a protein engineering technique for the improvement of alkaline stability. As a result, we found a number of novel variants of the C domain that have higher alkaline stability than that of the G29A variant of the C domain at a single domain level. Our study indicates that even a single mutation in the C domain of protein A results in a further significant improvement in alkaline stability.
Conclusions
The evidence presented in this report demonstrates the inherent advantage of the C domain in terms of alkaline stability. Our data also include the following novel findings: its unique sequence affords resistance to alkaline cleavage as well as the increased high thermostability of proteins. Furthermore, our mutational study using BIACORE indicates that even a single mutation in the C domain increases its alkaline stability. Surprisingly, we also found that some other amino acid mutations at the Gly-29 position increase alkaline resistance more than does G29A. The results of this study emphasise that a combination of a broad investigation of the C domain and an effective protein engineering approach via mutational screening is extremely useful from an industrial point of view.
In this article, we focused on improving the alkaline stability of the protein A ligand, while we have already conducted studies to improve the elution properties of the protein A ligand by introducing other mutations to the C domain.25 The IgG affinity matrix, which combines the superior properties of alkaline stability and IgG elution, should afford significant advantages for the industrial-scale purification of monoclonal antibodies.
Materials and Methods
DNA construction
The coding DNA sequence for each domain (B or C) of Staphylococcus aureus protein A was designed by reverse translation of its amino acid sequence. The dsDNA of each domain was prepared by a polymerase chain reaction (PCR) technique using synthetic oligonucleotides (SIGMA genosys). Each dsDNA fragment was restricted with BamHI and EcoRI (Takara), and inserted into a vector pGEX-6P (GE Healthcare) that had been restricted with the same enzymes. Expression plasmids encoding the C-domain mutants, in which the Gly-29 was replaced with Val, Leu, Ile, Tyr, Phe, Thr, Trp, Ser, Asp, Glu, Arg, His, or Met, were prepared using a QuickChange site-directed mutagenesis kit (Stratagene) according to the protocol recommended by the supplier using Pfu Turbo DNA polymerase and the methylated DNA (template DNA) cleavage enzyme DpnI (both available from Stratagene). For example, the expression plasmid for a G29V mutant of the C domain was prepared by a PCR reaction using two primers (5′-CAACGTAACGTGTTCATCCAAAG-3′ and 5′-CTTTGGATGAACACGTTACGTTG-3′) and the template plasmid of the wild-type C domain. Likewise, expression plasmids encoding the B-domain mutants, in which the Gly-29 was replaced with Val, Arg, or Trp, were produced using the wild-type B domain expression plasmid as a template. The expression plasmids for a tandem repeat of the single domain were prepared by the re-insertion of another dsDNA fragment into the expression plasmid for a single domain with the same sequence. The dsDNA was prepared by PCR methods using one primer with a NarI (TOYOBO) site and one with an EcoRI site. This NarI site is located in the coding DNA at around residue 55–57 of the 58 residues comprising the domain (B or C). This dsDNA fragment, restricted with NarI and EcoRI, is capable of insertion into the expression plasmid for a single domain restricted with NarI and EcoRI.
Protein expression and purification
Protein expression was carried out by transformation of each expression plasmid into E. coli HB101 (Takara). Proteins are expressed as glutathione S-transferase (GST) fusion proteins using the vector pGEX-6P. Fermentation was carried out in Sakaguchi flasks at 37°C in LB medium containing 50 µg/mL ampicillin. Protein expression was induced by 0.1M IPTG, and cells were harvested by centrifugation. Cell pastes were suspended in PBS (pH 7.4) containing 0.5 mM EDTA, and the suspended cells were disrupted using a probe sonicator. The supernatant separated from the sonicated mixtures by centrifugation was applied to a GSTrap column (GE Healthcare) equilibrated with 20 mM sodium phosphate (pH 7.4) containing 0.15M NaCl. After washing in equilibrating buffer, the target protein was eluted with 50 mM Tris–HCl (pH 8.0) containing 20 mM Glutathione. The GST-tag was cleaved by the addition of PreScission Protease (GE Healthcare) to the eluted fraction. The target protein was separated from the GST-tag through Gel-filtration chromatography using Superdex 75 (GE Healthcare). The purity of each protein sample was confirmed by SDS-PAGE analysis. Concentrations of the purified proteins were determined by absorbance measurements at 280 nm using the absorbance coefficients calculated with the ProtParam tool on the ExPASy Proteomics Server: http://expasy.org/.
Cleavage site analysis
Industrial samples of the recombinant protein A (XM region deleted) and CHO culture supernatant (CHO-K1, CD-DG44 medium) were produced in-house. In the alkaline cleavage site analysis, protein A solutions (40 mg/mL pure water) were diluted 1:20 in 0.05M NaOH, and incubated at 25°C for 5, 17, 31, and 90 h. SDS-PAGE was carried out on reaction mixture samples after neutralization. For the protease cleavage site analysis, protein A solutions were diluted 1:10 in CHO culture supernatant containing 25 mM citrate (pH 5.0), and incubated at 37°C for 64 h. Reaction mixture samples were loaded directly onto the SDS-PAGE gel. Isolated cleaved fragments of protein A were blotted onto an Immobilon-PSQ membrane (Millipore) in a blotting buffer. Protein bands indicating cleaved fragments in the membrane were excised, and the N-terminus of the protein was sequenced with a PPSQ-33A protein sequencer (Shimazu).
Circular dichroism (CD) analysis
CD spectra were recorded on a JASCO J-805 instrument (JASCO) using a 1-cm pathlength cell. Protein solutions (C and B domain) were added to 20 mM sodium phosphate (pH 7.4) to give a final concentration of 5 µM. The scans were recorded at 0.5-nm intervals and at a rate of 50 nm/min (in the range of 200–250 nm), with a sensitivity of 100 millidegrees and a response time of 1 s. Spectra were recorded at every five degrees from 25 to 85°C. CD spectra were then converted to mean residue molar ellipticities.
BIACORE analysis on alkaline stabilities
We monitored changes in the binding capacity of each purified recombinant protein for human IgG following exposure to NaOH using a BIACORE 3000 instrument (GE Healthcare). Human IgG (Gammagard, Baxter) was immobilized by amine coupling on the carboxylated dextran surface of a CM5 sensor chip (GE Healthcare) using N-hydroxysuccinimide (NHS) and N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide (EDC) chemistry according to the manufacturer's protocols. A reference lane was chemically blocked with ethanolamine immediately after activation with NHS/EDC. A 1 mg/mL human IgG solution was prepared by dilution in 20 mM sodium phosphate (pH 7.4) containing 0.15M NaCl. The IgG solution was then further diluted in 10 mM sodium acetate (pH 4.5) prior to use in this immobilization process. To analysis IgG-binding affinity, solutions of three different protein concentrations (of 10 to 1000 nM) were prepared for each protein using a running buffer (20 mM NaH2PO4-Na2HPO4, 150 mM NaCl, 0.005% P-20, pH 7.4), and each protein solution was added onto the sensor chip. To analyze their alkaline stability, each protein, adjusted to a common concentration (e.g., 26.2 µM), was mixed with a specific amount of 0. 625M NaOH to a final concentration of 0.5M (or 1.0M) and incubated for a specific time at 30°C (or r.t.). Subsequently, 0.5M HCl was added to each mixture to neutralize the solution. The solution was further diluted 1:1 in a running buffer (20 mM sodium phosphate, 0.15M NaCl, 0.005% P-20, pH 7.4). The protein solutions were also prepared in the same manner prior to alkali treatment. Each of the protein solutions before and after alkali treatment was applied to the sensor chip at a flow rate 20 µL/min, and 50 mM NaOH was used to regenerate the surface. The data was analyzed using BIA evaluation software. The sensorgrams of the control lane were subtracted from the sensorgrams of the IgG-immobilized lane. A 1:1 Langmuir model was used to calculate apparent values of the association rate constant ka (M−1 s−1), the dissociation rate constant kd (s−1), and the affinity constant KA (M−1, ka/kd). The global fitting method was used in the confirmation of the affinity constant of each mutant to IgG, while the local fitting method was used in the analysis of its alkaline stability. For the analysis of alkaline stability, the concentration was set to remain constant before and after treatment in the fitting analysis. The alkali stability was evaluated by calculating, for each mutant, the value of the Rmax after the alkali treatment relative to the Rmax before the alkali treatment [residual IgG binding activity (%)].
Glossary
- Mabs
monoclonal antibodies
- CIP
cleaning-in-place
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Hober S, Nord K, Linhult M. Protein A chromatography for antibody purification. J Chromatogr B. 2007;848:40–47. doi: 10.1016/j.jchromb.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 2.Uhlen M, Guss B, Nilsson B, Gatenbeck S, Philipson L, Lindberg M. Complete sequence of the staphylococcal gene encoding protein A. A gene evolved through multiple duplications. J Biol Chem. 1984;259:1695–1702. [PubMed] [Google Scholar]
- 3.Shukla AA, Hubbard B, Tressel T, Guhan S, Low D. Downstream processing of monoclonal antibodies: application of platform approaches. J Chromatogr B. 2007;848:28–39. doi: 10.1016/j.jchromb.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 4.Hahn R, Shimahara K, Steindl F, Jungbauer A. Comparison of protein A affinity sorbents III. Life time study. J Chromatogr A. 2006;1102:224–231. doi: 10.1016/j.chroma.2005.10.083. [DOI] [PubMed] [Google Scholar]
- 5.Nilsson B, Moks T, Jansson B, Abrahmsen L, Elmblad A, Holmgren E, Henrichson C, Jones TA, Uhlen M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987;1:107–113. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- 6.Bhatt NP, Patel K, Borchardt RT. Chemical pathways of peptide degradation. I. Deamidation of adrenocorticotropic hormone. Pharm Res. 1990;7:593–599. doi: 10.1023/a:1015862026539. [DOI] [PubMed] [Google Scholar]
- 7.Patel K, Borchardt RT. Chemical pathways of peptide fegradation. II. Kinetics of deamidation of asparaginyl residue in a model hexapeptide. Pharm Res. 1990;7:703–711. doi: 10.1023/a:1015807303766. [DOI] [PubMed] [Google Scholar]
- 8.Patel K, Borchardt RT. Chemical pathways of peptide fegradation. III. Effect of primary sequence on the pathways of deamidation of asparaginyl residues in hexapeptides. Pharm Res. 1990;7:787–793. doi: 10.1023/a:1015999012852. [DOI] [PubMed] [Google Scholar]
- 9.Linhult M, Gulich S, Graslund T, Simon A, Karlsson M, Sjoberg A, Nord K, Hober S. Improving the tolerance of a protein A analogue to repeated alkaline exposures using a bypass mutagenesis approach. Proteins. 2004;55:407–416. doi: 10.1002/prot.10616. [DOI] [PubMed] [Google Scholar]
- 10.Palmer B, Angus K, Taylor L, Warwicker J, Derrick JP. Design of stability at extreme alkaline pH in streptococcal protein G. J Biotechnol. 2008;134:222–230. doi: 10.1016/j.jbiotec.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Chadd HE, Chamow SM. Therapeutic antibody expression technology. Curr Opin Biotechnol. 2001;12:188–194. doi: 10.1016/s0958-1669(00)00198-1. [DOI] [PubMed] [Google Scholar]
- 12.Omasa T, Onitsuka M, Kim WD. Cell engineering and cultivation of Chinese hamster ovary (CHO) cells. Curr Pharm Biotechnol. 2010;11:233–240. doi: 10.2174/138920110791111960. [DOI] [PubMed] [Google Scholar]
- 13.Sandberg H, Lutkemeyer D, Kuprin S, Wrangel M, Almstedt A, Persson P, Ek V, Mikaelsson M. Mapping and partial characterization of proteases expressed by a CHO production cell line. Biotechnol Bioeng. 2006;95:961–971. doi: 10.1002/bit.21057. [DOI] [PubMed] [Google Scholar]
- 14.Gouda H, Torigoe H, Saito A, Sato M, Arata Y, Shimada I. Three-dimensional solution structure of the B domain of staphylococcal protein A: comparisons of the solution and crystal structures. Biochemistry. 1992;31:9665–9672. doi: 10.1021/bi00155a020. [DOI] [PubMed] [Google Scholar]
- 15.Starovasnik MA, Skelton NJ, O'Connell MP, Kelley RF, Reilly D, Fairbrother WJ. Solution structure of the E-domain of staphylococcal protein A. Biochemistry. 1996;35:15558–15569. doi: 10.1021/bi961409x. [DOI] [PubMed] [Google Scholar]
- 16.Tashiro M, Tejero R, Zimmerman DE, Celda B, Nilsson B, Montelione GT. High-resolution solution NMR structure of the Z domain of staphylococcal protein A. J Mol Biol. 1997;272:573–590. doi: 10.1006/jmbi.1997.1265. [DOI] [PubMed] [Google Scholar]
- 17.Gulich S, Uhlen M, Hober S. Protein engineering of an IgG-binding domain allows milder elution conditions during affinity chromatography. J Biotechnol. 2000;76:233–244. doi: 10.1016/s0168-1656(99)00197-2. [DOI] [PubMed] [Google Scholar]
- 18.Olszewski KA, Kolinski A, Skolnick J. Folding simulations and computer redesign of protein A three-helix bundle motifs. Proteins. 1996;25:286–299. doi: 10.1002/(SICI)1097-0134(199607)25:3<286::AID-PROT2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 19.Jendeberg L, Persson B, Andersson R, Karlsson R, Uhlen M, Nilsson B. Kinetic analysis of the interaction between protein A domain variants and human Fc using plasmon resonance detection. J Mol Recognit. 1995;8:270–278. doi: 10.1002/jmr.300080405. [DOI] [PubMed] [Google Scholar]
- 20.Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry. 1981;20:2361–2370. [PubMed] [Google Scholar]
- 21.MacArthur MW, Thornton JM. Influence of proline residues on protein conformation. J Mol Biol. 1991;218:397–412. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 22.Wong LS, Khan F, Micklefield J. Selective covalent protein immobilization: strategies and applications. Chem Rev. 2009;109:4025–4053. doi: 10.1021/cr8004668. [DOI] [PubMed] [Google Scholar]
- 23.Tyler-Cross R, Schirch V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J Biol Chem. 1991;266:22549–22556. [PubMed] [Google Scholar]
- 24.Cornish VW, Kaplan MI, Veenstra DL, Kollman PA, Schultz PG. Stabilizing and destabilizing effects of placing .beta.-branched amino acids in protein .alpha.-Helixes. Biochemistry. 1994;33:12022–12031. doi: 10.1021/bi00206a003. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida S, Murata D, Taira S, Iguchi K, Takano M, Nakano Y, Minakuchi K. Rational design and engineering of protein A to obtain the controlled elution profile in monoclonal antibody purification. Chem-Bio Informatics J. 2012;12:1–13. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.