

Abstract
Designed ankyrin repeat proteins (DARPins) are well-established binding molecules based on a highly stable nonantibody scaffold. Building on 13 crystal structures of DARPin-target complexes and stability measurements of DARPin mutants, we have generated a new DARPin library containing an extended randomized surface. To counteract the enrichment of unspecific hydrophobic binders during selections against difficult targets containing hydrophobic surfaces such as membrane proteins, the frequency of apolar residues at diversified positions was drastically reduced and substituted by an increased number of tyrosines. Ribosome display selections against two human caspases and membrane transporter AcrB yielded highly enriched pools of unique and strong DARPin binders which were mainly monomeric. We noted a prominent enrichment of tryptophan residues during binder selections. A crystal structure of a representative of this library in complex with caspase-7 visualizes the key roles of both tryptophans and tyrosines in providing target contacts. These aromatic and polar side chains thus substitute the apolar residues valine, leucine, isoleucine, methionine, and phenylalanine of the original DARPins. Our work describes biophysical and structural analyses required to extend existing binder scaffolds and simplifies an existing protocol for the assembly of highly diverse synthetic binder libraries.
Keywords: DARPins, synthetic binder libraries, library assembly, structure-based protein engineering, protein-protein interactions, in vitro selection, ribosome display
Introduction
Protein-protein interactions play an essential role in every living organism. B cells, for example, are a major component of the adaptive immune system and generate specific antibodies to combat invading pathogens and viruses. Starting in the 1970s, scientists have acquired technologies enabling the selection of monoclonal antibodies against desired protein targets initially using the hybridoma technology which relies on the immunization of mice.1 Over the years, selection methods have been expanded to in vitro approaches that became available with the development of phage display2 and ribosome display.3 Thereby, the binder selection process was transferred from animals into the test tube where experimental conditions can be controlled.
In vitro display methods couple the folded binding protein (phenotype) physically to the genetic information (genotype). This is achieved by either fusing binders to phage surface proteins in phage display or by forming stable ternary complexes of mRNA, ribosome, and the nascent polypeptide chain in ribosome display. Large combinatorial gene libraries typically encoding for 109 to 1012 possible binder molecules are the centerpiece of every in vitro selection technique. With the exception of binders based on small peptides (e.g. bicyclic peptides4), binding protein libraries are based on binding scaffolds. Traditionally, antibody fragments such as Fabs5 and scFvs6 served as scaffold which were either cloned from natural sources to form naïve libraries7 or constructed in vitro by genetic engineering into synthetic libraries.8 To overcome some of the undesirable biophysical properties of antibodies such as disulfide bonds, protein aggregation, and low expression yields, novel scaffolds have been exploited to introduce randomized surfaces (reviewed in Ref.9–11). Among the nonantibody scaffolds, designed ankyrin repeat proteins (DARPins) belong to the best characterized molecules.12 Naturally occurring ankyrin repeat proteins consist of tandem arrays of ankyrin repeat modules and mediate protein-protein interactions mainly in eukaryotic cells.13 Each repeat module consists of 33 amino acids and folds into a β-turn followed by two anti-parallel α-helices and a loop that connects to the next repeat. Ankyrin repeat modules assemble into elongated and slightly curved structures thereby shaping a concave surface that forms specific contacts with binding partners.14 The original DARPin library was constructed following a consensus design approach, in which naturally variable amino acid positions were subjected to randomization and conserved residues were defined as invariant framework residues.15 Typically, DARPins consist of two to three internal repeats that are flanked by an N- and a C-terminal capping repeat (N- and C-cap) which are then called N2C and N3C DARPins, respectively. DARPins are thermodynamically very stable due to tight and regular packing of the repeat segments and conserved hydrogen bonding networks.16,17 DARPins have been raised against a plethora of target proteins using ribosome and phage display.3,18–25 Of note, several successful in vitro selections of DARPins against integral membrane proteins have been reported, underscoring the suitability of the scaffold to target “difficult to handle” proteins.26–30
Target specific binders can be used in a multitude of applications: they carry drugs to the desired target in the body (e.g. in tumor targeting19,23,31), serve as tools in enzymology to inhibit kinases and proteases in the reducing milieu of the cell interior,22,32,33 are used as affinity probes to discover novel conformational states of membrane proteins,29 and have been shown to modulate the function of membrane transport proteins in vivo.28,30
In the context of membrane protein crystallography, the use of binders as crystallization aids has raised particular interest.28,34,35 Although a high-resolution structure of the membrane protein AcrB in complex with DARPins has been reported,28,36 the selection of such binders against membrane proteins often turned out to be difficult.26,29 A recurrent problem is the enrichment of hydrophobic binders that are prone to form higher oligomeric species and unspecifically bind to transmembrane proteins.29,30
Here we describe the design, construction, characterization, and successful application of a second-generation DARPin library that was built based on the information provided by the many target protein-DARPin complex structures obtained in the past decade. We analyzed the DARPin binding interfaces of all available structures to modify and extend the randomized surface. Regarding the framework, we reduced the surface entropy of the DARPin backside to be favorable for forming crystal contacts. To minimize problems in enriching unspecifically aggregating binders due to hydrophobic surface patches, apolar residues were omitted in the randomized positions. To compensate for the reduced hydrophobicity, the library is enriched in tyrosines that have been demonstrated to be particularly suited to mediate molecular contacts in protein-protein interactions.37 The library was validated in test selections against caspase-3, caspase-7, and AcrB and a crystal structure of a new DARPin in complex with caspase-7 was solved at 1.7 Å resolution.
Results
Analysis of the randomized DARPin surface based on crystal structures
Since the construction of the original DARPin library by Binz et al.15 [Fig. 1(A)], 18 crystal structures of DARPins in complex with their target proteins have been reported.12,21,22,28,36,38–46 We analyzed all DARPin-target protein interfaces of crystals solved at resolutions of 3 Å and better using the EPPIC server (http://www.eppic-web.org)47 (Supporting Information File “DARPin_interfaces.xls”). Twenty-two interfaces found in 13 crystal structures were taken into account for analysis (some interfaces were present twice in the asymmetric unit). The buried surface area (BSA) upon complex formation of every amino acid residue position was calculated as percentage of total BSA [Fig. 2(A), Supporting Information File “DARPin_interfaces.xls”)]. As intended by design, the majority (56 % of total BSA) of protein-protein interactions were mediated by randomized residues located at internal repeats. Interactions involving residues of the third repeat appear underrepresented because our dataset contained numerous N2C DARPins lacking a third repeat. The randomized residue at position 33 according to the ankyrin repeat sequence defined by Binz et al.15 [Fig. 1(B)] is only marginally involved in target contacts, because it points away from the concave randomized surface of the DARPin scaffold. In contrast, the aspartate of the DxxG motif (position 1 of the ankyrin repeat) and a leucine in the first α-helix of the ankyrin repeat (position 10) were found to be involved in target contacts, although these residues were not diversified in the original DARPin library [Fig. 2(A)]. Interestingly, invariant residues of the N- and C-cap were frequently involved in molecular contacts with target proteins providing 44% of total BSA. Our analysis further allowed us to plot the burial of each of the 20 amino acids as percentage of total BSA and to specifically highlight contacts mediated by diversified residues [Fig. 2(B)]. Major contacting residues (threshold 4% of total BSA) are D, K, R, N, L, I, M, F, Y, and W. Limiting this analysis to the diversified residues, the major players are L, I, F, Y, and W. Cysteines which are absent in DARPins and prolines which are completely buried in folded DARPins are not involved in target contacts. In contrast to a recent analysis of DARPin interfaces in which the number of tyrosines involved in target contacts were counted under the condition that burial is greater than 1 Å2,48 our quantitative analysis did not assign a special role to Y relative to L, I, F, and W. Furthermore, we found nonrandomized D, K, R, and N residues of the capping repeats to be frequently involved in target contacts.
Figure 1.
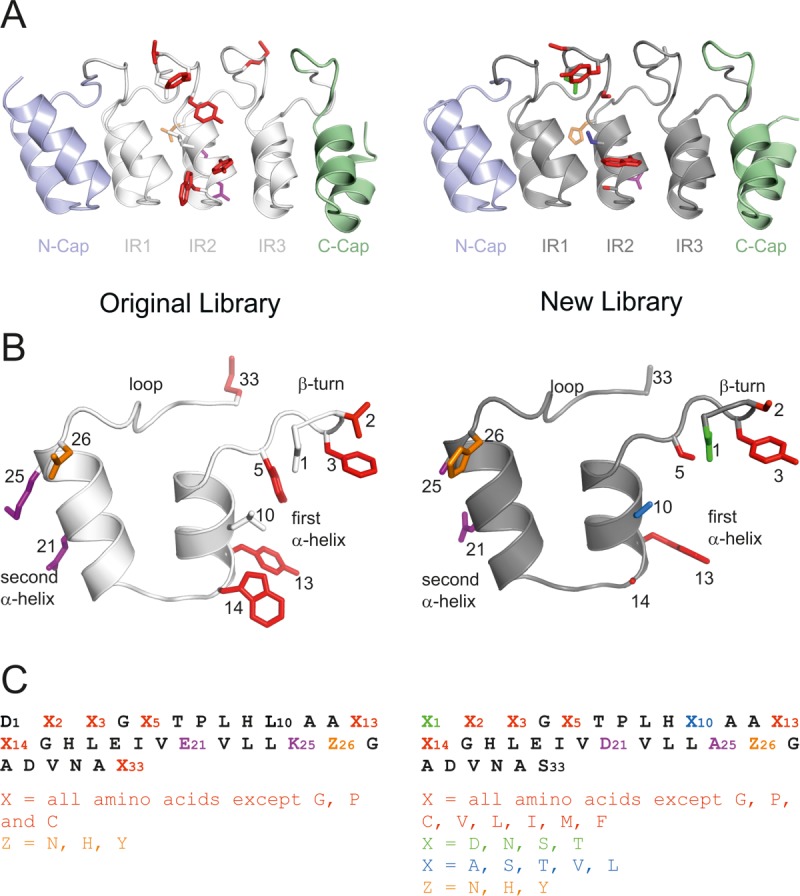
Randomization of the internal DARPin repeat. The second internal repeat (IR2) of the original (off7, left panel, PDB 1SVX) and new (C7_16, right panel, PDB 4JB8) library is shown in the context of the N3C DARPin (A) and in isolation (B). Diversified positions are highlighted as colored side chains shown as sticks. The repeat sequences and the permitted residues of the randomized positions are given in (C).
Figure 2.
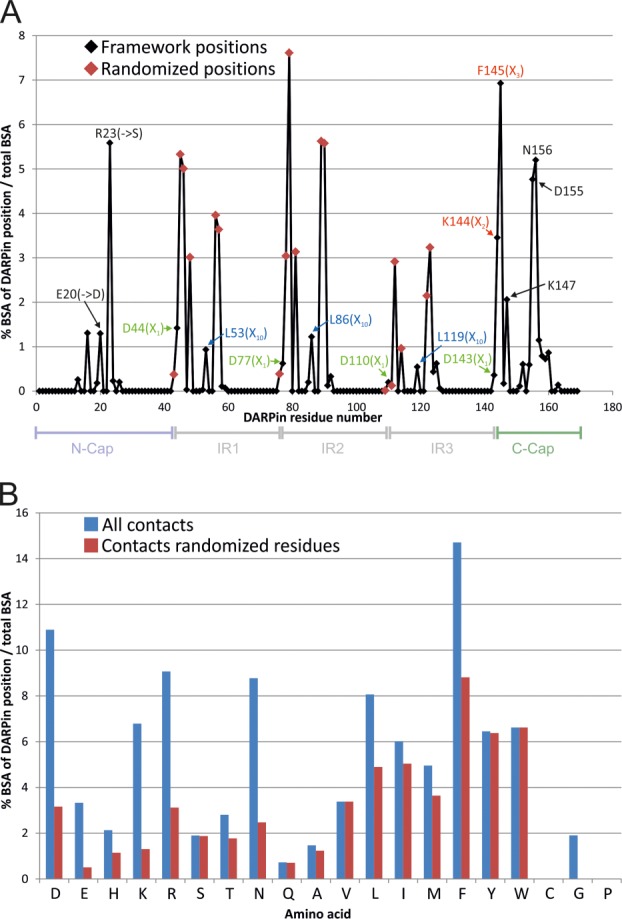
Analysis of target contacts by original DARPins based on 13 crystal structures. (A) The relative contribution of each DARPin sequence position to the total buried surface area (BSA) of all analyzed DARPins is plotted (i.e. the sum of all positions equals 100%). Randomized positions of the original library are indicated by red diamonds. Residues which were subjected to changes or randomization in the new library are highlighted using the same color scheme as in Figure 1. (B) Amino acid usage of the analyzed DARPins is visualized as % BSA of total BSA. Blue bars depict all contacting residues (total BSA of 100%) and red bars depict diversified residues only (total BSA of 56%).
Randomization scheme for the internal ankyrin repeat module of the new DARPin library
Taking these findings into account, the randomization pattern of the internal repeat defined by Binz et al. was modified. As in the original DARPin library, the ankyrin repeat positions 2, 3, 5, 13, and 14 were subjected to randomization [Fig. 1(B)]. Randomized position 33 of the original library was rarely involved in protein interactions and thus defined as invariant serine. In addition, the aspartate at position 1 and the leucine at position 10 of the ankyrin repeat module were randomized. Because these two residues are important for the structural integrity of the DARPin framework,49 the randomization was restricted to a more narrow range of permitted amino acid residues (see sections below). By using trinucleotides phosphoramidites for the synthesis of oligonucleotides, the amino acid composition for each residue position was adjusted.50,51 In the original DARPin library, the randomized positions were permitted to contain any amino acid except P, G, and C.15 Small and polar/charged amino acids (A, D, E, H, K, N, Q, R, S, T) were overrepresented with 7% each compared with bulkier and hydrophobic residues (F, I, L, M, V, W, Y) with 4.3% each. During DARPin selections against membrane proteins we repeatedly observed the enrichment of hydrophobic binders containing multiple apolar residues in the randomized positions (not shown). Such DARPins were found to promiscuously bind to diverse transmembrane proteins and were prone to form soluble aggregates during DARPin expression and purification.29,30 For the new DARPin library, we therefore limited the number of permitted residues in the randomized positions and excluded, besides G, P, and C also the hydrophobic amino acids I, L, V, M, and F. With these measures, we aimed at generating more hydrophilic DARPins that are expected to be less prone to unspecific hydrophobic interactions. The composition of permitted amino acids in the five randomized positions 2, 3, 5, 13, and 14 was biased towards small and polar amino acids and tyrosine and contain the following amino acid composition: A, S, T, N, and Y; 12% each and D, E, R, K, Q, W, and H; 5.7% each.
Randomization of residue D1 in the β-turn
In the analyzed DARPin structures, the β-turn aspartate contributes 2.6% of total BSA [Fig. 2(A)] and in naturally occurring ankyrin repeat proteins, this position is not highly conserved.15 However, the carboxyl moiety of this residue stabilizes the β-turn of the DARPin scaffold by interacting via two hydrogen bonds [Fig. 3(C)].49 To warrant stability of the β-turn of the new DARPin library, only residues D, N, T, and S were permitted at position 1. N, T, and S are expected to establish hydrogen bonds similar to the ones formed by aspartate. To verify our assumption experimentally, the β-turn aspartate in the middle repeat of the previously characterized unselected DARPin E3_5 was mutated to serine (D77S). Although serine is shortened by one methylene group compared with the original aspartate, E3_5_D77S was found to be more stable in guanidinium hydrochloride (GdnHCl) unfolding experiments as compared with wild type E3_5 and the midpoint of unfolding (Dm) was increased by 0.5M GdnHCl [Fig. 3(B)]. E3_5_D77S was crystallized and its structure was solved at a resolution of 1.7 Å (Table I). A superimposition of E3_5_D77S with wild type E3_5 revealed only marginal structural changes at the β-turn of the middle repeat [Fig. 3(C)]. As expected, the hydroxyl group of S77 stabilizes the β-turn by virtue of two hydrogen bonds. Tyrosine at position 5 of the first internal repeat (Y48) fills the space that became vacated by the D to S mutation. As a consequence, the entire β-turn of the first repeat is shifted towards the middle repeat by 0.5 to 1 Å (measured at the Cα positions). The more intimate contacts between the first and the second internal repeat might explain the increased stability of the E3_5_D77S mutant compared with wild type E3_5.
Figure 3.
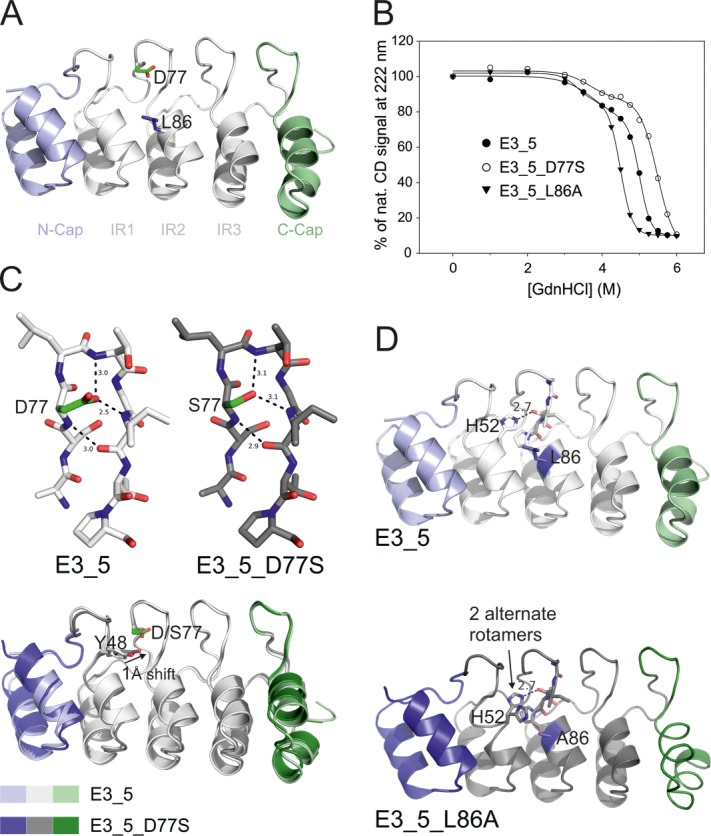
Biophysical and structural characterization of E3_5 mutants. (A) E3_5 was mutated at the second internal repeat at residues D77S or L86A representing randomized positions 1 and 10 in the new DARPin library. (B) The stability of the mutants was analyzed by GdnHCl equilibrium unfolding monitored by CD spectroscopy. Data were fitted to a biphasic sigmoidal equation and the midpoints of the second transition (Dm,2) were as follows: wild type, 5.01M; D77S mutant, 5.48M; L86A mutant, 4.49M. (C and D) Structures of the D77S and the L86A mutants were solved by X-ray crystallography. (C) S77 in the D77S mutant establishes two hydrogen bonds similar to the ones of the wild type E3_5 and thereby stabilizes the β-turn (top). A superimposition of both structures reveals a shift of Y48 toward the middle repeat in the D77S mutant (bottom). (D) In the structure of the L86A mutant, H52 of the first repeat adopts two alternate rotamer positions, one of which occupying the space vacated by the L to A mutation (lower panel). In wild-type E3_5, H52 stabilizes the DARPin fold by establishing a hydrogen bond with the internal repeat (upper panel).
Table I.
Data Collection and Refinement Statistics
| E3_5_D77S (PDB: 4J8Y) | E3_5_L86A (PDB: 4J7W) | C7_16-caspase-7 (PDB: 4JB8) | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 (19) | P212121 (19) | C2 (5) |
| Cell dimensions | |||
| a, b, c (Å) | 34.88; 53.63; 78.38 | 44.37; 78.59; 82.23 | 143.88; 52.51; 60.09 |
| α, β, γ (°) | 90.00; 90.00; 90.00 | 90.00; 90.00; 90.00 | 90.00; 96.92; 90.00 |
| Resolution (Å) | 50–1.7 (1.74–1.7) | 50–1.6 (1.64–1.6) | 50–1.7 (1.74–1.7) |
| Rmerge (%) | 6.0 (39.5) | 9.4 (32.6) | 4.0 (56.8) |
| I/σI | 22.21 (5.37) | 19.20 (11.87) | 23.21 (3.11) |
| Completeness (%) | 99.4 (99.4) | 98.9 (99.4) | 99.9 (99.9) |
| Redundancy | 5.6 (5.1) | 8.2 (8.8) | 6.8 (6.9) |
| Refinement | |||
| Resolution (Å) | 50-1.7 | 50-1.6 | 50-1.7 |
| No. reflections (work/test) | 15869/836 | 36343/1913 | 47187/1967 |
| Rwork/Rfree | 17.16/20.76 | 16.79/19.19 | 17.22/19.37 |
| No. atoms | |||
| Protein | 1192 | 2393 | 3107 |
| Water | 167 | 414 | 257 |
| B-factors | |||
| Total | 21.94 | 11.56 | 41.06 |
| R.M.S deviations | |||
| Bond lengths (Å) | 0.004 | 0.006 | 0.006 |
| Bond angles (°) | 0.874 | 1.033 | 1.008 |
Randomization of residue L10
The leucine residue at position 10 was kept invariant in the original DARPin library mainly due to stability considerations15 although it is not conserved in natural ankyrin repeat proteins. In DARPin-target protein co-crystal structures, L10 is frequently found to mediate molecular contact and contributes 2.7% of total BSA [Fig. 2(A)]. For this second-generation DARPin library, we therefore permitted the five residues A, S, T, V, and L at this position. To test for potential biochemical and structural consequences of randomizing this residue, L10 of the middle repeat of DARPin E3_5 was replaced by alanine (L86A) and analyzed by GdnHCl unfolding experiments. Dm of the E3_5_L86A mutant was decreased by 0.5M GdnHCl [Fig. 3(B)]. The crystal structure of the E3_5_L86A mutant was solved at a resolution of 1.6 Å (Table I) containing two E3_5_L86A molecules per asymmetric unit. In one DARPin of the asymmetric unit (chain B), the highly conserved histidine in position 9 of the first internal repeat (H52) adopts two rotamer conformations. One rotamer establishes a hydrogen bond with the consecutive middle repeat as in wild-type E3_5, while the second rotamer fills the space that was occupied by the bulky L86 in wild-type E3_5 [Fig. 3(D)]. As a consequence, the hydrogen bonding contact between the first and the second internal repeat segments appears to be weakened which might explain the decreased stability of the E3_5_L86A mutant. However, in the other E3_5_L86A molecule (chain A) of the asymmetric unit, H52 shows a position identical to that of wild type E3_5 (not shown).
Modification and randomization of the capping repeats
Two non-randomized and bulky side chains of the N-cap (E20 and R23) were frequently found to interact with the target protein and contribute to 1.3% and 5.6% of total BSA, respectively [Fig. 2(A)]. We chose not to randomize the N-cap, but to replace these two residues with smaller ones (E20D and R23S), in order to reduce sterical interference with DARPin binding [Fig. 4(A,C)]. Although the C-cap was not randomized in the original library, it matches the internal repeats with regard to surface burial upon complex formation [Fig. 2(A)]. Furthermore, the C-cap of the original library was found to be the least stable repeat of DARPins. C-cap packing to the internal repeat was stabilized employing combined computational and experimental methods.52 For this second generation DARPin library, we chose the sequence for an improved C-cap corresponding to the stabilized “mutant 5” described previously.52,53 In contrast to the original DARPin library, the β-turn of the C-cap was subjected to randomization using amino acid compositions identical to the ones of the internal repeats [Fig. 4(A,C)].
Figure 4.
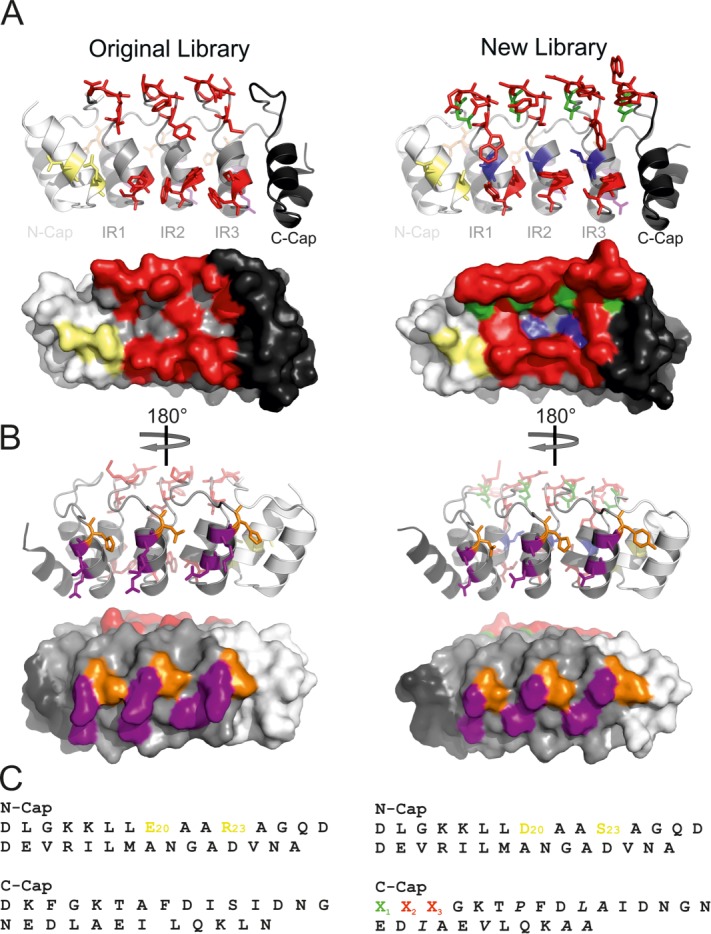
Modifications and randomizations at the capping repeats and surface entropy reduction of the DARPin backside. DARPins of the original (off7, left, PDB 1SVX) and new (C7_16, right, PDB 4JB8) library are shown in cartoon representation with diversified/modified residues highlighted as colored sticks or in surface representation. (A and C) Bulky E20 and R23 residues of the N-cap are frequently involved in target contacts [Fig. 2(A)] and were exchanged for the smaller D20 and S23 in the new library. The positions 1–3 of the C-cap (β-turn) were subjected to randomization in the new library following the same scheme as depicted in Figure 1(C). (B) DARPins viewed from their backside. By mutating E21D and K25A in each internal repeat [Fig. 1(C)], the surface entropy of the DARPin backside was reduced in the new library, which is expected to facilitate crystallization. (C) Sequences of the N- and C-caps are shown and modifications of the original library are highlighted.
Surface entropy reduction in the new DARPin library
Derewenda and colleagues have postulated that bulky charged side chains at the surface of proteins, in particular lysines and glutamates form an entropic barrier that counteracts the formation of crystal contacts.54 The original DARPin library has a glutamate at position 21 (E21) and a lysine at position 25 (K25) at the DARPin backside [Figs. 1(B,C) and 4(B)]. These residues were chosen to keep DARPins soluble. In addition, the lysine exhibits a high α-helical propensity,55 which appears important for the integrity of the DARPin scaffold. In the new library, we chose to replace E21 with a shorter aspartate (thereby maintaining the negative charge at this position) and K25 with an alanine, whose α-helical propensity is even higher than the one of lysine. To investigate whether the loss of these changes have any adverse influence on the oligomeric state of a N3C DARPin, we introduced these mutations in DARPin_55, a specific binder of the ABC transporter MsbA.29 Both wild-type and mutant DARPin_55 were found to be monomers as judged from size exclusion chromatography [Supporting Information Fig. S1(A)] and thus the loss of three positive charges due to the K25A mutations did not lead to the formation of DARPin oligomers in this test case. Mutant DARPin_55 was still co-migrating with MsbA on SEC (not shown). Equilibrium unfolding using GdnHCl revealed a stabilization of DARPin_55 upon introduction of these mutations [Supporting Information Fig. S1(A)].
Assembly and initial quality check of the new DARPin library
An N3C DARPin library containing three internal repeats flanked by a N- and a C-terminal cap was assembled as described previously for the original DARPin library.15 The caps and internal repeats were generated by PCR followed by ligating the fragments repeat by repeat using type IIS restriction enzymes (a detailed protocol is given in Materials and Methods). The theoretical library diversity of the N3C library is calculated as follows. Each of the three internal repeats contains five randomized positions with 12 permitted residues, position 1 with four allowed residues, position 10 with five permitted residues, and position 26 with three permitted residues. Each internal repeat therefore exhibits a theoretical diversity of 125 × 4 × 5 × 3 = 1.5 × 107 variants. The diversity of the C-cap is 576 owing to two randomized positions with 12 permitted residues (122 variants) and position 1 of the β-turn (four variants). The N-cap is not randomized. The theoretical diversity of the new N3C DARPin library is therefore 1.9 × 1024 and lies in a similar range as the original DARPin library, whose theoretical diversity amounted to 3.8 × 1023.15 The practical diversity of the library was estimated to be 1011 variants (see Materials and Methods). To evaluate the quality of the new library at the DNA level, the assembled library was cloned into an expression vector (pQE30) and 26 randomly picked library members were sequenced and named DARPin D1-D26. Twenty out of the 26 members were found to be in frame (77%), which indicates that the primers used to perform the assembly PCR were of good quality (Supporting Information Fig. S2). The codon frequencies of the randomized positions were found to be in decent agreement with the intended mixture (Fig. 5). Eighteen of the 20 in-frame DARPins were tested for expression in Escherichia coli and 16 DARPins were purified at high yields. Fifteen out of these 16 DARPins were separated by SEC (Supporting Information Fig. S3) and further analyzed by equilibrium unfolding using GdnHCl (see below).
Figure 5.
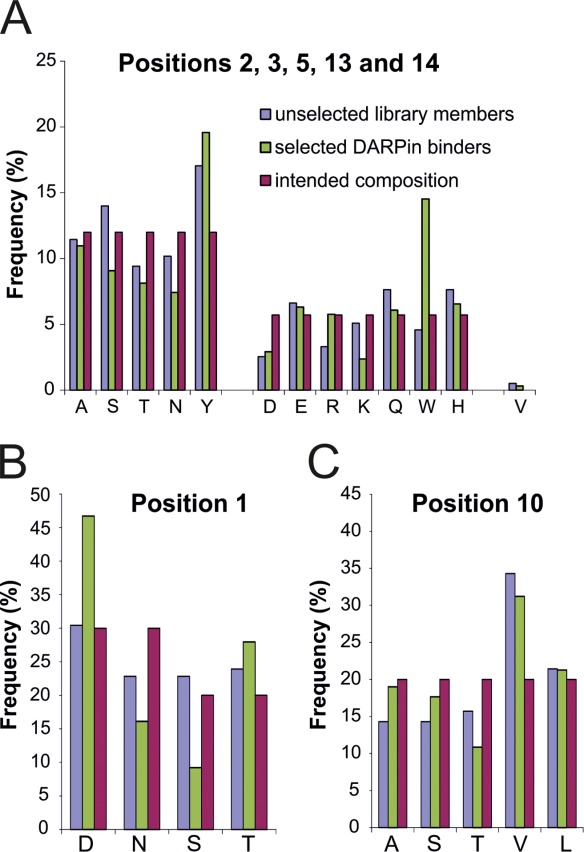
Amino acid composition in diversified positions of unselected library members (based on 20 DARPins, Supporting Information Fig. S2) and selected DARPin binders (based on 50 DARPins, Supporting Information Figs. S5–S7). The frequencies (%) are given for ankyrin repeat positions 2, 3, 5, 13, and 14 (A), position 1 (B) and position 10 (C) and are compared with the intended frequencies.
DARPin selections against two human caspases and membrane transporter AcrB
As a proof of principle we decided to select binders of the new library against human caspase-3 and caspase-7 and multidrug transporter AcrB from E. coli, which served as protein targets in previous DARPin selections in our laboratory using the original library (Refs.28 and40 and unpublished results). The first three selection rounds were carried out using the surface panning method, followed by a fourth round of solution panning. In order to increase the specificity during selection, a pre-panning step with biotinylated maltose binding protein (MBP) was performed in every round. Starting from round two, a cross-panning analysis, in which pre-panning on target protein was followed by panning on MBP was introduced. In this analysis, target-specific binders are removed in the pre-panning step and remaining unspecific binders are captured by MBP in the panning step. In case of binder enrichment against the desired target, comparatively low amounts of mRNA are expected to be recovered in such cross-panning experiments. Already in the second selection round, mRNA recovery was higher in the normal panning procedure (pre-panning on MBP followed by panning on target) compared with the cross-panning analysis, indicating that the enriched DARPin pools were target specific [Fig. 6(A)].
Figure 6.
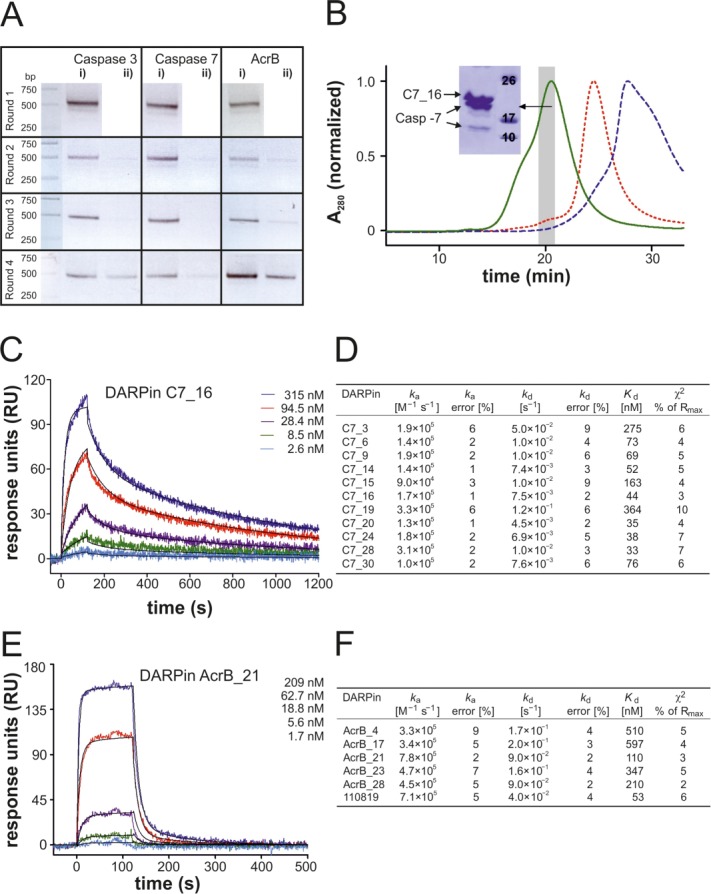
Validation of the new library with test selections against caspase-3, caspase-7, and AcrB (A) RT-PCR of ribosome display selection rounds 1 to 4 with applied cross-panning in rounds 2 to 4. Retrieved mRNA of potential binders were reverse transcribed and amplified by PCR (45, 40, 30, and 25 cycles for rounds 1, 2, 3, and 4, respectively) resulting in a 500 bp PCR product. “i)” indicates pre-panning against MBP followed by panning against target protein and “ii)” vice versa. (B) Analysis of ELISA-positive DARPin C7_16 (Supporting Information Fig. S4) by analytical SEC. Complex formation with caspase-7 leads to a distinct peak shift (green, solid line) compared with caspase-7 alone (red, short dashed line) and DARPin alone (blue, dashed line). The SDS-polyacrylamide gel of the corresponding complex peak fractions (grey bar) is shown (inset). (C and D) The affinities of eleven caspase-7 DARPins were determined by SPR. Exemplary SPR traces are shown for DARPin C7_16 (C) and kinetic binding parameters as well as dissociation constants are shown in (D). (E and F) The affinities of five AcrB DARPins of the new library were determined by SPR alongside with the previously described AcrB DARPin 110819.28 Exemplary SPR traces are shown for DARPin AcrB_21 (E) and kinetic binding parameters as well as dissociation constants are shown in (F).
ELISA - analysis and sequencing of caspase- and AcrB- specificDARPins
From the 3rd and 4th selection round against caspase-3, caspase-7, and AcrB, DARPins were screened for binding by crude extract ELISA26,30 (Supporting Information Fig. S4). For caspase-3, 53% of all tested DARPins of selection round 3 and 45% of round 4 were found to be specific binders, whereas the hit rate was lower for caspase-7 and AcrB (13% and 16% of round 3 and 17% and 30% of round 4, respectively). Binders exhibiting the strongest signals in the crude extract ELISA were subjected to DNA sequencing (top 20 hits of the third selection round and top 10 hits of the fourth round). The binders were named C3_1–30, C7_1–30, and AcrB_1–30, respectively. Seventy-five out of the 90 chosen clones exhibited unambiguous sequencing results and unique sequences (Supporting Information Figs. S5–S7). Forty-nine out the 75 DARPins contained at least one unintended framework mutation. In three AcrB DARPins a frameshift mutation occurred, resulting in a change of the NGADVNAS loop motif, connecting helix 2 with the β-turn, to TVLTLTLL, and thereby rendering these DARPins considerably more hydrophobic. All caspase-3 DARPins harbor a conserved TXYGE motif in the β-turn region of the second internal repeat, suggesting that these binders recognize one single epitope (Supporting Information Fig. S5). In contrast, the caspase-7 and AcrB binders appeared more diverse and therefore are likely to recognize different epitopes (Supporting Information Figs. S6 and S7). By comparing the sequences of selected and unselected DARPins, the influence of binder selection on the amino acid composition of the randomized positions can be followed (Fig. 5). In positions 2, 3, 5, 13, and 14, residue frequency changes greater than 40% were observed for tryptophan (+217%), arginine (+74%) and lysine (−53%) [Fig. 5(A)]. In position 1, aspartate (+53%) was enriched and serine (−60%) was depleted during the selection process [Fig. 5(B)]. No major changes were observed at position 10 [Fig. 5(C)].
Biochemical characterization of caspase- and AcrB- specific DARPins
Twenty DARPins each specific for caspase-3, caspase-7, and AcrB were purified and their oligomeric states were analyzed by SEC (Supporting Information Fig. S3). The binders yielded between 4 and 66 mg pure protein per liter of culture. Eighteen out of 20 caspase-3 DARPins, 15 out of 20 caspase-7 DARPins, and 16 out of 20 AcrB DARPins eluted as monodisperse proteins with a retention volume corresponding to DARPin monomers as judged from a SEC column calibration run (not shown). Complex formation with target proteins was confirmed for monomeric caspase-3 and -7 DARPins by a peak shift on analytical SEC and subsequent SDS-PAGE analysis of the complex peak fractions [Fig. 6(B), Supporting Information Fig. S8(A,B)]. Eleven caspase-7 DARPins and all monomeric AcrB DARPins were further analyzed by surface plasmon resonance (SPR). A typical sensogram is shown for C7_16 and AcrB_21 [Fig. 6(C,E)]. For all caspase-7 binders, the binding constants were determined from the binding and dissociation kinetics. Binding of caspase-7 DARPins was characterized by rather slow on-rates and slow off-rates resulting in dissociation constants ranging from 33 nM to 364 nM [Fig. 6(D)]. Only five of the tested AcrB DARPins elicited SPR binding curves suitable for fitting and exhibited Kds in the range of 110 nM to 597 nM [Fig. 6(F)]. Their affinities were therefore considerably lower than an original DARPin selected against AcrB (DARPin 110819 described in Ref.28) whose Kd amounted to 53 nM in our analysis [Fig. 6(F)]. Notably, the original DARPin binders against AcrB were identified by an in vivo assay in E. coli cells. This allowed for screening a much larger number of clones, which explains the better affinities observed. The five high affinity AcrB DARPins were then analyzed for complex formation with AcrB by SEC. They all co-migrated with the targeted transporter [Supporting Information Fig. S8(C)], but AcrB_4 and AcrB_23 appear to dissociate during SEC due to low binding affinity.
Equilibrium unfolding of DARPins
To assess whether the second-generation library has retained the high thermodynamic stability of the original DARPins, equilibrium unfolding curves for 11 original DARPins and 25 second-generation DARPins (15 unselected DARPins and 5 DARPins each specific for caspase-3 and caspase-7, respectively) were determined (Supporting Information Fig. S9). With the exception of unselected DARPin D20, equilibrium unfolding curves for the new DARPins appeared monophasic. On the other hand, the unfolding curves of several original DARPins were clearly biphasic due to the early unfolding of the nonoptimized C-cap.52 DARPin unfolding and folding is a complicated process involving several folding intermediates as detailed previously using a consensus repeat approach.56 Here, we will confine our analysis to midpoints of denaturation (Dm) for clarity of discussion. The Dm values range from 2.3M to 5.1M GdnHCl for the original DARPins and 1.9M to 4.3M GdnHCl for the second-generation DARPins. On average, the new DARPins were less stable than the original ones with average Dm values of 3.0M and 3.8M GdnHCl, respectively (Fig. 7). This loss of stability might be attributed to the randomization of position 10 in the second-generation DARPins. For stability considerations, this position was kept as invariant leucine in the original library, and we found in this study that the E3_5_L86A mutant was less stable than wild type E3_5 [Fig. 3(B)].
Figure 7.
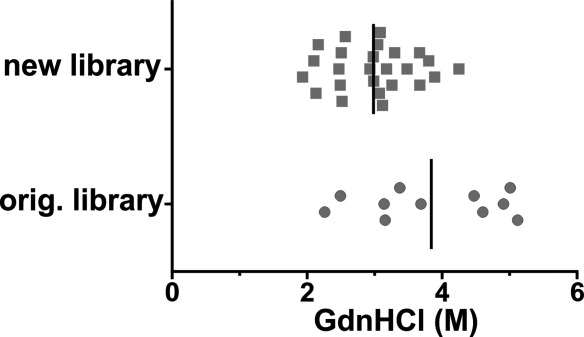
Equilibrium unfolding of DARPins. GdnHCl unfolding of a set of DARPins of the original and new library was determined by CD spectroscopy (Supporting Information Fig. S9) which allowed the determination of midpoints of unfolding (Dm). Dm values of individual DARPins are depicted together with the mean Dm (vertical line) for original and new DARPins, respectively.
Crystal structure of DARPin C7_16 in complex with caspase-7
DARPin C7_16 was purified in complex with caspase-7 by SEC, crystallized and the structure was determined at 1.7 Å resolution (Table I). The structure reveals one DARPin and one caspase-7 monomer in the asymmetric unit (Fig. 8). DARPin C7_16 superimposes well with the structure of a full-consensus DARPin containing the identical optimized C-cap53 indicating that the changes in the framework and the extended randomization did not alter the scaffold structure [Supporting Information Fig. S10(A)]. DARPin C7_16 interacts exclusively with the large subunit of caspase-7 involving residues mainly located at the second and third internal repeat and the C-cap. The binding interface covers an area of 843 Å2 on the DARPin and is stabilized by 13 hydrogen bonds and four salt bridges. Four tryptophans and two tyrosines are prominently involved in mediating molecular contacts to caspase-7 [Fig. 8(B)] and they account for 63% of the interaction surface and establish five hydrogen bonds. Residues of the C-cap account for approximately 49% of the binding surface. In contrast, residues in the randomized ankyrin positions 1 and 10 play a minor role in this particular DARPin-caspase-7 complex accounting for 1.6% and 0.1% of total BSA, respectively. Based on this particular crystal structure we cannot yet draw conclusions whether randomization of the two additional ankyrin repeat positions 1 and 10 was beneficial or not. The crystal structure nevertheless provides evidence that crystal contacts are mediated between two DARPins involving residues with reduced entropy, namely A68, A134, and D130 [Supporting Information Fig. S10(B)]. More crystal structures will be needed to analyze the consequences of surface entropy reduction in a systematic manner.
Figure 8.
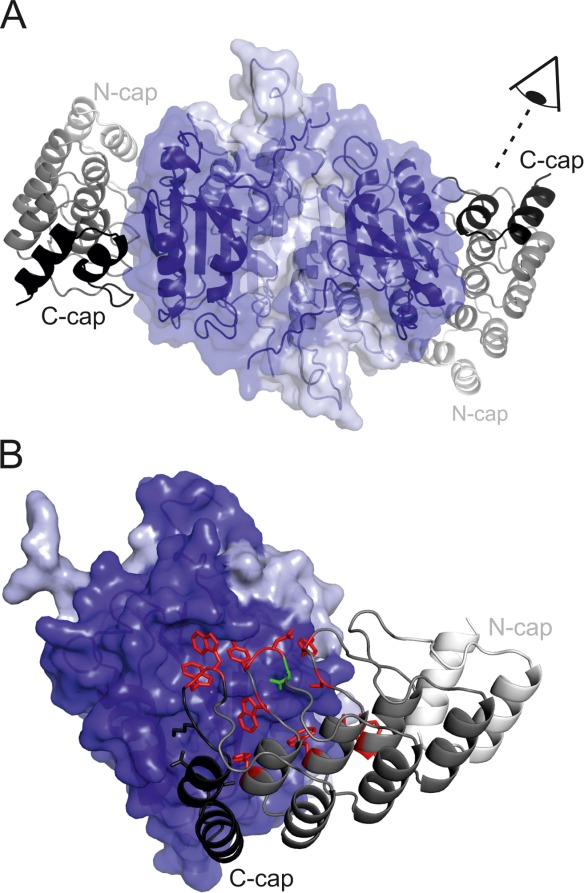
Crystal structure of the C7_16-caspase-7 complex. (A) Standard view of caspase-7 (surface representation, light and dark blue corresponding to the small and large subunits) in complex with DARPin C7_16 (cartoon representation, shades of grey). The DARPin binds side-ways to the large subunit of the enzyme. (B) Close-up view of the binding interface. Interacting DARPin residues (distance <4.5 Å) are highlighted as sticks and colored according to the scheme of Figure 1. Four tryptophans and two tyrosines play a dominant role in providing target contacts.
Discussion
In vitro selected binders have become popular tools in structural, cell, and chemical biology. Amongst such binders, nonantibody scaffolds offer unprecedented advantages due to their novel shapes of randomized surfaces and their favorable biophysical properties such as high stability and the absence of cysteines.11 Virtually any protein surface can be subjected to randomization which explains the development of numerous binding scaffolds in past years and certainly many more in the future. Here, we chose to refine the properties of an already established scaffold, namely the well-characterized DARPins.
Features of the second-generation DARPins
The presented second-generation library builds on an optimized scaffold and the randomized surface was extended based on the analysis of 13 crystal structures. Ankyrin repeat position 33 was no longer randomized in the second-generation library and positions 1 and 10 were subjected to focused randomization including a small number of permitted residues which lead to a slight reduction of thermodynamic stability. However, the average midpoint of denaturation still amounts to 3M GdnHCl and therefore, DARPins from the library described here are based on a very stable scaffold. The C-cap was found to be the Achilles heel of the original DARPins and therefore we added an optimized C-cap,52 which in contrast to the original DARPins was subjected to randomization. Moreover, we exchanged a lysine and a glutamate at the backside of each ankyrin repeat by alanine and aspartate, respectively, in order to reduce the surface entropy of DARPins for crystallization purposes.
Library assembly and validation
At the center of the binder selection technology stands the binder library, which encodes the sum of all randomized scaffolds in form of DNA. Its quality is characterized by (i) the library diversity (typically limited by the amount of DNA obtained in the last step of library assembly), (ii) the sequence quality (i.e. the percentage of library members encoded in frame containing the intended residues), and (iii) the biochemical property of the randomized scaffold (i.e. the percentage of in-frame library members that fold into monomeric proteins). For the library presented here, these three parameters are excellent. Due to the large theoretical library diversity of 1.9 × 1024, the practical diversity of this library was limited by the amount of assembled DNA and corresponds to approximately 1011 different library members. Seventy-seven percent of assembled library members are devoid of frame shifts and 83% of randomly picked in-frame DARPins could be purified and run as monomers on SEC. Currently, the practical diversity of the library is limited by the final ligation of the library into the in vitro transcription vector pRDV.12 In future work, the library diversity can be further increased by using type IIS restriction enzymes not only for the assembly of the repeats, but also for the cloning of the flanking elements required for in vitro transcription and translation of the library for ribosome display.57
Strong enrichment of tryptophan residues during binder selection
In previous studies, degenerate codons were used to construct a more hydrophilic synthetic scFv library based on a stable antibody scaffold.58 In addition, selections with Fab and monobody libraries containing only serines and tyrosines in the randomized positions were reported.59,60 Tyrosines were highlighted to play a key role in such minimalist synthetic libraries to culminate in a review article entitled “The Importance of Being Tyrosine”.37 In order to reduce the accumulation of “sticky” binders during the in vitro selection process, our second-generation DARPin library was diversified omitting the hydrophobic amino acids V, L, I, M, and F (except for position 10, where V and L are permitted). The diversified positions were enriched with tyrosine expecting that they will substitute for the hydrophobic amino acids excluded in the new library. In test selections against caspase-3, caspase-7 and AcrB, we observed specific enrichment of binders starting from round 2. A large proportion of binders were monomers, including those selected against the membrane protein AcrB, which underscores the advantage of working with a more hydrophilic DARPin library. The affinities of eleven caspase-7 and five AcrB DARPins measured by SPR range between 33 nM and 597 nM, which is lower than the affinities of DARPins of the original library selected against the same targets. However, current selections against other targets using the new DARPin library revealed binding affinities in the picomolar range (not shown). In our selections we did not observe substantial further enrichment of tyrosines, but strikingly, the number of tryptophans tripled. This result suggests that synthetic libraries depleted of apolar amino acids in diversified positions should be enriched in tyrosine and tryptophan in the randomization mixtures.
Crystal structure of the DARPin C7_16-caspase-7 complex
The crystal structure of DARPin C7_16 in complex with caspase-7 provides three important insights. First, the scaffold structure is unchanged. Moreover, the optimized C-cap superimposes very well with the recently published full-consensus DARPin structure containing this C-cap.53 Second, tyrosines and tryptophans in the randomized positions play a key role in mediating contacts to the target as they contribute 23% and 40%, respectively to the buried DARPin surface and five hydrogen bonds. Third, the residues subjected to surface entropy reduction were indeed found to be involved in crystal contacts.
Future prospects
A key motivator in designing this new library is its future use to select binders against difficult targets such as membrane proteins and soluble proteins exhibiting hydrophobic surfaces. Here, we could demonstrate successful selections against the membrane protein AcrB. Selections and binder analysis against a number of challenging proteins are ongoing, but will require several years of data accumulation to allow meaningful conclusions. The technical quality of this DARPin library is excellent and the test selections and characterizations presented in this study are encouraging. Library design and binder characterization is an iterative process.48 Thanks to the numerous available crystal structures, DARPins represent an ideal playground to improve synthetic library design. We hope that in the future more DARPin libraries will be created to identify the best possible randomization and selection strategy for this scaffold.
Materials and Methods
Reagents
Primers containing incorporated trinucleotide phosphoramidites for the targeted randomization were obtained from Ella Biotech (Martinsried, Germany). The other oligonucleotides were purchased from Microsynth (Balgach, Switzerland) at PAGE-purified quality. Restriction enzymes (if not stated otherwise), Vent polymerase and Phusion polymerase were from NEB. T4 DNA ligase and BpiI was purchased from Thermo Scientific. PCR purification kit, gel extraction kit, and plasmid miniprep kit were from Qiagen. Chemicals were from Sigma/Fluka, if not stated otherwise.
Analysis of the crystal interfaces
For the analysis of DARPin contact residues, the following 13 crystal structures (PDB entries) containing 22 interfaces in their asymmetric units were analyzed using the EPPIC server (http://www.eppic-web.org):47 1SVX, 2BKK, 2V4H, 2V5Q, 2XZD, 2Y1L, 3HG0, 3NOC, 3ZU7, 3ZUV, 4DRX, 4DX5, 4F6R. Crystal structures 2P2C, 3NOG and 4GRG were omitted due to resolutions limits (>3 Å) and 2J8S (solved at 2.5 Å) is represented here by the identical 1108_19-AcrB complex 4DX5 (solved at 1.9 Å). Accessible surface areas (ASA) and buried surface areas (BSA) upon complexation were retrieved from the EPPIC log files and compiled in an Excel sheet (Supporting Information File “DARPin_interfaces.xls”). In case several chains of the target protein were involved in the binding interface, the respective surfaces were summed up. At the transition between two target protein chains, there were around 10 cases in which the BSA of a DARPin residue was larger than its ASA. In these cases, the burial was set to 100% (BSA = ASA).
Generation of E3_5 mutants and surface entropy reduced DARPin_55
The plasmid pQE30_E3_515 served as template to introduce single mutations (D77S and L86A) using QuikChange® site-directed mutagenesis. The primer sequences were as follows: E3_5_D77S_for (5'-GTT AAC GCT TCT AGC CTT ACT GGT ATT AC), E3_5_D77S_rev (5'-GTA ATA CCA GTA AGG CTA GAA GCG TTA AC), E3_5_L86A_for (5'-CTC CGC TGC ACG CGG CTG CTG CTA C) and E3_5_L86A_rev (5'-GTA GCA GCA GCC GCG TGC AGC GGA G). The coding sequence of mutant DARPin_55 was custom-synthesized by Geneart (Regensburg) and cloned into pQE30.
DARPin purification and crystallization of E3_5_D77S and E3_5_L86A
E3_5 wild type and mutants as well as DARPin_55 and its mutant were expressed and purified as previously described.15 E3_5 mutants were applied on SEC (Superdex 200 5/150 GL, 20 mM Tris/HCl (pH 7.4), 150 mM NaCl) for buffer exchange, concentrated to 15 mg/mL and subjected to crystallization trials. For the E3_5_D77S mutant, crystals appeared in 1.6M citrate pH 6.5 and for the E3_5_L86A mutant, crystal grew in 0.1M citrate pH 5.6, 20% 2-propanol, 20% PEG4000. Crystals were directly picked from the screening plate without further cryo-protection and flash-frozen in liquid nitrogen.
PCR amplification of DARPin capping repeats for library assembly
PCR reactions for the amplification of the caps were carried out on a Bio-Rad PCR machine using the temperature protocol: 2 min, 95°C; 30 cycles of 30 sec 95°C (melting), 30 sec 60°C (annealing) and 2 min/kb amplified DNA 72°C (elongation); a final elongation step 10 min 72°C; cooling to 10°C. The E20D_R23S double mutation at the N-Cap was introduced into pQE30_E3_5 using QuikChange® site-directed mutagenesis using primers NCap_E20D_R23S_for (5'-GCT GGA TGC TGC TAG TGC TGG TCA GG) and NCap_E20D_R23S_rev (5'-CCT GAC CAG CAC TAG CAG CAT CCA GC). This mutant served as template to PCR amplify the N-cap using primers NCap1 (5'- ATG GCG CGC CAT GGG TAT GAG AGG ATC G) and NCap2 (5'-ATA AGC TTG GTC TCA CGT CAG CAC CGT TAG CCA TCA GTA TAC G). The C-Cap was amplified from the vector pQE30_NI3C_Mut553 using primers CCap1 (5'-ATG GCG CGC CGA AGA CCT GAC GTC AAC GCT AAC XXX NNN NNN GGT AAG ACC CCG TTC GAC TTA GCG, in which XXX stands for a codon mix for D, N; 30% each and S, T; 20% each and NNN stands for a codon mix for A, S, T, N, and Y; 12% each and D, E, R, K, Q, W, and H; 5.7% each) and NC_shorter (5'-ATA AGC TTC GCC GCT TTT TGC AGC AC). Both caps were amplified using Phusion polymerase (amounts used as suggested by the manufacturer), 0.5 μM of each primer and 0.2 mM of dNTPs resulting in a single band on agarose gel (1.5%) and yielding around 3 μg of DNA per 50 μL of PCR reaction.
PCR amplification of the internal repeats for library assembly
The internal repeats were amplified in a single step using a simple assembly PCR protocol.61 Low amounts (0.05 μM final conc, each) of inner primers Rep2 (5'-GA CCT GAC GTT AAC GCT TCT XXX NNN NNN GGT NNN ACT CCG CTG CAC YYY GCT GCT NNN NNN GGT CAC CTG GAA ATC GTC G, in which XXX and NNN denote codons for the same amino acid mixtures as specified above and YYY stands for a codon mix for A, S, T, V, and L, 20% each), Rep3 (5'-ATA AGC TTG TCA GGT CTC ACG TCA GCA CCG TDA GCC AGC AGA ACA TCG ACG ATT TCC AGG TG, in which D stands for a mixture of the bases A, G, and T) were mixed with an excess (1 μM final conc, each) of outer primers Rep1 (5'-ATG GCG CGC CGA AGA CCT GAC GTT AAC GC) and Rep4 (5'- GTA CCA AGC TTG TCA GGT CTC ACG TC). PCR was carried out using Vent polymerase (1 µL per 100 µL PCR mix), 0.4 mM dNTPs and 5% DMSO. The cycle protocol was: 2 min, 95°C; 35 cycles of 30 sec 95°C (melting), 30 sec 60°C (annealing) and 30 sec 72°C (elongation); a final elongation step 3 min 72°C; cooling to 10°C. The PCR product ran as a single band of the expected size on a 1.5% agarose gel. The DNA was purified with a PCR purification kit (Qiagen) to yield around 1.2 μg DNA per 50 μL of PCR reaction volume. In order to make the individual internal repeats of a fully assembled DARPin distinguishable by DNA sequence the glycine at position 27 of the ankyrin repeat module was encoded with three different triplets in each repeat of a N3C DARPin. To this end, the second internal repeat was assembled as described above, but instead of primer Rep3, Rep3_v2 (5'-ATA AGC TTG TCA GGT CTC ACG TCA GCG CCG TDA GCC AGC AGA ACA TCG ACG ATT TCC AGG TG, the nucleotide that differs is underscored) was used. Rep3_v3 (5'-ATA AGC TTG TCA GGT CTC ACG TCA GCC CCG TDA GCC AGC AGA ACA TCG ACG ATT TCC AGG TG) was used accordingly to assemble the third internal repeat.
Library assembly
The DARPin library was assembled repeat by repeat as first described by Binz et al.15 In contrast to previously described protocols for DARPin and Armadillo library assemblies.15,62 we did not subclone the repeat segments into a plasmid in a first step, but rather assembled them directly after PCR. Moreover, we provide here a comprehensive protocol for library assembly, which includes DNA and enzyme amounts as well as reaction volumes. In the first assembly step the N-Cap and the first internal repeat (1.5 μg each) were digested with BsaI and BpiI (20 units each), respectively, for 2 h in a volume of 100 μL. The small fragments containing the typeII S restriction sites were removed using the PCR purification kit (Qiagen) and cut N-cap and repeat were eluted in volumes of 50 μL elution buffer (Qiagen) each. The fragments were then ligated in a volume of 120 μL using 20 units of T4 ligase for 1 h. The ligation product was separated on a 1.5% agarose gel and the ligation efficiency was found to exceed 90%. The fragment consisting of the N-cap and the first internal repeat (called NI1) was retrieved by gel extraction (elution in 50 μL elution buffer, Qiagen) yielding 0.8 μg of DNA. DNA (0.3 μg) thereof was PCR-amplified in 200 μL using the primers NCap1 and Rep4 and Vent polymerase following the same PCR parameters as for the amplification of internal repeats (see above) and yielding 8 μg of DNA after PCR purification. Then 2.5 μg thereof and 1.25 μg of the second internal repeat were digested using BsaI and BpiI followed by PCR purification, ligation and gel extraction under identical conditions as for the assembly of NI1 to yield 0.86 μg NI2. Finally 0.43 μg thereof was PCR-amplified in a 400 μL reaction mixture to yield 17.8 μg of DNA, half of which were digested in 300 μL reaction volume using 80 units of BsaI. The third internal repeat (3 μg) was digested with 25 units BpiI in 150 μL. After ligation (30 units T4 ligase in 220 μL) and gel extraction, 3.6 μg NI3 were obtained. Half of it was amplified by PCR in 800 μL reaction mixture to yield 21.2 μg DNA which was digested in a volume of 600 μL using 80 units BsaI. C-cap (3 μg) was digested using 25 units of BpiI in 150 μL. The fragments were again ligated (50 units T4 ligase in 500 μL) and separated by gel electrophoresis to yield 6.5 μg assembled N3C DARPins, which corresponds to 1.2 × 1013 DNA molecules. Since the theoretical library diversity is 1.9 × 1024 and the last step of the assembly is a ligation (i.e. there was no amplification of the N3C fragment by PCR at this point) it is highly unlikely that two of these 1.2 × 1013 N3C DARPins are identical. Hence the practical diversity of the assembled N3C DARPins is limited by the amount of ligated DNA and corresponds to 1.2 × 1013. One third of this ligated N3C product (thereby limiting the practical diversity to 4 × 1012) was amplified by PCR in a volume of 2400 μL using primers NCap1 and NC_shorter to yield 84 μg of DNA. Ten micrograms thereof were digested with 50 units each of NcoI and BamHI in a volume of 300 μL for 1.5 h. Using the same restriction enzymes, 60 μg of pRDV12 was digested. Both fragments were purified by gel extraction. About 1.25 µg digested N3C were ligated into 9 µg of digested pRDV (25 units T4 ligase in 300 μL for 3 h). Assuming that 5% of the digested N3C is being integrated into pRDV,57 the final practical diversity of the library is estimated to amount to 1011 library members. The ligation product served as template for PCR amplification using primers T7 and tolAk12 in a total volume of 3000 μL to yield DNA ready for in vitro transcription.63 An estimated 5 μg DNA was used to generate 720 μg of mRNA, which was then used to perform test selections.
Ribosome display
Ribosome display selections were carried out following the established standard protocol with some minor modification as indicated.63 Biotinylated caspase-3 and caspase-7 were prepared as described previously.64 AcrB containing a C-terminal avi-tag was biotinylated enzymatically as described.30 Each selection round included a pre-panning step using MBP. Biotinylated MBP12 and caspases were immobilized on NeutrAvidin coated surfaces as described63 (surface panning method) or in the fourth and last round using magnetic beads (solution panning method). The washing times were as follows: first round, three washes w/o incubation; second round, 3 × 10 min; third round, 3 × 15 min; fourth round, 3 × 30 min. Retrieved mRNA of DARPins were reverse transcribed and amplified by PCR with 45, 40, 30, and 25 cycles for rounds 1, 2, 3, and 4, respectively. In the initial round, the amount of in vitro translation ribosomal complexes was increased five times compared with the standard protocol and Nunc MaxiSorp Immuno Tubes (Thermo Fisher Scientific) were used to immobilize MBP (for prepanning) and caspases (for panning). The number of ternary ribosome complexes of the first round was estimated to correspond to at least 3.5 × 1011 displayed DARPin molecules.63 This number is approximately three times higher than the practical diversity of the DARPin library used, thus the in vitro translation step did not represent the bottleneck of the selection procedure. Due to an exchanged C-cap in the second-generation DARPin library, NC_shorter was used instead of the standard primer WTC4.12
Crude cell extract ELISAs
Crude cell extract ELISAs were carried out as initially established by Huber et al.26 and exactly as described in Seeger et al.30 with one notable exception. Namely, instead of using the DARPin expression vector pQE30_myc5,26 which adds five consecutive myc-tags to the C-terminus of the DARPin leading to DARPin aggregation,30 we constructed a pQE30_myc1 vector adding a single myc-tag as follows. pQE30_myc5 containing the LmrCD-specific DARPin α-LmrCD#530 was digested using HindIII and Bpu1102I to excise the myc5 tag, followed by gel extraction of the backbone. The myc1-tag was generated by annealing primers myc1_ for (5'-AGC TTG GTT CTG GAA GTA TGG AGC AAA AGC TCA TTT CTG AAG AGG ACT TGA ATG AAT AAT GAG C) and myc1_rev (5'-CTC AGC TCA TTA TTC ATT CAA GTC CTC TTC AGA AAT GAG CTT TTG CTC CAT ACT TCC AGA ACC A) to a double-stranded DNA product, which contains overhangs compatible with cloning into HindIII/Bpu1102I of the pQE30 backbone. Binding of each DARPin was probed against the respective caspase and as a control against MBP. Binders yielding strong ELISA signals (at least 10 times above MBP background) were classified as specific. Positive clones were sequenced at Microsynth.
Small-scale purification of DARPins
Purified DARPins containing a single myc-tag were indistinguishable by SEC from the same DARPins without a myc-tag (not shown). Therefore, binder characterization was carried out with DARPins containing an N-terminal RGSHis6-tag and a C-terminal myc-tag. DARPins were expressed overnight at 37°C in E. coli XL-1 blue cells in 100 mL auto inducing medium containing ampicillin (100 μg/mL). After harvest, cells were resuspended in 20 mL lysis buffer (50 mM Tris/HCl (pH 7.4), 500 mM NaCl, 20 mM imidazole, 10% glycerol, 100 mg/L lysozyme, and 10 mg/L DNaseI) and incubated 1 h at 4°C before cell disruption by sonication for 3 min. Cell lysate was clarified by centrifugation (10,000g, 5 min) and filtration (0.45 μm). Ni2+-NTA columns (0.5 mL resin) were equilibrated with wash buffer (50 mM Tris/HCl (pH 7.4), 500 mM NaCl, 20 mM imidazole, 10% glycerol) and cell lysate was loaded on the column. The columns were washed with 30 mL wash buffer and the proteins eluted with 2 mL elution buffer (same as wash buffer, but containing 250 mM imidazole). SEC was used to analyze the oligomeric state of selected DARPin binders as well as the formation of stable DARPin/target protein complexes in solution. SEC was performed at 4°C on an Agilent 1200 Series HPLC system. Sample volumes of 100 μL were injected on a Superdex 200 5/150 GL column (GE Healthcare) using SEC buffer (20 mM Tris/HCl (pH 7.4), 150 mM NaCl, 0.05% Tween-20) at a flow rate of 0.08 mL/min. To study DARPin-caspase complexes, DARPins (added in excess in concentrations ranging from 8 to 24 μM) were preincubated with caspase-3 or caspsase-7 before SEC analysis. For the equilibrium unfolding experiments, DARPins were purified devoid of a myc-tag. To this end, the C-terminal myc-tag of DARPin C7_16 was removed via an introduction of two stop-codons in front of the tag using QuikChange® site-directed mutagenesis. The primer sequences were as follows: 5'-AAA GCG GCG AAG CTT TGA TAA GGA AGT ATG GAG CAA and 5'-TTG CTC CAT ACT TCC TTA TCA AAG CTT CGC CGC TTT.
Equilibrium unfolding
DARPins were purified as described above and separated by SEC in 20 mM Tris/HCl (pH 7.4), 150 mM NaCl. DARPins were diluted in increasing concentrations of GdnHCl dissolved in SEC buffer to obtain a final protein concentration of 10 μM and incubated for at least 12 h in the dark at 20°C. The disappearance of the native CD signal at 222 nm with increasing GdnHCl concentrations was determined using a Jasco J-810 instrument (Jasco, Japan). As far as possible, a monophasic sigmoidal equation [Eq. (1)] was fitted to the experimental data (SigmaPlot10, standard 4 parameter sigmoidal fit):
| (1) |
where x0 denotes the inflection point corresponding to Dm.
In cases of two distinct unfolding transition phases, a biphasic sigmoidal equation [Eq. (2)] was fitted to the data:
| (2) |
with the constraints x01 < x02 and a1 <0.5 × a2.
Here, inflection points x01 and x02 correspond to midpoints of transition Dm,1 and Dm,2. This biphasic model was only applied if Dm,1 and Dm,2 differed by at least 0.9M GdnHCl.
SPR analysis
The affinity of a set of caspase-7 DARPins was determined by surface plasmon resonance analysis on a ProteOn XPR36 protein interaction array system (Bio-Rad). Biotinylated caspase-7 (1200 RU) were immobilized on a NeutrAvidin coated NLC sensor chip in SPR buffer (20 mM Tris/HCl (pH 7.4), 150 mM NaCl, 0.05% Tween-20). DARPins were injected at six different concentrations (threefold dilution series) appropriate for analysis of the interaction with immobilized target proteins. Binding kinetics were performed with 120 s contact time followed by 600 to 1200 s dissociation time at a flow rate of 100 μL/min. Referenced binding signals were fitted using a 1:1 binding model (ProteOn Manager Software) and the dissociation constant Kd was calculated using Eq. (3) in which ka is the association rate constant and kd the dissociation rate constant:
| (3) |
Crystallization of the C7_16-caspase-7 complex
Myc-tag free C7_16 was expressed and purified as mentioned above. Caspase-7 expression and purification is described elsewhere.64 Caspase-7 and DARPin C7_16 were mixed in a molar ratio of 1:3 and separated by SEC (50 mM Tris/HCl (pH 7.4), 150 mM NaCl; Superdex 200 10/300 GL). Fractions corresponding to the C7_16-caspase-7 complex were concentrated to 11 mg/mg and subjected to crystallization trials. Crystals appeared in 100 mM Tris/HCl (pH 8.3), 200 mM MgCl2, 9% PEG 1000, and 9% PEG 8000. Upon cryo-protection using ethylene glycol (20%), they were frozen in liquid nitrogen.
Crystallographic methods
Crystals were measured at the protein crystallography beamline X06SA at the Swiss Light Source (SLS) and processed using the program XDS.65 Structures of E3_5_D77S, E3_5_L86A and the C7_16-caspase-7 complex were solved by molecular replacement using Phaser66 and search models 1MJ0 to solve the structures E3_5 mutants and search models 3IBC (Caspase-767) and 2P2C (Caspase-2 DARPins22) to solve the structure of the C7_16-caspase-7 complex. Models were built manually using Coot68 and refined using PHENIX69 including TLS refinement.70 Structural pictures were generated using PyMOL (http://www.pymol.org).
Acknowledgments
The authors thank Beat Blattmann and Céline Stutz-Ducommun from the NCCR crystallization facility for crystal screening, and the staff of the X06SA beamline at the Swiss Light Source of the Paul Scherrer Institute for support during data collection. We thank Andreas Plückthun for providing the ribosome display plasmid pRDV and DARPin expression vectors. The authors acknowledge Stefan Schauer from the Functional Genomics Center Zürich (FGCZ) for support during SPR measurements.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 2.Clackson T, Hoogenboom HR, Griffiths AD, Winter G. Making antibody fragments using phage display libraries. Nature. 1991;352:624–628. doi: 10.1038/352624a0. [DOI] [PubMed] [Google Scholar]
- 3.Hanes J, Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci USA. 1997;94:4937–4942. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angelini A, Cendron L, Chen S, Touati J, Winter G, Zanotti G, Heinis C. Bicyclic peptide inhibitor reveals large contact interface with a protease target. ACS Chem Biol. 2012;7:817–821. doi: 10.1021/cb200478t. [DOI] [PubMed] [Google Scholar]
- 5.Röthlisberger D, Pos KM, Plückthun A. An antibody library for stabilizing and crystallizing membrane proteins—selecting binders to the citrate carrier CitS. FEBS Lett. 2004;564:340–348. doi: 10.1016/S0014-5793(04)00359-X. [DOI] [PubMed] [Google Scholar]
- 6.Silacci M, Brack S, Schirru G, Marlind J, Ettorre A, Merlo A, Viti F, Neri D. Design, construction, and characterization of a large synthetic human antibody phage display library. Proteomics. 2005;5:2340–2350. doi: 10.1002/pmic.200401273. [DOI] [PubMed] [Google Scholar]
- 7.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–314. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 8.Prassler J, Thiel S, Pracht C, Polzer A, Peters S, Bauer M, Norenberg S, Stark Y, Kolln J, Popp A, Urlinger S, Enzelberger M. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol. 2011;413:261–278. doi: 10.1016/j.jmb.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Skerra A. Alternative non-antibody scaffolds for molecular recognition. Curr Opin Biotech. 2007;18:295–304. doi: 10.1016/j.copbio.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 10.Boersma YL, Plückthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotech. 2011;22:849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 12.Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, Grütter MG, Plückthun A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22:575–582. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- 13.Lambert S, Yu H, Prchal JT, Lawler J, Ruff P, Speicher D, Cheung MC, Kan YW, Palek J. cDNA sequence for human erythrocyte ankyrin. Proc Natl Acad Sci USA. 1990;87:1730–1734. doi: 10.1073/pnas.87.5.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batchelor AH, Piper DE, de la Brousse FC, McKnight SL, Wolberger C. The structure of GABPa/b: an ETS domain- ankyrin repeat heterodimer bound to DNA. Science. 1998;279:1037–1041. doi: 10.1126/science.279.5353.1037. [DOI] [PubMed] [Google Scholar]
- 15.Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol. 2003;332:489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 16.Binz HK, Kohl A, Plückthun A, Grütter MG. Crystal structure of a consensus-designed ankyrin repeat protein: implications for stability. Proteins. 2006;65:280–284. doi: 10.1002/prot.20930. [DOI] [PubMed] [Google Scholar]
- 17.Merz T, Wetzel SK, Firbank S, Plückthun A, Grütter MG, Mittl PR. Stabilizing ionic interactions in a full-consensus ankyrin repeat protein. J Mol Biol. 2008;376:232–240. doi: 10.1016/j.jmb.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 18.Steiner D, Forrer P, Stumpp MT, Plückthun A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat Biotechnol. 2006;24:823–831. doi: 10.1038/nbt1218. [DOI] [PubMed] [Google Scholar]
- 19.Boersma YL, Chao G, Steiner D, Wittrup KD, Plückthun A. Bispecific designed ankyrin repeat proteins (DARPins) targeting the epidermal growth factor receptor inhibit A431 cell proliferation and receptor recycling. J Biol Chem. 2011;286:41273–41285. doi: 10.1074/jbc.M111.293266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zahnd C, Pecorari F, Straumann N, Wyler E, Plückthun A. Selection and characterization of Her2 binding-designed ankyrin repeat proteins. J Biol Chem. 2006;281:35167–35175. doi: 10.1074/jbc.M602547200. [DOI] [PubMed] [Google Scholar]
- 21.Kohl A, Amstutz P, Parizek P, Binz HK, Briand C, Capitani G, Forrer P, Plückthun A, Grütter MG. Allosteric inhibition of aminoglycoside phosphotransferase by a designed ankyrin repeat protein. Structure. 2005;13:1131–1141. doi: 10.1016/j.str.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 22.Schweizer A, Roschitzki-Voser H, Amstutz P, Briand C, Gulotti-Georgieva M, Prenosil E, Binz HK, Capitani G, Baici A, Plückthun A, Grütter MG. Inhibition of caspase-2 by a designed ankyrin repeat protein: specificity, structure, and inhibition mechanism. Structure. 2007;15:625–636. doi: 10.1016/j.str.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 23.Stefan N, Martin-Killias P, Wyss-Stoeckle S, Honegger A, Zangemeister-Wittke U, Plückthun A. DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J Mol Biol. 2011;413:826–843. doi: 10.1016/j.jmb.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 24.Martin-Killias P, Stefan N, Rothschild S, Plückthun A, Zangemeister-Wittke U. A novel fusion toxin derived from an EpCAM-specific designed ankyrin repeat protein has potent antitumor activity. Clin Cancer Res. 2011;17:100–110. doi: 10.1158/1078-0432.CCR-10-1303. [DOI] [PubMed] [Google Scholar]
- 25.Steiner D, Forrer P, Plückthun A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J Mol Biol. 2008;382:1211–1227. doi: 10.1016/j.jmb.2008.07.085. [DOI] [PubMed] [Google Scholar]
- 26.Huber T, Steiner D, Röthlisberger D, Plückthun A. In vitro selection and characterization of DARPins and Fab fragments for the co-crystallization of membrane proteins: the Na+-citrate symporter CitS as an example. J Struct Biol. 2007;159:206–221. doi: 10.1016/j.jsb.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Milovnik P, Ferrari D, Sarkar CA, Plückthun A. Selection and characterization of DARPins specific for the neurotensin receptor 1. Protein Eng Des Sel. 2009;22:357–366. doi: 10.1093/protein/gzp011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 2007;5:e7. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mittal A, Böhm S, Grütter MG, Bordignon E, Seeger MA. Asymmetry in the homodimeric ABC transporter MsbA recognized by a DARPin. J Biol Chem. 2012;287:20395–20406. doi: 10.1074/jbc.M112.359794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seeger MA, Mittal A, Velamakanni S, Hohl M, Schauer S, Salaa I, Grütter MG, van Veen HW. Tuning the drug efflux activity of an ABC transporter in vivo by in vitro selected DARPin binders. PLoS One. 2012;7:e37845. doi: 10.1371/journal.pone.0037845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon M, Zangemeister-Wittke U, Plückthun A. Facile double-functionalization of designed ankyrin repeat proteins using click and thiol chemistries. Bioconjug Chem. 2012;23:279–286. doi: 10.1021/bc200591x. [DOI] [PubMed] [Google Scholar]
- 32.Parizek P, Kummer L, Rube P, Prinz A, Herberg FW, Plückthun A. Ankyrin repeat proteins (DARPins) as novel isoform-specific intracellular inhibitors of c-Jun N-terminal kinases. ACS Chem Biol. 2012;7:1356–1366. doi: 10.1021/cb3001167. [DOI] [PubMed] [Google Scholar]
- 33.Amstutz P, Binz HK, Parizek P, Stumpp MT, Kohl A, Grütter MG, Forrer P, Plückthun A. Intracellular kinase inhibitors selected from combinatorial libraries of designed ankyrin repeat proteins. J Biol Chem. 2005;280:24715–24722. doi: 10.1074/jbc.M501746200. [DOI] [PubMed] [Google Scholar]
- 34.Sennhauser G, Grütter MG. Chaperone-assisted crystallography with DARPins. Structure. 2008;16:1443–1453. doi: 10.1016/j.str.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 35.Koide S. Engineering of recombinant crystallization chaperones. Curr Opin Struct Biol. 2009;19:449–457. doi: 10.1016/j.sbi.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eicher T, Cha HJ, Seeger MA, Brandstatter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grütter MG, Diederichs K, Pos KM. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc Natl Acad Sci USA. 2012;109:5687–5692. doi: 10.1073/pnas.1114944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koide S, Sidhu SS. The importance of being tyrosine: lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem Biol. 2009;4:325–334. doi: 10.1021/cb800314v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grubisha O, Kaminska M, Duquerroy S, Fontan E, Cordier F, Haouz A, Raynal B, Chiaravalli J, Delepierre M, Israel A, Veron M, Agou F. DARPin-assisted crystallography of the CC2-LZ domain of NEMO reveals a coupling between dimerization and ubiquitin binding. J Mol Biol. 2010;395:89–104. doi: 10.1016/j.jmb.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 39.Bandeiras TM, Hillig RC, Matias PM, Eberspaecher U, Fanghanel J, Thomaz M, Miranda S, Crusius K, Putter V, Amstutz P, Gulotti-Georgieva M, Binz HK, Holz C, Schmitz AA, Lang C, Donner P, Egner U, Carrondo MA, Muller-Tiemann B. Structure of wild-type Plk-1 kinase domain in complex with a selective DARPin. Acta Crystallogr D Biol Crystallogr. 2008;64:339–353. doi: 10.1107/S0907444907068217. [DOI] [PubMed] [Google Scholar]
- 40.Schroeder T, Barandun J, Flütsch A, Briand C, Mittl PR, Grütter MG. Specific inhibition of caspase-3 by a competitive DARPin: molecular mimicry between native and designed inhibitors. Structure. 2013;21:277–289. doi: 10.1016/j.str.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 41.Veesler D, Dreier B, Blangy S, Lichiere J, Tremblay D, Moineau S, Spinelli S, Tegoni M, Plückthun A, Campanacci V, Cambillau C. Crystal structure and function of a DARPin neutralizing inhibitor of lactococcal phage TP901-1: comparison of DARPin and camelid VHH binding mode. J Biol Chem. 2009;284:30718–30726. doi: 10.1074/jbc.M109.037812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monroe N, Sennhauser G, Seeger MA, Briand C, Grütter MG. Designed ankyrin repeat protein binders for the crystallization of AcrB: plasticity of the dominant interface. J Struct Biol. 2011;174:269–281. doi: 10.1016/j.jsb.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 43.Kummer L, Parizek P, Rube P, Millgramm B, Prinz A, Mittl PR, Kaufholz M, Zimmermann B, Herberg FW, Plückthun A. Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proc Natl Acad Sci USA. 2012;109:E2248–E2257. doi: 10.1073/pnas.1205399109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pecqueur L, Duellberg C, Dreier B, Jiang Q, Wang C, Plückthun A, Surrey T, Gigant B, Knossow M. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proc Natl Acad Sci USA. 2012;109:12011–12016. doi: 10.1073/pnas.1204129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim B, Eggel A, Tarchevskaya SS, Vogel M, Prinz H, Jardetzky TS. Accelerated disassembly of IgE-receptor complexes by a disruptive macromolecular inhibitor. Nature. 2012;491:613–617. doi: 10.1038/nature11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mignot I, Pecqueur L, Dorleans A, Karuppasamy M, Ravelli RB, Dreier B, Plückthun A, Knossow M, Gigant B. Design and characterization of modular scaffolds for tubulin assembly. J Biol Chem. 2012;287:31085–31094. doi: 10.1074/jbc.M112.383869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duarte JM, Srebniak A, Schärer MA, Capitani G. Protein interface classification by evolutionary analysis. BMC Bioinform. 2012;13:334. doi: 10.1186/1471-2105-13-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilbreth RN, Koide S. Structural insights for engineering binding proteins based on non-antibody scaffolds. Curr Opin Struct Biol. 2012;22:413–420. doi: 10.1016/j.sbi.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohl A, Binz HK, Forrer P, Stumpp MT, Plückthun A, Grütter MG. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc Natl Acad Sci USA. 2003;100:1700–1705. doi: 10.1073/pnas.0337680100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Virnekas B, Ge L, Plückthun A, Schneider KC, Wellnhofer G, Moroney SE. Trinucleotide phosphoramidites: ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucleic Acids Res. 1994;22:5600–5607. doi: 10.1093/nar/22.25.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kayushin AL, Korosteleva MD, Miroshnikov AI, Kosch W, Zubov D, Piel N. A convenient approach to the synthesis of trinucleotide phosphoramidites—synthons for the generation of oligonucleotide/peptide libraries. Nucleic Acids Res. 1996;24:3748–3755. doi: 10.1093/nar/24.19.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Interlandi G, Wetzel SK, Settanni G, Plückthun A, Caflisch A. Characterization and further stabilization of designed ankyrin repeat proteins by combining molecular dynamics simulations and experiments. J Mol Biol. 2008;375:837–854. doi: 10.1016/j.jmb.2007.09.042. [DOI] [PubMed] [Google Scholar]
- 53.Kramer MA, Wetzel SK, Plückthun A, Mittl PR, Grütter MG. Structural determinants for improved stability of designed ankyrin repeat proteins with a redesigned C-capping module. J Mol Biol. 2010;404:381–391. doi: 10.1016/j.jmb.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 54.Derewenda ZS. Rational protein crystallization by mutational surface engineering. Structure. 2004;12:529–535. doi: 10.1016/j.str.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 55.O'Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 56.Wetzel SK, Settanni G, Kenig M, Binz HK, Plückthun A. Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J Mol Biol. 2008;376:241–257. doi: 10.1016/j.jmb.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 57.Ng DT, Sarkar CA. Model-guided ligation strategy for optimal assembly of DNA libraries. Protein Eng Des Sel. 2012;25:669–678. doi: 10.1093/protein/gzs019. [DOI] [PubMed] [Google Scholar]
- 58.Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol. 2004;338:299–310. doi: 10.1016/j.jmb.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 59.Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci USA. 2004;101:12467–12472. doi: 10.1073/pnas.0401786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S. High-affinity single-domain binding proteins with a binary-code interface. Proc Natl Acad Sci USA. 2007;104:6632–6637. doi: 10.1073/pnas.0700149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiong AS, Yao QH, Peng RH, Duan H, Li X, Fan HQ, Cheng ZM, Li Y. PCR-based accurate synthesis of long DNA sequences. Nat Protoc. 2006;1:791–797. doi: 10.1038/nprot.2006.103. [DOI] [PubMed] [Google Scholar]
- 62.Varadamsetty G, Tremmel D, Hansen S, Parmeggiani F, Plückthun A. Designed Armadillo repeat proteins: library generation, characterization and selection of peptide binders with high specificity. J Mol Biol. 2012;424:68–87. doi: 10.1016/j.jmb.2012.08.029. [DOI] [PubMed] [Google Scholar]
- 63.Zahnd C, Amstutz P, Plückthun A. Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat Methods. 2007;4:269–279. doi: 10.1038/nmeth1003. [DOI] [PubMed] [Google Scholar]
- 64.Roschitzki-Voser H, Schroeder T, Lenherr ED, Frölich F, Schweizer A, Donepudi M, Ganesan R, Mittl PR, Baici A, Grütter MG. Human caspases in vitro: expression, purification and kinetic characterization. Protein Expr Purif. 2012;84:236–246. doi: 10.1016/j.pep.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 65.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystrallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Agniswamy J, Fang B, Weber IT. Conformational similarity in the activation of caspase-3 and −7 revealed by the unliganded and inhibited structures of caspase-7. Apoptosis. 2009;14:1135–1144. doi: 10.1007/s10495-009-0388-9. [DOI] [PubMed] [Google Scholar]
- 68.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.