

Abstract
Many regulatory proteins are homo-oligomeric and designing assays that measure self-assembly will provide novel approaches to study protein allostery and screen for novel small molecule modulators of protein interactions. We present an assay to begin to define the biochemical determinants that regulate dimerization of the cancer-associated oncoprotein AGR2. A two site-sandwich microtiter assay (2SMTA) was designed using a DyLight800-labeled monoclonal antibody that binds to an epitope in AGR2 to screen for synthetic self-peptides that might regulate dimer stability. Peptides derived from the intrinsically disordered N-terminal region of AGR2 increase in trans oligomer stability as defined using the 2SMTA assay. A DSS-crosslinking assay that traps the AGR2 dimer through K95-K95 adducts confirmed that Δ45-AGR2 was a more stable dimer using denaturing gel electrophoresis. A titration of wt-AGR2, Δ45-AGR2 (more stable dimer), and monomeric AGR2E60A revealed that Δ45-AGR2 was more active in binding to Reptin than either wt-AGR2 or the AGR2E60A mutant. Our data have defined a functional role for the AGR2 dimer in the binding to its most well characterized interacting protein, Reptin. The ability to regulate AGR2 oligomerization in trans opens the possibility for developing small molecules that regulate its' biochemical activity as potential cancer therapeutics. The data also highlight the utility of this oligomerization assay to screen chemical libraries for ligands that could regulate AGR2 dimer stability and its' oncogenic potential.
Keywords: oligomerization, allostery, protein interactions, monoclonal antibody
Introduction
The discovery of protein–protein interactions is a key, fundamental goal in life sciences research and protein–protein interactions are emerging as compelling landscapes in the drug discovery field.1,2 The fundamental nature of a protein–protein interaction has changed since the realization that intrinsically unstructured proteins form important components of a large number of protein–protein interactions.3 This view of a protein-protein interaction provides approaches for using structural and computational sciences to develop lead molecules that disrupt a given protein–protein interaction.4 In addition, the realization that proteins exist in large oligomeric signaling complexes provides a relatively large surface area for identifying protein-interaction hotspots that might be amenable to fragment-based lead molecule discovery.5,6
Structure-based lead molecule discovery remains a fundamental mechanism to exploit protein structure, develop small molecule drug leads, and manipulate allostery in proteins.7 Amongst the many emerging models in the cancer field, for example, small molecules exist that stabilize the tetramer–dimer/monomer conversion of pyruvate kinase that has potential to regulate metabolism in cancer cells.8,9 In addition, the structure of porphobilinogen synthase has led to models on the interconversion of octomers to hexamers through dimer assembly.10 Small molecules have been developed that stabilize the hexameric form of the enzyme and inhibit enzyme activity11 that highlight the ability to exploit the allosteric regulatory nature of many proteins by developing small molecule tools or drug leads.
One limitation of structure-based drug discovery in human disease-associated proteins is that a large proportion of polypeptide sequence information in higher eukaryotes is composed of intrinsically disordered regions that make some domains refractory to structural solutions.12,13 These disordered regions can acquire structure by an induced-fit mechanism upon binding to their targets thus providing a specificity in the protein–protein interaction.14 Proteins with disordered domains and oligomeric properties can form prototypes for developing innovative approaches to measure allostery in protein control.15 The oncoprotein MDM2 has formed such a prototype model whose full-length properties have been refractory to structural biology solutions.16–18 We had used MDM2 as a model to develop a nanoparticle binding assay using surface-enhanced RAMAN spectroscopy (SERS) that can measure the oligomerization state of small amounts of protein when bound to ligand thus providing a nano-sensing assay to measure protein allostery.19 Although one methodological advantage of SERS as a nanoparticle binding assay is its use of small amounts of protein, a major limitation of SERS in measuring protein oligomerization is that it requires the target protein to be complexed to its ligand.
In this study, we present an alternative to the ligand-dependent SERS oligomerization assay by using a fluorescently labeled monoclonal antibody based microtiter assay that measures oligomerization of a ligand-free protein. The prototype protein used was the pro-oncogenic protein Anterior Gradient-2, that is an emerging cancer drug target in the endoplasmic-reticulum secretory system.20–22 Anterior-Gradient-2 has an intrinsically disordered N-terminal region and the protein has recently been reported to be dimeric.23 This fluorescent-based microtiter assay measured changes in AGR2 dimer stability in response to synthetic peptides derived from its intrinsically disordered domain in trans. This methodology provides a robust assay that can be used to screen chemical libraries for molecules that can alter AGR2 dimer stability and that can in turn be tested for inhibition of the oncogenic function of AGR2 in cells. In addition, this methodological concept is amenable in principle to other oligomeric proteins and can be used to screen for and isolate chemicals that alter oligomeric protein stability.
Results
Size exclusion chromatography demonstrates that AGR2 can exist as a homodimer
Gel-filtration size exclusion chromatography of recombinant mature AGR221–175 was used to define its oligomeric structure [Fig. 1(A)]. At highest concentrations (2.5 mg mL−1; 136 µM), the protein eluted with a calculated molecular mass of 34.843 kDa [Fig. 1(B)], significantly greater than the theoretical monomeric molecular mass of 18252.1 kDa (Expasy Compute pI/MW tool, http://web.expasy.org/compute_pi/), more closely resembling a dimer structure relative to the theoretical mass of 36,504 kDa. The elution profiles of globular proteins used for calibration are summarized in Figure 1(C,E). By comparison, a prior analysis of AGR221–175 protein using SEC-MALLS gave a dimeric mass of 30.5 kDa from a protein with a monomeric mass of 17.8 kDa and predicted dimeric mass of 32.2 kDa.23
Figure 1.
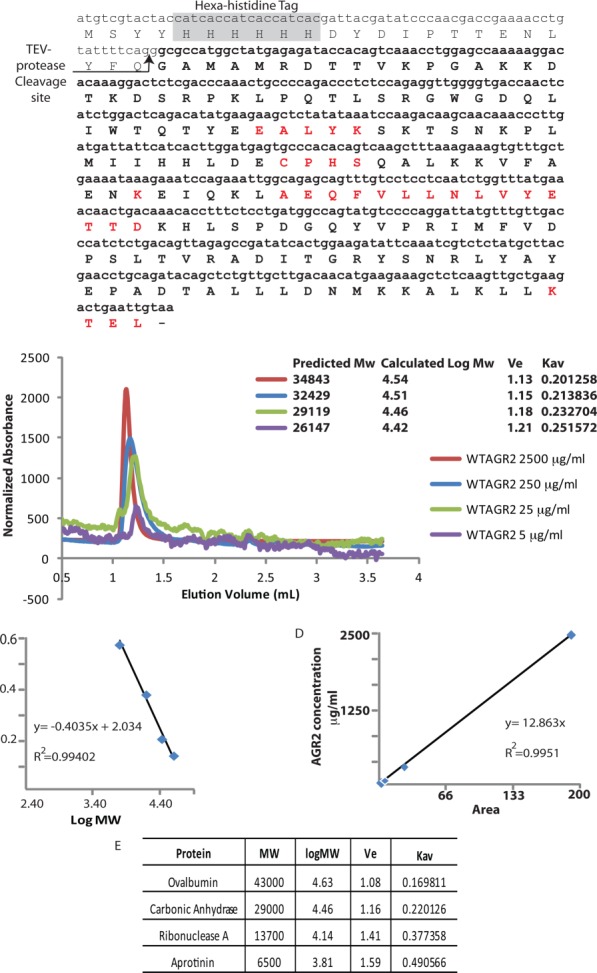
Molecular mass of native AGR221–175 protein. (A) The amino acid sequence of the recombinant “mature” AGR221–175 protein lacking the N-terminal leader sequence, highlighting the: (i) hexa-histidine motif and TEV protease cleavage site in the tag, (ii) 60-EALYK-64 dimerization motif, (iii) 81-CPHS-84 thioredoxin motif, (iv) K95, (v) 101-AEQFVLLNLVYETTD-115 hydrophobic Reptin binding site, and (vi) degenerate 172-KTEL-175 ER retention site. (B) Gel filtration size exclusion chromatography of AGR221–175 protein, injected at the indicated concentrations, using an analytical Superdex PC 75 column. The data are plotted as relative absorbance to allow superimposition of the elution profiles. (C and E). Calibration of the Superdex PC 75 column was performed with the indicated gel filtration markers (GE) in SEC buffer (50 mM Tris pH 7.5, 200 mM NaCl. (D). Displays a plot of the peak area of the AGR2 peak (from B) as a function of AGR2 protein concentration at the time of injection, to highlight the linearity between protein absorbance upon elution (at 214 nm) and protein (concentration) injected. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
We diluted AGR2 protein from 136 µM down to 13.6, 1.36, and 0.27 µM prior to injection on the Sephadex-75 column to determine whether there is a concentration-dependence to dimerization [Fig. 1(B)]. AGR2 protein injected at a concentration of 13.6 µM eluted with an estimated mass of 32.429 kDa, protein injected at 1.36 µM exhibited a slower eluting species with an estimated mass of 29.119 kDa, and injection at a concentration of 0.27 µM displayed a mass of 26.147 kDa, suggesting that the protein can exist in a dimer-monomer equilibrium as it approaches predicted monomeric mass of 18.2 kDa at lower concentrations. The observed absorbance upon elution after each injection (214 nm; data not shown) corresponds to the starting concentration, as after integration of the peaks of each trace and plotting against the concentration, the values appear linear with R2 = 0.9951 [Fig. 1(D)]. The traces in Figure 1(B) have been normalized for ease of visual comparison. These data are consistent with data suggesting that a stable dimeric species of AGR2 requires an in vitro concentration of the pure protein in the low µM range.23
Developing a quantitative microtiter assay to measure AGR2 oligomerization
It is not known whether the oligomeric (e.g., dimeric) structure of AGR2 is required for any of its protein-interaction functions.23 To develop quantitative assays to measure AGR2 dimerization, we aimed to first determine whether a quantitative two-site “sandwich” microtiter assay (2SMTA) could be used to quantify oligomerization (e.g., dimerization). We had previously published a panel of monoclonal antibodies generated to the AGR2 orthologue, AGR3. Like AGR2,24 AGR3 can mediate cisplatin resistance25 in xenografts. Of these monoclonal antibodies,25 one (MAB3.4), cross-reacted with AGR2 [Fig. 2(D)]. The AGR2 epitope recognized by MAB3.4 was fine mapped to a short linear 5 amino acid residue motif of 76-HHLED-80 [Fig. 2(C)];25 that is out with the dimerization site [Fig. 2(A)] and therefore the antibody can be used in the 2SMTA. The premise of the 2SMTA is that the same immobilized MAB can both capture and detect the target protein only if the protein was oligomeric; for example, monomers cannot be detected by this assay [Fig. 2(E)]. Fluorescent labeling would allow quantitative detection of oligomers over monomers in real time [Fig. 2(F)]. As we cannot distinguish a dimer (based on gel filtration) from an oligomer using the 2SMTA, we prefer to name the species we observe an oligomer.
Figure 2.
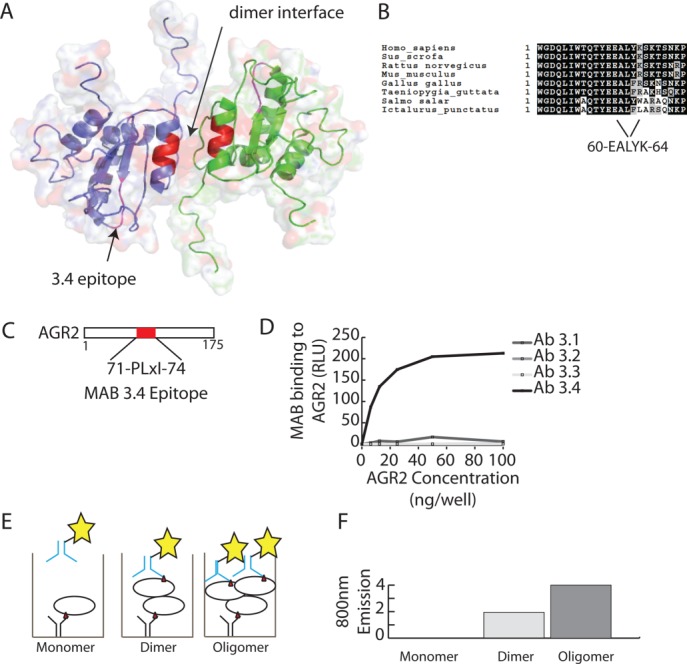
Localization of the epitope for MAB3.4 on AGR2 protein. (A) Cartoon of the dimeric structure of AGR2 (PDB; 2LNS;23) highlighting the dimer interface (B) and the MAB3.4 epitope (C). (D) ELISA-based assay analyzing the specific reactivity a panel of monoclonal antibodies raised against AGR325 toward AGR2. One of the AGR3-targetting monoclonal antibodies binds to AGR2 (MAB3.4). (E and F) Theory of analyzing and quantifying the oligomeric nature of AGR2 using the monoclonal antibody 3.4; (E) If AGR2 was monomeric and captured in the solid phase with MAB3.4, then the detection of this immune complex with the same MAB would not give a signal as there is not a second epitope (red triangle) present in the monomeric protein. However, a dimeric or oligomeric protein could be captured and detected with a monoclonal antibody that binds to the same epitope as there would be more than one epitope exposed in the immune complex. Although we can presume that AGR2 is a dimer in solution (as from Fig. 1 and from Patel et al.,23), this assay cannot distinguish between a dimer and higher oligomer, so we use oligomer throughout the text. (F) Predicted generic emission of an assay to measure AGR2 oligomerization using the fluorescently tagged MAB3.4. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Labeling the monoclonal antibody and optimization of the 2SMTA
To determine whether MAB3.4 can be used to measure AGR2 oligomerization, it was used to capture AGR2 and then a DyLight800-(DyL800)-labeled version of MAB3.4 was used to detect the captured AGR2. If AGR2 was only a monomer, then there could not be any detection of AGR2 protein using DyL800-MAB3.4 in the solution phase. However, if AGR2 were oligomeric (e.g., dimeric or larger order assemblies), the epitope would, in principle, be presented to DyL800-MAB3.4 in the liquid phase and the extent of oligomerization would be proportional to the amount of DyL800-MAB3.4 bound [Fig. 2(E,F)].
To develop this 2SMTA, we needed to first label the MAB3.4 with a fluorescent probe under conditions in which the labeled MAB retained functionality. Labeling of MAB3.4 was done with the far-infrared fluorophore DyL800. The labeling reaction proceeds through modification of primary amines and if an essential lysine in the complimentary determining region (CDR) was important for AGR2 binding, then labeling might inactive the MAB [Fig. 3(A)]. A titration of MAB3.4 and DyL800-MAB3.4 reveals that the labeled antibody produces a binding activity in RLU ∼1.8–2.5 lower fold lower than the unlabeled monoclonal antibody [Fig. 3(B)]. These data indicate that the DyL800-labeled monoclonal antibody retains significant affinity for its epitope in AGR2 protein and that binding could be quantified in a dose-dependent manner. The AGR2 protein-MAB interaction in Figure 3(B) measured the binding of the MAB3.4 or DyL800-MAB3.4 using peroxidase-labeled anti-mouse IgG secondary antibody and detection using chemiluminescence [Fig. 3(A)]. Following this, we evaluated binding using Licor far-infrared detection by excitation at 750 nm and fluorophore emission detected at 800 nm [Fig. 3(C)]. The DyL800-MAB3.4 binding to AGR2 adsorbed on the solid phase could be detected in a dose-dependent manner [Fig. 3(D)], relative to the unlabeled MAB3.4 that did not give rise to any signal. The binding reaction appeared more linear using DyL800-MAB3.4 in the direct excitation-emission assay [Fig. 3(D)] than in the indirect peroxidase-labeled secondary antibody [Fig. 3(B)]. There are two explanations for this phenomenon. First, the chemiluminescence assay [in Fig. 3(B)] uses “enhanced” enzymatic conversion by the antibody-peroxidase conjugate of substrate to create light that is quantified as a function of a fixed time. As a result, the data do not reflect a reaction rate but a final product accumulation. This “enhancement” of the primary signal may produce results that deviate from linearity. By contrast, the fluorescently conjugated monoclonal antibody emits a signal that is detected directly and the results are presumably more linear. Second, the indirect enhanced chemiluminescence assay incorporates an additional 75 min of incubation with secondary antibody, washings, and chemiluminescence substrate addition and this incorporates an unquantified effect of antibody off rate on the signal intensity. By contrast, after incubations with the fluorescently conjugated antibody, the reaction is washed rapidly, and fluorescence intensity is read immediately thus minimizing effects of the antibody off rate on the signal intensity. These data validate the DyL800-MAB3.4 excitation-emission assay as a bioactive probe. As such, we next evaluated whether DyL800-MAB3.4 could detect oligomeric forms of AGR2 in assay as a quantitative assay to measure oligomerization (oligomerization) of AGR2 protein [as outlined in Fig. 2(E)]. An outline of approach using DyL800-MAB3.4 to detect an oligomeric (potentially dimeric) species of AGR2 is outline in Figure 3(E). Reactions were assembled without or with adsorption of unlabeled MAB3.4 on the solid phase. Upon titration of DyL800-MAB3.4, a dose-dependent signal can be observed [Fig. 3(F)], suggesting that this assay can indeed measure oligomeric AGR2 protein.
Figure 3.
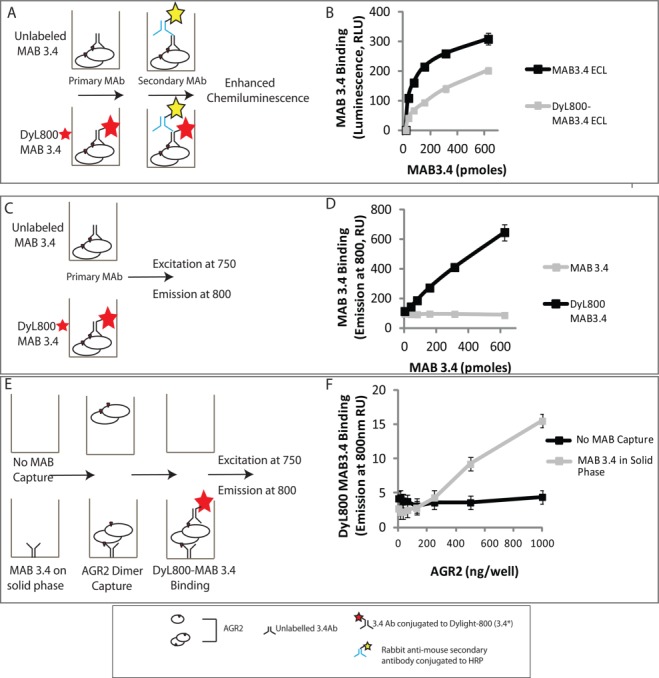
Developing a quantitative microtiter assay to measure AGR2 oligomerization. (A) Evaluation of the bioactivity of fluorescently labeled MAB3.4 in a luminescent-based ELISA. (B) MAB3.4 was left unconjugated or conjugated to DyL800 and after purification of the latter; the monoclonal antibodies were titrated into reactions containing AGR2 protein on the solid phase. The binding of MAB3.4 to AGR2 was measured using an anti-mouse IgG secondary antibody conjugated to peroxidase. The bioactivity of the monoclonal antibody (in relative luminescent units) is measured as a function of increasing MAB3.4 concentration. (C) Evaluation of the bioactivity of fluorescently labeled MAB3.4 in a fluorescence detection assay. (D) MAB3.4 was left unconjugated or conjugated to DyL800 and the monoclonal antibodies were titrated into reactions containing AGR2 protein on the solid phase. The bioactivity of the monoclonal antibody was measured as levels of emission at 800 nm a function of increasing MAB3.4 concentration. (E) Evaluation of the bioactivity of fluorescently labeled MAB3.4 to detect a potential AGR2 oligomer. (F) MAB3.4 was coated onto the solid phase and increasing amounts of oligomeric AGR2 were added to allow capture onto the solid phase. Fixed amounts of DyL800 MAB were added into reactions and the binding of DyL800-MAB3.4. The extent of oligomerization was quantified as levels of emission at 800 nm a function of increasing AGR2 protein concentration and is presented as an average from triplicate titrations. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Self-peptides that stabilize or disrupt the oligomer equilibrium
We next used the 2SMTA to determine whether AGR2 oligomer stability can be attenuated using dimer interface peptides or indeed stabilized in trans by regulatory motifs from the intrinsically disordered region of the protein [Fig. 4(B)]. The predicted disorder of polypeptides regions in AGR2 protein (using Disprot) are shown in Figure 4(B). The assay [Fig. 4(A)] included a pre-incubation stage where AGR2 oligomers were incubated with overlapping peptides derived from the open reading frame of AGR2 [Fig. 4(C)]. These overlapping AGR2-derived peptides (numbered 1–5) are from the N-terminal leader sequence, disordered region, and the dimeric interface [Fig. 4(C)]. Peptides 2 and 3 derived from the disordered region stabilize the AGR2 oligomer in trans, with peptide 3 inducing the most stabilized oligomer [Fig. 4(D)]. There is a dose-dependence in oligomer stabilization with a concentration of peptide as little as 3 µM increasing stability [Fig. 4(D)]. As controls, peptides overlapping the dimer interface do not stabilize the oligomer. Rather, these peptides 4 and 5 reduce the fluorescence [Fig. 4(E)] with peptide 5 containing the interface sequence 60-EALYK-64 inducing the greatest degree of oligomer de-stabilization. The ability of peptide 4 to similarly de-stabilize the oligomer, although to a lesser extent than peptide 5, suggests other motifs in AGR2 can be targeted to affect the dimer stability.
Figure 4.
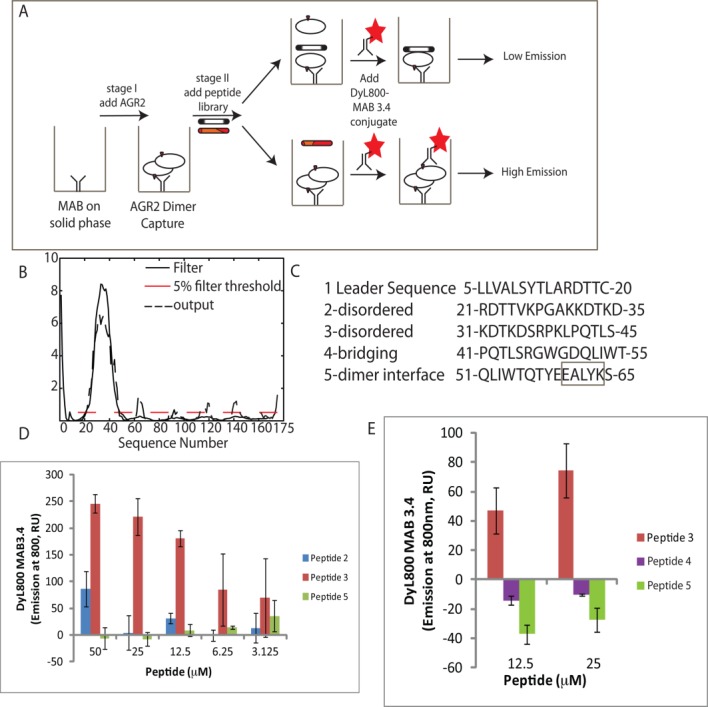
Effects of self-peptides on the extent of AGR2 multimerization. (A) Staging the effects of self-peptides on the oligomerization of AGR2. (B) Plot of the predicted disorder of AGR2 as a function of the amino acid sequence (Disprot). (C). Overlapping peptide sequences from the disordered and dimeric region of AGR2. (D) Effects of the “disordered” peptides 2 and 3 on AGR2 oligomerization. The 2SMTA was set up as in Figure 3(E) except that AGR2 protein (5.5 pmoles) was preincubated with the overlapping peptides 2 and 3 at the indicated concentrations from 3 to 50 µM for 1 h. After washing the DyL800-MAB3.4 probe was added for 1 h and the extent of oligomerization was measured as a function of emission at 800 nm. The data are depicted as DyL800-MAB binding (emission at 800 nm) as a function of peptide 2 and 3 concentration with subtraction of the inert peptide control. (E). Effects of the dimer-interface peptides 4 and 5 on AGR2 oligomerization. AGR2 protein (5.5 pmoles) was preincubated with the overlapping peptides 3, 4, and 5 at the indicated concentrations for 1 h. After washing the DyL800-MAB3.4 probe was added for 1 h and the extent of oligomerization was measured as a function of emission at 800 nm. The data are depicted as DyL800-MAB binding (emission at 800 nm) as a function of peptide 3, 4, and 5 concentration after subtraction of the inert peptide control background. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
The common amino acid sequences of peptides 4 and 5 that attenuate oligomer stability include 51-QLIWT-55 [Fig. 5(A)]. The more bioactive peptide 5 has the additional 56-QTYEEALYK-64 comprising the dimer interface [Fig. 5(A)], which suggests why peptide 5 is more active at disrupting the AGR2 oligomer using the 2SMTA. We created a set of AGR2 mutants to determine whether the 2SMTA can be used on full-length mutants to observe changes in oligomer stability. We chose to mutate Y58, since it protrudes into the adjacent internal β-sheet and might be required for stabilizing the dimer interface [Fig. 5(C)]. We also mutated E59 as it protrudes outward into solvent [Fig. 5(C)], whilst E60 forms a salt bridge with K64 across the adjacent monomer forming an important feature of the dimer interface [Fig. 5(A)]. The mutant forms of AGR2 created to determine how these affect oligomer stability, included Y58A, E59A, E60A, and Δ45 (deletion of amino acids 21–45) [Fig. 5(B)]. A titration of AGR2-E59A revealed its' oligomer stability is similar to wt-AGR2 [Fig. 5(B)]. By contrast, AGR2-E60A was in an apparent monomeric state, as judged by the absence of a signal in the oligomer assay [Fig. 5(B)]. The AGR2-Y58A was attenuated as an oligomer with a signal in the 2SMTA only seen at the highest concentration of the protein [Fig. 5(B)]. By contrast to these loss-of-function mutants, Δ45-AGR2 was more stable as a dimer [Fig. 5(B)]. The enhanced stability of the oligomer by deletion of the disordered region is consistent with prior research showing that deletion of amino acids 21–40 increases dimer stability.23
Figure 5.
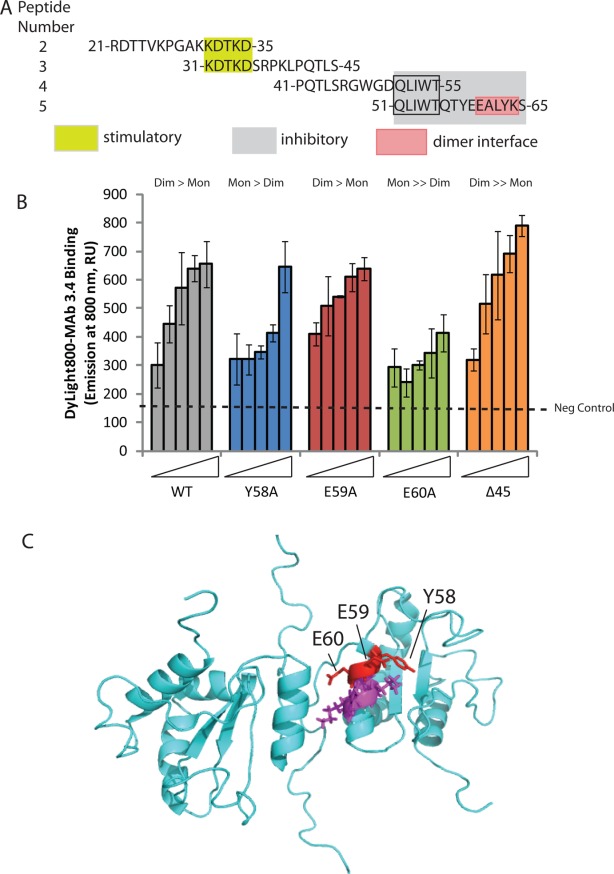
Effects of single point mutations in AGR2 dimer interface on multimerization using 2SMTA. (A) Sequence of bioactive peptides (from Fig. 4) derived from the disordered and dimerization region of AGR2 protein. Highlighted are minimal motifs predicted to drive stimulation (KDTKD from peptides 2 and 3) and inhibition (QLIWT from peptides 4 and 5). The dimer interface (from Ref.23) in peptide 5 is highlighted as EALYK. (B). Oligomer stability in AGR2 mutant panel. Increasing amounts of wild-type and mutant AGR2 proteins (from left, 0, 0.69, 1.38, 2.75, and 5.5 pmoles) were captured using MAB3.4 as indicated in Figure 3(E). Following the addition of DyL800-MAB3.4, the extent of oligomerization was quantified by emission at 800 nm and the data is presented as an average from triplicate titrations. No protein background signal is represented on the far left (157 U). The ratio of dimer to monomer is highlighted above each series: wt: dimmer > monomer; Y58A: monomer > dimer; E59: as wild-type; E60A: monomeric; and Δ45: more stable dimeric over monomeric. (C). Summary of the positions of Y58, E59, and E60 across the dimer interface region. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Mapping the crosslinked AGR2 dimer
We sought to develop an independent assay to validate the stability of the AGR2 dimer through the use of a DSS crosslinker that can trap a covalent dimeric species [Fig. 6(A)]. Mapping of this crosslink using mass spectrometry identified K95 as the dominant cross-link [Fig. 6(B,C)]. Mutagenesis of K95 to create AGR2-K95R confirmed that this is the dominant cross-link site [Fig. 6(D)]. An image of the position of K95 relative to the dimer interface is depicted in Figure 6(E). We used this assay to determine whether the AGR2 mutant panel exhibited altered dimerization (Fig. 7). A titration of DSS into reactions containing wt-AGR2, AGR2-E60A, and Δ45-AGR2 demonstrated that AGR2-E60 is attenuated in dimer formation [Fig. 7(B)]. In addition, Δ45-AGR2 exists in a more stable, spontaneous dimer in the absence of cross link [Fig. 7(C), lane 1], suggesting that deletion of the N-terminus can allow it to form a stable dimer in the presence of the 0.1% SDS buffer. These data together are consistent with the quantitative 2SMTA that demonstrated Δ45-AGR2 is a more stable dimer whilst AGR2-E60A is predominantly monomeric.
Figure 6.
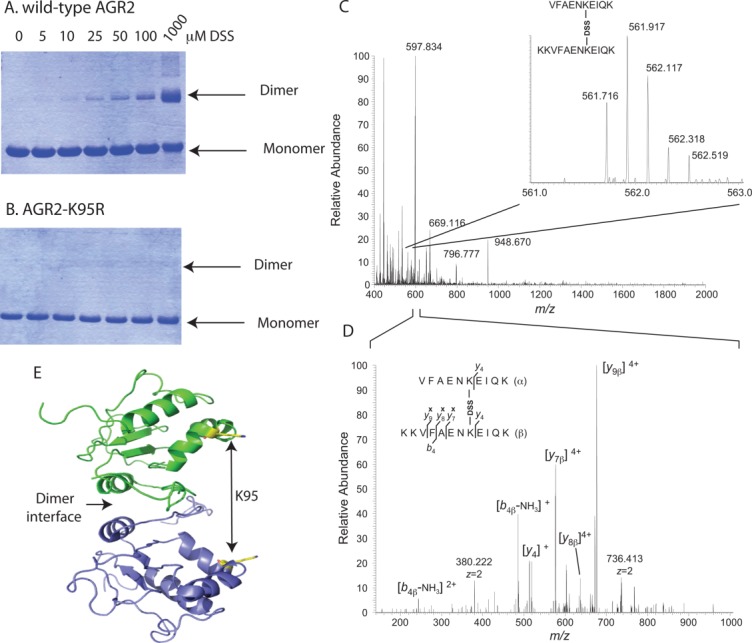
Mapping the sites of DSS dimer crosslinking to Lysine-95. (A) Crosslinking of AGR2 into a homodimeric species using the covalent crosslinker DSS. (B) Absence of crosslinking of AGR2-K95R mutation into a homodimeric species using the covalent crosslinker DSS, based on the K95-K95 crosslinks identified in C and D. (C). Mass spectrum of interprotein crosslinked peptides VFAENKEIQK and KKVFAENKEIQK (the bold K corresponds to the DSS-linked amino-acid), at [M+5H]5+ = 561.716 m/z units. (D) The lower panel shows the annotated tandem mass spectrum after coalitional-induced dissociation and subsequent Orbitrap mass analysis. Crosslinked peptides were identified as indicated in the materials and methods. (E) Cartoon of the interdomain orientation of the K95 amino acid residues with respect to the dimeric interface. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 7.
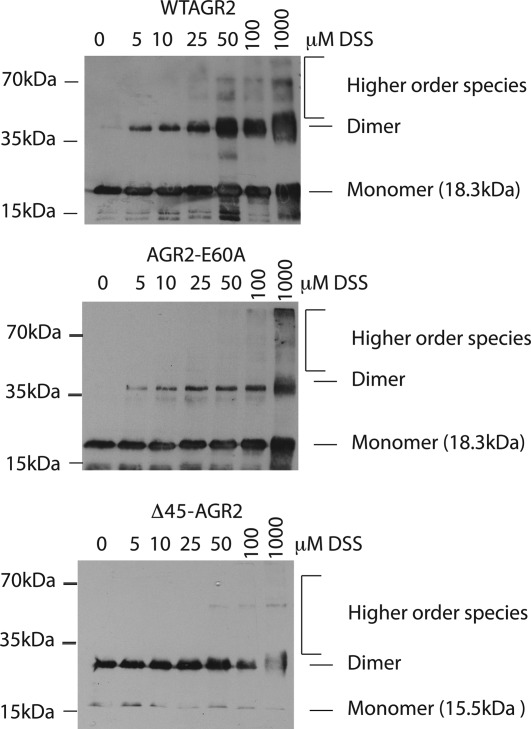
Effects of DSS on the dimerization of AGR2 mutants. The indicated proteins; (A) wt-AGR2, (B), AGR2-E60A, or (C) Δ45-AGR2, were incubated with increasing concentrations of DSS and separated by electrophoresis on an SDS gel (containing 0.1% SDS). AGR2 was detected using MAB3.4 in an immunoblot. The positions of monomeric and dimeric proteins are highlighted.
Evaluating the specific activity of the AGR2 oligomer
There is no known biochemical function for the AGR2 dimer. Having created a series of gain-of-function or loss of function AGR2 mutants, we evaluated the role of oligomerization in molecular interaction of AGR2 with two of its most well-characterized substrates: peptides containing the [T/S]-x-I-[YF]-[Y/F] consensus motif20 and the AAA+ chaperone protein, Reptin.26 The former protein-interaction presumably represents docking sites in its client proteins (manuscript in preparation). The latter protein interaction is with the AAA+ protein Reptin, which itself functions as a molecular machine able to assemble multiprotein complexes. The in vivo impact of AGR2 protein binding on client proteins containing the [T/S]-x-I-[Y/F]-[Y/F] motif and on Reptin complex assembly is currently undefined. Nevertheless, we wanted to determine whether AGR2 binding to either of these two targets was influenced by its' dimer-monomer state. A titration of wt-AGR2, AGR2-E60A, and Δ45-AGR2 indicates that all three exhibited relatively similar activity in binding to this peptide substrate [Fig. 8(A)]. By contrast, the Reptin substrate is substantially stabilized using the gain-of-function more stable dimer mutant Δ45-AGR2, whilst AGR2-E60A is attenuated [Fig. 8(B)]. These data provide the first biochemical evidence that dimerization of AGR2 protein can play a role in its affinity for a client protein. As there are over one dozen binding proteins identified for AGR2 protein using yeast-two hybrid,21 it will be interesting to see whether this panel of AGR2 oligomerization mutants can be used to access the role of the AGR2 dimer in these protein-protein interactions.
Figure 8.

The effects of AGR2 oligomerization on its protein-interaction activity. The indicated AGR2 proteins (wt-AGR2, AGR2-E60A, or Δ45-AGR2) were incubated in reactions containing (A) biotinylated TxIYY containing peptide and (B) Reptin. AGR2 bound to either peptide or Reptin was detected using MAB3.4 and quantified using anti-secondary IgG coupled to peroxidase and chemiluminescence. The data are depicted as AGR2 binding activity (in RLU) as a function of increasing AGR2 protein concentration (in triplicate and presented as an average). [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Discussion
Developing assays that measure oligomerization and how allosteric effects can be measured in parallel will provide important tools in the emerging field of drugging protein-protein interactions. We evaluate here an approach to measure oligomerization and allostery using Anterior Gradient-2. The mechanism whereby AGR2 mediates its' functions is thought to include its ability to chaperone proteins in the ER27 and to mediate induction of oncogenic signals like EGF or p53 tumor suppressor silencing.28,29 The majority of AGR2 protein interactions have been identified using yeast-two hybrid,21 with the only well-validated protein being the AAA+ pro-metastatic protein Reptin.26 Characterizing these various protein–protein interactions will likely shed light on its mechanism of action. Toward this aim, we focused on characterizing the AGR2 oligomer (dimer) as a model, since our initial biochemical characterizations demonstrated that its apparent mass was consistent with a homodimeric quaternary structure (Fig. 1). This is consistent with a recent report showing that recombinant AGR2 (AGR221–175 or AGR241–175) lacking the N-terminal leader sequence or disordered region, respectively, can form homodimers.23 Showing that AGR2 can exist as a dimer in vitro, we then wanted to determine whether the dimeric structure was important for two of the most well-characterized interactions; (i) the [T/]-x-I-[Y/F]-[Y/F] pentapeptide consensus binding site; and (ii) the AAA+ protein Reptin. Toward this aim, we needed to create a set of mutants in AGR2 with altered oligomeric state and we needed robust assays to define changes in its dimeric structure as a function of mutation.
The assay designed to measure oligomer stability was a two-site microtiter assay in which an oligomeric protein captured in solid phase can be detected in small volume liquid phase using the same monoclonal antibody; for example a “sandwich assay” (2SMTA). In this assay it is not possible for a monomer to give a signal. The 2SMTA detected attenuation of AGR2 oligomerization using self-peptides derived from the dimer interface (60-EALYK-64). Amino acids outwith this motif also attenuated oligomer stability (e.g., peptide 4; Fig. 4) suggesting that the dimer motif requires the stability of the adjacent peptide chain. In addition, we also surprisingly identified peptide motifs that can stimulate AGR2 oligomerization in trans. These peptides are derived from the intrinsically disordered region in the N-terminal domain of AGR2 (Fig. 4) and their ability to act in trans as positive effectors of oligomerization suggests they disrupt the function of the natural N-terminal sequences in cis. There are interesting paradigms where peptides and mimetics can allosterically stimulate protein–protein interactions30,31 and it will be interesting to determine whether these AGR2 stimulatory peptides can be used as tools to manipulate the activities of the protein. Using 2SMTA as a rapid screen for oligomerization capability, we confirmed that AGR2-E60A was largely monomeric (Fig. 5) and Δ45-AGR2 had an enhanced oligomerization capacity (Fig. 5). This was confirmed using a DSS crosslinking assay (Fig. 7) that was mapped to the K95 residue using mass spectrometry (Fig. 6).
Having developed a set of well-characterized oligomerization mutants, the effect of subunit structure on biochemical activity was finally evaluated. The AGR2 monomeric mutant E60A bound as stably as wild-type AGR2 or the enhanced dimer mutant Δ45-AGR2 to the [T/S]-x-I-[Y/F]-[Y/F] pentapeptide motif. This data indicates that if the TxIYY docking site represents a cellular binding interface for AGR2 client proteins, then the monomeric form of AGR2 has the potential to be equally active. By contrast, the binding of AGR2 to Reptin was stabilized by creating an enhanced dimer (Δ45-AGR2) and attenuated using the monomeric E60A mutant AGR2. Thus the determinant in AGR2 that interacts with Reptin (primarily within amino acids 104–111;26) exploit its' dimeric subunit structure. Solving the interface of AGR2-Reptin might facilitate developing biologics that disrupt this protein interaction to determine how it might contribute to cancer cell growth.
In conclusion, we have developed a microtiter-based assay that measures in small volumes the oligomerization state of AGR2 protein. Such an assay was used to identify ligands like self-peptides that can allosterically regulate multimer stability thus forming a proof-of-concept assay for screening for cellular proteins or drug-leads that might regulate of AGR2 dimer stability in vivo. The use of microtiter assays like the “sandwich”-type assay (2SMTA) that measures ligand-free oligomerization provides a robust small volume methodology that can be adapted to screen for ligands that regulate the stability of oligomeric proteins implicated in human disease.
Experimental
Purification of recombinant AGR2 protein and analytical gel filtration size-exclusion chromatography
AGR221–175 was cloned into pEHISTEV and transformed into BL21 (DE3).20,32 Bacteria were grown to OD600 of 0.6 at 37°C and 1 mM IPTG was added for a further 3 h, followed by harvesting and resuspending in 30 mL of lysis buffer (50 mM NaH2PO4 pH 7.5, 400 mM NaCl, 10 mM Imidazole) on ice. Cells were subjected to high-pressure (25k PSI) followed by centrifugation at 40,000g for 50 min to isolate the soluble fraction. The supernatant was applied to a Ni-charged HisTrap FF 5-mL column (GE Life Sciences). The immobilized protein was subject to a step-elution with 30 mM then 300 mM Imidazole. The eluted protein was pooled and cleaved with recombinant tobacco etch virus protease overnight at 20°C. The resultant solution was desalted into protein buffer (50 mM Tris pH 7.5, 200 mM NaCl) with a HiPrep desalt 26/10 column (GE Life Sciences) before reloading onto the IMAC to remove protease and uncleaved protein. Purified protein was analyzed by gel-filtration size exclusion chromatography on an analytical Superdex PC 75 3.2/30'' column (GE Healthcare) at titrated concentrations ranging from 2.5 mg mL−1 to 5 µg mL−1 (injection volume of 20 µL). Detection was by absorption at 214 nm at a flow rate of 0.5 mL min−1. The column was calibrated using protein standards (GE Healthcare).
Monoclonal antibody purification and DyLight800 labeling
Monoclonal antibody MAB3.425 was affinity purified on a Protein G column by elution using a buffer containing 0.1 M Glycine (pH 2.5) and then neutralized with 1/10th volume of a 1 M Tris HCl (pH 8.8) buffer. The IgG was dialyzed against PBS and was conjugated to DyLight800 fluorophore (excitation at 770 nm and emission at 794 nm) using DyLight800 Microscale Antibody Labeling Kit (53063; Thermo Scientific). Briefly, 50 μL of MAB3.4 (1.3 mg mL−1) was diluted to 100 µL final volume with 0.67M Borate buffer and the protein as added a vial of DyLight800 reagent to initiate coupling and the reaction is continued in the dark for 60 min at room temperature. The labeled protein was added to a spin column in microfuge format and the labeled antibody was separated from the label by collecting the eluate after centrifugation.
Measuring AGR2 oligomerization using a two-site sandwich microtiter assay (2SMTA)
Immunoglobulins (100 ng/50 ml per well) were adsorbed onto 96-well Costar opaque (white) wells, diluted in 100 mM NaHCO3 pH 8.6 and incubated at 4°C, with agitation, for 14–16 h. Wells were washed 3–5× with PBS-0.05% Tween-20, prior to blocking with 200 µL of 3% bovine serum albumin in PBS-0.05% Tween-20 per well for 1 h at room temperature. Protein was titrated in triplicate to the desired concentration in blocking buffer to 50 µL per well and incubated on the plate for 1 h at room temperature and washed 3–5× with PBS-0.05% Tween-20 prior to antibody detection. Primary antibody was diluted 1:1000 (1 µg mL−1) and 50-µL per well incubated at room temperature for 1 h, plates were then washed 3–5× with PBS-0.05% Tween-20. For ECL, 50 µL secondary antibody (rabbit anti-mouse, 1:1000 in 3% BSA-PBST) was incubated for 1 h, prior to a final 3–5× washes with PBS-0.05% Tween-20 and relevant detection. To detect dimerization: (i) enhanced chemiluminescence was carried out using white 96-well plates and secondary antibody conjugated to horseradish peroxidase followed by detection on a Fluoroskan Accent FL (ii) alternatively, a 800 nm near-IR label (DyLight 800, Thermo) was used to label the anti-AGR2 MAB3.4 and, using black walled, clear bottomed 96-well microtiter plates (Costar), antibody binding was quantified using Licor Odyssey.
Oligomer disruption assays using 2SMTA
The capture MAB (100 ng per well of MAB3.4) was diluted in 50 µL of NaHCO3 (pH 9.0) buffer and adsorbed on to the black walled, clear bottomed 96-well microtiter plate overnight at 4°C. Plates were then washed 3–5× with PBS-0.05% Tween-20, and blocked with 3% BSA in PBS-0.05% Tween-20 for 1 h. Concurrently, 100 ng (up to 5.5 pmoles) of purified AGR2 protein was diluted to 50 µL of protein buffer, and incubated with the indicate amounts of biotinylated overlapping peptide (in triplicate) for 1 h. The blocking solution was removed from the plate and replaced by peptide–protein solution and incubated for 1 h. Nearly 150 ng of DyLight800-labeled MAB3.4-labeled was then added to each well and allowed to bind for 1 h. Unbound or weakly bound antibody was then removed by 5× washes with PBS-0.05% Tween-20 before directly detected on the Licor Odyssey with 750 nm laser excitation and emission at 800 nm. Images were analyzed and quantified with the Odyssey Sa software.
Site directed mutagenesis of AGR221–175-pEHISTEV to produce Y58A, E59A, and E60A mutants and cloning for Δ45 mutant
The indicated primers (Y58A, E59A, and E60A) were used for mutagenesis using GENEART mutagenesis kit (Invitrogen).
| Mutation | Forward primer | Reverse primer |
| Y58A | ATCTGGACTCAGACAGCTGAAGAAGCTCTATAT | ATATAGAGCTTCTTCAGCTGTCTGAGTCCAGAT |
| E59A | TGGACTCAGACATATGCAGAAGCTCTATATAAA | TTTATATAGAGCTTCTGCATATGTCTGAGTCCA |
| E60A | ACTCAGACATATGAAGCAGCTCTATATAAATCC | GGATTTATATAGAGCTGCTTCATATGTCTGAGT |
The Δ45 mutant was amplified by PCR using forward (CCATGGCTATGAGAGGTTGGGGTGACCAAC) and reverse (CTCGAGTTACAATTCAGTCTTCAGCAACTT). The PCR products were cloned into pEHISTEV vector digested with NcoI and XhoI restriction enzymes to generate the final vectors.
Measuring mutant AGR2 protein oligomerization status using 2SMTA
About 500 mL pellets of bacteria induced to express wild-type and mutant AGR2 were resuspended in 10 mL lysis buffer and subject to 3 × 10 s sonication on ice. Protein mutants included Y58A, E59A, E60A, and Δ45-AGR2. The lysed bacteria were centrifuged to isolate the soluble fraction. About 10 µL fractions of each mutant crude lysate was separated by SDS-PAGE, transferred to nitrocellulose and probed with the DyLight800-labeled MAB3.4. Using the Licor Odyssey system an immunoblot was used to quantify and normalize AGR2 protein concentrations (as in Fig. 5). Nearly 100 ng of MAB3.4 antibody was adsorbed onto a clear-bottomed 96-well plate and the plate was washed and blocked as described previously. A titration of the normalized AGR2 protein (in triplicate) was applied to the plate in the mobile phase before washing 5× PBS-0.05% Tween-20 and quantification using the DyLight800-labeled MAB3.4.
Measuring the AGR2 oligomerization status using the K95-dependent DSS cross-linking assay
For protein crosslinking, AGR2 protein was exposed to DSS (Pierce 21555) and separated by SDS gel electrophoresis. The DSS crosslinked protein was dissolved in 50 mM ammonium bicarbonate and 6 M urea, reduced with 1 µL 50 mM DTE (dithioerythritol) for 60 min at 37°C, and alkylated by adding 1 µL of 400 mM iodoacetamide for 60 min at room temperature. The sample was diluted three times and digested overnight at 37°C with 0.1 µg of sequencing grade trypsin (Promega, Madison, WI). The remaining methodology for identification of crosslinked peptides has been described,33–35 including the use of an LTQ Orbitrap velos mass spectrometer (Thermo Electron; San Jose, CA) equipped with a NanoAcquity system (Waters). Mutagenic primers for K95R mutant generation were as follows: K95R forward: 5′-gtgtttgctgaaaatagagaaatccagaaattg, and reverse: 5′-caatttctggatttctctattttcagcaaacac.
Measuring AGR2 protein–protein interactions
AGR2-Reptin
Human Reptin expressed in BL21 AI (Invitrogen) E. coli was purified as described.26 To measure the binding of AGR2 (and mutants), purified Reptin (100 ng per well) was adsorbed to the well in carbonate buffer (0.1 M pH 9.0) overnight. Following blocking with PBS containing 0.05% Tween-20 and 3% BSA, the indicated AGR2 proteins were titrated in triplicate and incubated for 1 h at room temperature followed by detection with an AGR2 monoclonal antibody (Abnova) and HRP conjugated rabbit anti-mouse secondary antibody (Dako).
AGR2-TxIYY peptide
The TxIYY containing peptide (Biotin-SGSG-HLPTTIYYGPPG;20) was captured on streptavidin-coated wells and incubated for 1h, then plates were washed 5× with PBS-0.05% Tween-20 and blocked. AGR2 protein and the indicated mutants were titrated in triplicate onto the plates and incubated for 1 h at room temperature followed by detection using the AGR2 monoclonal antibody and HRP conjugated rabbit anti-mouse secondary antibody.
References
- 1.Morelli X, Hupp T. Searching for the Holy Grail; protein–protein interaction analysis and modulation. EMBO Rep. 2012;13:877–879. doi: 10.1038/embor.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thangudu R, Bryant SH, Panchenko R, Madej T. Modulating protein–protein interactions with small molecules: the importance of binding hotspots. J Mol Biol. 2012;415:443–453. doi: 10.1016/j.jmb.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure–function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 4.Jubb H, Higueruelo AP, Winter A, Blundell TL. Structural biology and drug discovery for protein–protein interactions. Trends Pharmacol Sci. 2012;33:241–248. doi: 10.1016/j.tips.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Winter A, Higueruelo AP, Marsh M, Sigurdardottir A, Pitt WR, Blundell TL. Biophysical and computational fragment-based approaches to targeting protein–protein interactions: applications in structure-guided drug discovery. Q Rev Biophys. 2012;45:383–426. doi: 10.1017/S0033583512000108. [DOI] [PubMed] [Google Scholar]
- 6.Smith MC, Gestwicki JE. Features of protein–protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev Mol Med. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basse MJ, Betzi S, Bourgeas R, Bouzidi S, Chetrit B, Hamon V, Morelli X, Roche P. 2P2Idb: a structural database dedicated to orthosteric modulation of protein–protein interactions. Nucleic Acids Res. 2013;41:D824–D827. doi: 10.1093/nar/gks1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, Yang H, Mattaini KR, Metallo CM, Fiske BP, Courtney KD, Malstrom S, Khan TM, Kung C, Skoumbourdis AP, Veith H, Southall N, Walsh MJ, Brimacombe KR, Leister W, Lunt SY, Johnson ZR, Yen KE, Kunii K, Davidson SM, Christofk HR, Austin CP, Inglese J, Harris MH, Asara JM, Stephanopoulos G, Salituro FG, Jin S, Dang L, Auld DS, Park HW, Cantley LC, Thomas CJ, Vander Heiden MJ. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan HP, O'Reilly FJ, Wear MA, O'Neill JR, Fothergill-Gilmore L, Hupp T, Walkinshaw MD. M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc Natl Acad Sci USA. 2013;110:5881–5886. doi: 10.1073/pnas.1217157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaffe EK, Lawrence SH. Allostery and the dynamic oligomerization of porphobilinogen synthase. Arch Biochem Biophys. 2012;519:144–153. doi: 10.1016/j.abb.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawrence SH, Ramirez UD, Selwood T, Stith L, Jaffe EK. Allosteric inhibition of human porphobilinogen synthase. J Biol Chem. 2009;284:35807–35817. doi: 10.1074/jbc.M109.026294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 13.Tompa P. On the supertertiary structure of proteins. Nat Chem Biol. 2012;8:597–600. doi: 10.1038/nchembio.1009. [DOI] [PubMed] [Google Scholar]
- 14.Kovacs D, Szabo B, Pancsa R, Tompa P. Intrinsically disordered proteins undergo and assist folding transitions in the proteome. Arch Biochem Biophys. 2012;531:80–89. doi: 10.1016/j.abb.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztanyi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough JD. database of disordered protein predictions. Nucleic Acids Res. 2013;41:D508–D516. doi: 10.1093/nar/gks1226. (2)P(2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poyurovsky MV, Priest C, Kentsis A, Borden KL, Pan ZQ, Pavletich N, Prives C. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 2007;26:90–101. doi: 10.1038/sj.emboj.7601465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kostic M, Matt T, Martinez-Yamout MA, Dyson HJ, Wright PE. Solution structure of the Hdm2 C2H2C4 RING, a domain critical for ubiquitination of p53. J Mol Biol. 2006;363:433–450. doi: 10.1016/j.jmb.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 18.Maslon MM, Hupp TR. Drug discovery and mutant p53. Trends Cell Biol. 2010;20:542–555. doi: 10.1016/j.tcb.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Robson AF, Hupp TR, Lickiss F, Ball KL, Faulds K, Graham D. Nanosensing protein allostery using a bivalent mouse double minute two (MDM2) assay. Proc Natl Acad Sci USA. 2012;109:8073–8078. doi: 10.1073/pnas.1116637109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray E, McKenna EO, Burch LR, Dillon J, Langridge-Smith P, Kolch W, Pitt A, Hupp TR. Microarray-formatted clinical biomarker assay development using peptide aptamers to anterior gradient-2. Biochemistry. 2007;46:13742–13751. doi: 10.1021/bi7008739. [DOI] [PubMed] [Google Scholar]
- 21.Chevet E, Fessart D, Delom F, Mulot A, Vojtesek B, Hrstka R, Murray E, Gray T, Hupp T. Emerging roles for the pro-oncogenic anterior gradient-2 in cancer development. Oncogene. 2013;32:2499–2509. doi: 10.1038/onc.2012.346. [DOI] [PubMed] [Google Scholar]
- 22.Fourtouna A, Murray E, Nicholson J, Maslon MM, Pang L, Dryden DTF, Hupp TR. The anterior gradient-2 pathway as a model for developing peptide-aptamer anti-cancer drug leads that stimulate p53 function. Curr Chem Biol. 2009;3:124–137. [Google Scholar]
- 23.Patel P, Clarke C, Barraclough DL, Jowitt TA, Rudland PS, Barraclough R, Lian LY. Metastasis-promoting anterior gradient 2 protein has a dimeric thioredoxin fold structure and a role in cell adhesion. J Mol Biol. 2012;425:929–943. doi: 10.1016/j.jmb.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Hengel SM, Murray E, Langdon S, Hayward L, O'Donoghue J, Panchaud A, Hupp T, Goodlett DR. Data-independent proteomic screen identifies novel tamoxifen agonist that mediates drug resistance. J Proteome Res. 2011;10:4567–4578. doi: 10.1021/pr2004117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray TA, MacLaine NJ, Michie CO, Bouchalova P, Murray E, Howie J, Hrstka R, Maslon MM, Nenutil R, Vojtesek B, Langdon S, Hayward L, Gourley C, Hupp TR. Anterior gradient-3: a novel biomarker for ovarian cancer that mediates cisplatin resistance in xenograft models. J Immunol Methods. 2012;378:20–32. doi: 10.1016/j.jim.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 26.Maslon MM, Hrstka R, Vojtesek B, Hupp TR. A divergent substrate-binding loop within the pro-oncogenic protein anterior gradient-2 forms a docking site for Reptin. J Mol Biol. 2010;404:418–438. doi: 10.1016/j.jmb.2010.09.035. [DOI] [PubMed] [Google Scholar]
- 27.Higa A, Mulot A, Delom F, Bouchecareilh M, Nguyen DT, Boismenu D, Wise MJ, Chevet E. Role of pro-oncogenic protein disulfide isomerase (PDI) family member anterior gradient 2 (AGR2) in the control of endoplasmic reticulum homeostasis. J Biol Chem. 2011;286:44855–44868. doi: 10.1074/jbc.M111.275529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta A, Dong A, Lowe AW. AGR2 gene function requires a unique endoplasmic reticulum localization motif. J Biol Chem. 2012;287:4773–4782. doi: 10.1074/jbc.M111.301531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pohler E, Craig AL, Cotton J, Lawrie L, Dillon JF, Ross P, Kernohan N, Hupp TR. The Barrett's antigen anterior gradient-2 silences the p53 transcriptional response to DNA damage. Mol Cell Proteomics. 2004;3:534–547. doi: 10.1074/mcp.M300089-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Bouchecareilh M, Higa A, Fribourg S, Moenner M, Chevet E. Peptides derived from the bifunctional kinase/RNase enzyme IRE1alpha modulate IRE1alpha activity and protect cells from endoplasmic reticulum stress. FASEB J. 2011;25:3115–3129. doi: 10.1096/fj.11-182931. [DOI] [PubMed] [Google Scholar]
- 31.Nicholson J, Neelagandan K, Huart AS, Ball K, Molloy MP, Hupp TR. An iTRAQ proteomics screen reveals the effects of the MDM2 binding ligand Nutlin-3 on cellular proteostasis. J Proteome Res. 2012;11:5464–5478. doi: 10.1021/pr300698d. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Naismith JH. A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr Purif. 2009;63:102–111. doi: 10.1016/j.pep.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh P, Shaffer SA, Scherl A, Holman C, Pfuetzner RA, Larson Freeman TJ, Miller SI, Hernandez P, Appel RD, Goodlett DR. Characterization of protein cross-links via mass spectrometry and an open-modification search strategy. Anal Chem. 2008;80:8799–8806. doi: 10.1021/ac801646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsutsui Y, Wintrode PL. Hydrogen/deuterium exchange-mass spectrometry: a powerful tool for probing protein structure, dynamics and interactions. Curr Med Chem. 2007;14:2344–2358. doi: 10.2174/092986707781745596. [DOI] [PubMed] [Google Scholar]
- 35.Marin M, Thallmair V, Ott T. The intrinsically disordered N-terminal region of AtREM1.3 remorin protein mediates protein–protein interactions. J Biol Chem. 2012;287:39982–39991. doi: 10.1074/jbc.M112.414292. [DOI] [PMC free article] [PubMed] [Google Scholar]