

Abstract
Bacterial response regulators (RRs) can regulate the expression of genes that confer antibiotic resistance; they contain a receiver and an effector domain and their ability to bind DNA is based on the dimerization state. This is triggered by phosphorylation of the receiver domain by a kinase. However, even in the absence of phosphorylation RRs can exist in equilibrium between monomers and dimers with phosphorylation shifting the equilibrium toward the dimer form. We have determined the crystal structure of the unphosphorylated dimeric BaeR from Escherichia coli. The dimer interface is formed by a domain swap at the receiver domain. In comparison with the unphosphorylated dimeric PhoP from Mycobacterium tuberculosis, BaeR displays an asymmetry of the effector domains.
Keywords: unphosphorylated response regulator, domain swap receiver domain, asymmetric dimer, effector domain
Introduction
Multidrug resistance is a growing problem in the treatment of infectious diseases and pathogenic bacteria have developed several mechanisms to confer resistance to antibiotics. Bacteria respond to antibiotics by regulating the expression of genes that code for various membrane bound multidrug resistant pumps and transporters. These pumps and transporters are able to extrude structurally unrelated antibiotics and drugs and their expression is regulated on the transcription level.1–6 One of the mechanisms that bacteria utilize for regulating the gene expression is the two-component signal transduction system (TCS).7 The TCS comprises of a histidine kinase (HK) and a response regulator (RR).
BaeSR belongs to the TCS that activates transcription of the mdtABC and the acrD multidrug efflux pump genes8,9 and increases resistance to deoxycholate, indole, and novobiocin in Escherichia coli10,11 and in addition to oxacillin, cloxacillin, and nafcillin in an ΔacrAB strain of Salmonella enterica.12 The BaeSR system can also sense copper and zinc suggesting a possible role for detoxification in S. enterica through the MdtABC and the AcrD pumps.13 The S. enterica BaeSR and CpxAR influence the outer membrane protein OmpD and confer resistance to ceftriaxone.14 The spy gene which encodes for a periplasmic chaperone is also under the same influence.15
BaeR belongs to the OmpR/PhoB family of transcription factor RRs.16 The RR consists of a receiver and an effector domain that are linked together through a flexible linker. The N-terminal receiver domain contains the conserved aspartate and the C-terminal effector domain is a DNA-binding domain involved in the transcriptional regulation of genes. There are many structures of isolated receiver and effector domains from the OmpR/PhoB family but only a few full length ones.17–22 Based on the phosphorylation state of the key Asp and orientation of the switch residues, Tyr/Phe and Ser/Thr, in the receiver domain, they can be classified as active or inactive23 and this is linked with dimerization which is essential for binding DNA. RRs exist in equilibrium between active and inactive states and the equilibrium is shifted toward the active state upon phosphorylation. Phosphorylation of the RR triggers dimerization at their α4-β5-α5 face of the receiver domain24,25 and it has been shown to be essential for regulation. In monomeric inactive RR structures, OmpR/PhoB family, the receiver and effector domains show interdomain contacts involving the switch residues with the exception of DrrD.17 The interdomain contacts in the inactive RRs have been proposed to represent an inhibitory state since the effector domains cannot dimerize without steric clashes. Activation would require disruption of this interface and it has been proposed that phosphorylation of the receiver domain and rotation of the switch residues would relieve the interdomain contacts, allowing formation of the dimer interface and subsequently transmitting changes to the effector domain.20 In the inactive dimeric structure of PhoP from Mycobacterium tuberculosis there are no interdomain contacts and the dimer is formed by the α4-β5-α5 face.22 To-date there is no structure of a full length active RR from the OmpR/PhoB family.
In this study, we determined the crystal structure of the full length BaeR from E. coli at 3.3 Å resolution. The crystal structure shows a very compact domain swap dimer in the receiver domain. The unphosphorylated BaeR structure is a dimer and does not display any interdomain contacts.
Results
Overall structure of BaeR
The structure of full length BaeR from E. coli was determined using experimental phases from single wavelength anomalous dispersion dataset (λ = 1.255 Å) from a holmium derivatized protein co-crystal soaked in 20 mM Ta6Br12 cluster at 3.3 Å resolution in the C2 space group (Supporting Information Fig. S1A). The asymmetric unit contains 12 copies of BaeR related by a sixfold non-crystallographic symmetry (NCS) axis. We found that it was necessary to co-crystallize the protein with holmium in order to improve the diffraction quality of the crystals. The diffraction quality improved further after soaking them in the tantalum cluster. Data at low resolution, 3.8–4.0 Å resolution, were also collected for the native, selenomethionine, and holmium derivatized proteins. The holmium derivatized protein and tantalum cluster soaks resulted in breaking the symmetry from P3121 to C2 symmetry which improved the diffraction by slight rearrangements in the crystal packing; comparison of the BaeR molecules between the two different space groups did not reveal any structural differences. During the refinement and model building, we noticed extra electron density for all the cysteines, and we assigned it to arsenic (anomalous difference maps) from the cacodylate buffer (Supporting Information Fig. S1B).
In the crystal structure, BaeR is in dimeric form (Fig. 1) and the two protomers are related by a twofold rotational symmetry. The overall structure of BaeR resembles other full-length RR proteins of the OmpR/PhoB family; it contains an N-terminal receiver domain (residues 10–126) and a C-terminal effector DNA-binding domain (residues 141–240). The two domains are connected together by a linker of 15 residues in length; in our electron density maps we can observe some density for the linker but the resolution is not sufficient to be built (Supporting Information Fig. S2). Receiver domains of RRs comprise of a five-stranded β-core flanked by five α-helices and have a characteristic β1-α1-β2-α2-β3-α3-β4-α4-β5-α5 topology22 [Fig. 2(A)]. The receiver domain of BaeR is incomplete with topology β1-α1-β2-α2-β3-α3-β4-α5. The complete receiver domain is formed from a domain swap with the second protomer that provides the α4-β5 elements. Domain swapping has been reported before for another RR, RegX3 that also forms a domain-swap dimer in its receiver domain21; the complete receiver domain is formed from the β1-α1-β2-α2-β3-α3-β4 of the first molecule and the α4-β5-α5 elements are provided from the domain swap with the second molecule. In BaeR, the domain swapped region has a buried surface of 1370 Å2/protomer whereas RegX3 has 2357 Å2/protomer. β5 of the incomplete receiver domain aligns parallel to the domain-swapped four β-sheet core and forms a complete five stranded core. In BaeR structure, helix α4 (residues 92–101) has completely unwound; for consistency with the other published structures and for discussion purposes, it will be referred as “helix α4.” The unwound “helix α4” sits adjacent to the α3 of the domain swap dimer. The two receiver domains of the BaeR dimer can be superimposed with an rmsd of 2.55 Å over 110 Cα atoms. Closer inspection of the phosphorylation site β3-α3 reveals that the two protomers show structural differences; residues 62–68 have been flipped over by 80° and then displaced by 8.0 Å relative to each other.
Figure 1.
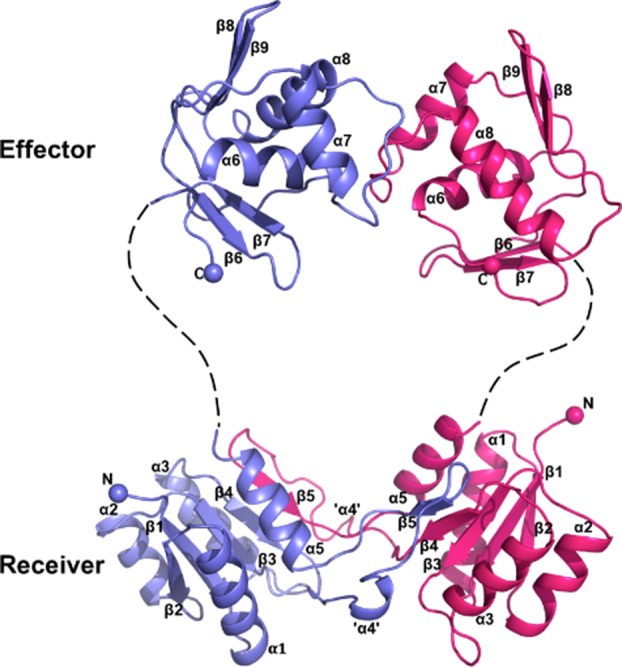
Ribbon representation of the BaeR dimer. One protomer is shown in light blue and the other in magenta. The linker region is missing from the final structure but for visualization purposes we have drawn it as dotted line (Supporting Information Fig. S2). The complete receiver domain is formed from a domain swap between the β-core domain and α4-β5. An interactive view is available in the electronic version of the article. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 2.
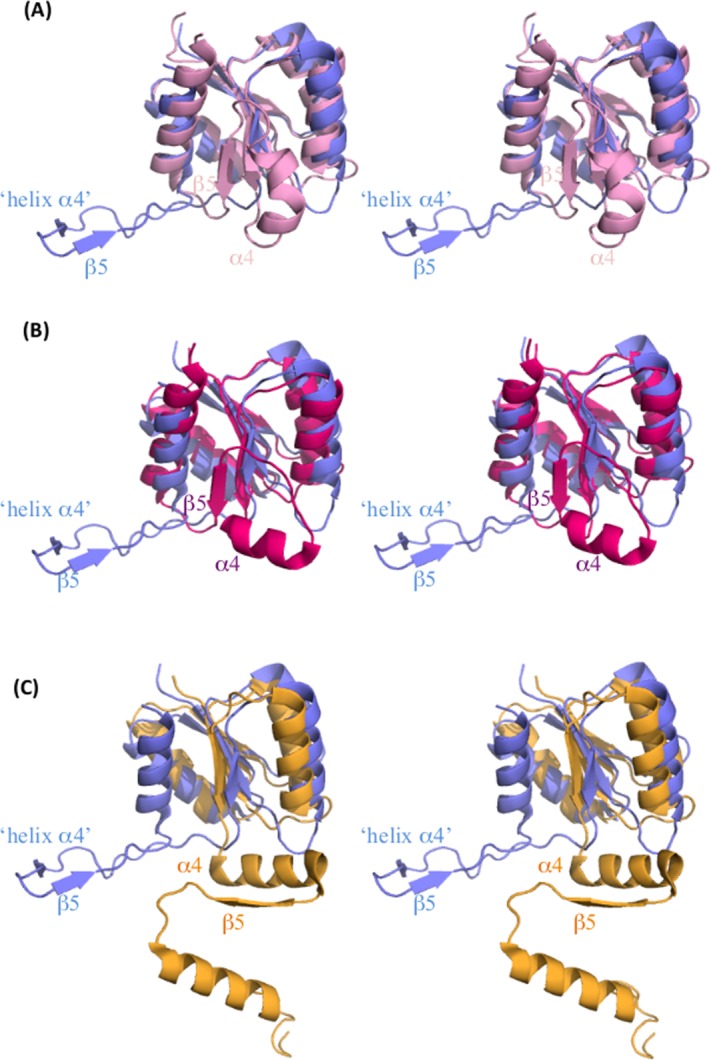
Structural differences in the receiver domain of RRs. Stereo-representation of the superimposed receiver domain of BaeR (blue) with (A) PhoP (pink, PDB ID 3R0J), (B) DrrD (magenta, PDB ID 1KGS), and (C) RegX3 (yellow, PDB ID 2OQR). Only the “helix α4”- β5 and equivalent elements are labeled. α4 of DrrD is sitting perpendicular to the β-core domain. In BaeR, rotation along the N-terminus of α5 has allowed the “helix α4”-β5 elements to swing out of the core domain. The α4- β5-α5 elements of RegX3 are involved in the domain swap. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
The overall structure of the BaeR effector domain is very similar to other effector domains of full length RRs of the OmpR/PhoB family.17 It has a winged helix-turn-helix (wHTH) motif consisting of a two-stranded antiparallel β-sheet followed by a three-helix bundle and a C-terminal β-hairpin (Fig. 1). Helix α8 is the recognition helix that interacts with the DNA and its accessibility to bind DNA is determined by the conformation of the transactivation loop (involved in transcription activation) between α7 and α8; in BaeR, the current conformation prevents it from binding DNA. The effector domain is linked to the receiver domain through a long linker but the two domains do not have any interdomain contacts.
Unwound “helix α4”
A striking feature of BaeR is that helix α4 of the receiver domain is completely unwound in both protomers (Fig. 1). The structural plasticity of α4 has been previously reported.26 Helix α4 is usually positioned parallel to the five-stranded β-sheet core of the receiver domain but in DrrD it has been displaced and adopted an almost perpendicular configuration relative to the five-stranded β-sheet core as a result of the rotation of the α4 helical segment about its own axis17 [Fig. 2(B,C)]. In BaeR, the unwound “helix α4” is sitting perpendicular to the β-sheet core domain; the hydrophobic residues are pointing toward the hydrophobic core of the adjacent domain swapped dimer compensating for the unwinding. In turn, β5 is also forced to adopt the same perpendicular orientation and is displaced from the core domain. Interactions from α3 of the domain swapped dimer place the unwound helix parallel to the hydrophobic core to form the complete receiver domain. This hydrophobic pocket is formed from Ile94, Leu97, and Leu100 of the unwound α4 and Leu62 and Leu70 from the domain swap α3. The side chain of Arg96 and main chain of Asp105 form a hydrogen bond with the main chain of Leu62 and the side chain of the domain swapped Arg77, respectively. Glu93 forms a hydrogen bond with Tyr29 from helix α1 of the same molecule. The interactions from the main molecule and the domain swapped protomer appear to stabilize the unwound helix (Supporting Information Fig. S3).
Discussion
BaeR represents the second example of a domain swapped dimer of a full length RR of the OmpR/PhoB family. The formation of RR domain swapped dimers in RegX3 has been previously associated with high protein concentrations or crystallization conditions.21,25 Both RegX3 and BaeR protein structures have a complete active site for phosphorylation which can be linked to the mechanism of activation via the switch residues (Supporting Information Fig. S4). The intrinsic plasticity of helix α4 has been previously reported and in our structure it is completely unwound. We cannot exclude that formation of the domain swapped dimer is due to the flexible nature of α4. Upon phosphorylation α4 could be stabilized and adopt a more helical conformation which would also bring β5 to the core domain and disrupt the domain swap.
In order for RRs to bind their target DNA, they need to dimerize. Unphosphorylated RRs exist in equilibrium between monomers and dimers, and phosphorylation pushes the equilibrium toward dimer, which is associated with DNA binding. Dimerization of the RRs, OmpR/PhoB family, usually involves the α4-β5-α5 face of the receiver domain. The BaeR dimer interface is formed between the α4 and β5 face elements from the domain swapped dimers. Isolated beryllium fluoride activated receiver domains usually dimerize in a face-to-face orientation. The BaeR dimers sit almost 90° from one another as a result of the domain swap suggesting a novel way of dimerization but with the same elements as observed in other structures. The fully phosphorylated BaeR might result in a tighter dimer interface; the inactive PhoP dimerizes at the α4-β5-α5 face whereas the isolated activated receiver domain has a more compact interface.22 Some inactive RRs display interdomain contacts and are believed to be inhibitory in forming dimers; rotation of the switch residues would relieve these contacts and allow dimerization upon phosphorylation and bind DNA. The current domain organization of the BaeR dimer does not allow for any interdomain contacts since it would create steric clashes. In addition, the BaeR dimer has an asymmetric conformation between the receiver and effector domain relative to the PhoP dimer. This domain organization probably explains the ability of unphosphorylated BaeR to bind its target DNA.10
In our structure, the two effector domains form weak bonds through their transactivation loop, stabilizing the extended receiver–effector interface (crystal contacts). From the crystal structure of the isolated effector domain of PhoB bound to DNA, the current conformation would prevent the protein from accessing the major groove. The PhoB binds to tandem repeat DNA half-sites with translational symmetry in a head-to-tail orientation.27 For BaeR to bind DNA it would require re-orientation of the effector domains; the rather flexible linker and absence of interdomain contacts would easily allow re-orientation of the domain in the presence of DNA. Another possible mode of binding would be in a face-to-face similar to the OmpR-DNA model.28 The unwinding of the last turn of α5 permits the effector domains to freely rotate.
Materials and Methods
Cloning
Full length baeR from E. coli MG1655 was cloned into the pEHis/TEV vector. The sequenced clone had a Thr71Met mutation that we decided not to correct since we could use it in selenomethionine derivatization and phasing.
Overexpression and purification
BaeR was transformed in BL21(DE3) cells. Cells were grown in LB medium supplemented with 34 µg/L kanamycin to an OD600 0.8 at 300 K and expression was induced by adding 1 mM isopropyl-1-thio-β-d-galactopyranoside for further 4 h. Cells were harvested by centrifugation at 6,000g.
All purification steps were carried out at 277 K. Cells were broken using a Constant Cell Disruption system. The unbroken cells and cell membranes were removed by centrifugation at 150,000g for 1 h. The supernatant was adjusted to 20 mM imidazole and passed over a 5 mL His-trap column HP, equilibrated with three column volumes (CV) of 20 mM imidazole in phosphate buffered saline (PBS). The column was washed with five CV of 30 mM imidazole in PBS. The protein was eluted with five CV of 500 mM imidazole in PBS. The His6-tag was cleaved from the protein using Tobacco Etch Virus (TEV) protease at a 1:100 ratio and dialyzed in 2 L of 20 mM Tris pH 7.5, 150 mM NaCl overnight with stirring. The dialyzed protein was passed over a 5 mL His-trap column equilibrated in dialysis buffer. The flow through was collected and injected over a Superdex 75 10/300 equilibrated in 20 mM Tris pH 8.0, 50 mM NaCl, and 5 mM dithiothreitol (DTT). The fractions containing BaeR were collected and concentrated using a 10 kDa concentrator.
Crystallization
Initial vapor diffusion sitting-drop crystallization trials were carried out using a Mosquito crystallization robot (TTP LabTech) from a protein solution at 30 mg/mL at 277 K and 293 K; 100 nL protein solution was mixed with 100 nL crystallization buffer. BaeR crystals appeared after 5 days in 1.2 M sodium acetate and 0.1 M sodium cacodylate, pH 8.4 at 277 K. Crystal quality and size were improved by seeding pre-equilibrated hanging-drops (mixing 1 µL protein with 1 µL crystallization buffer) overnight at 277 K; the precipitant concentration was reduced by 5%. The diffraction quality of the crystals was further improved by co-crystallizing the protein with 1 mM Ho (III). Crystals were derivatized by adding 20 mM Ta6Br12 cluster (Jena Biosciences) to the crystallization drop and were soaked for 24 h.
Data collection and structure determination
Crystals were transferred into crystallization solution containing 20% glycerol and were flash frozen into liquid nitrogen prior to data collection. Data were collected at beamline ID14-4 at ERSF, I02 and I04 at Diamond Light Source. Data were processed with xia2.29 The data collection statistics are summarized in Supporting Information Table S1.
Initial Ta heavy atom positions were determined by SHELXD30 using a SAD dataset from a single crystal containing both Ta6Br12 and Ho at a wavelength of 1.255 Å; the sites were refined and initial phases were calculated using autoSHARP31 assuming solvent content of 70% and 12 copies of BaeR in the asymmetric unit. The selenomethionine data were used to calculate anomalous difference maps and the sites were used for correct tracing of the sequence (Supporting Information Table S2). At this stage data from a crystal containing only Ta6Br12 was obtained at 3.3 Å resolution and the current model was used as a search model for molecular replacement in Phaser.32 The model underwent multiple rounds of model building using Coot and refinement using Phenix refine.33 In the last stages of refinement, the six dimers were restrained by sixfold NCS. The final model was refined to 3.3 Å resolution with an Rfactor of 22.52% and Rfree of 24.48%. The Ramachandran has 95.1% in favored regions, 4.66% in allowed, and 0.23% outliers as calculated by MolProbity. The Ramachandran outliers are in a loop region with poor density and correspond to a Gly.
The coordinates and structure factors have been deposited with the RCSB Protein Data Bank with PDB ID code 4B09.
Acknowledgments
Authors would like to acknowledge the Diamond Light Source and ESRF synchrotrons for beamtime. They would also like to thank Dr Alexander Cameron for critical reading of the manuscript.
Glossary
- HK
histidine kinase
- RR
response regulator
- TCS
two-component signal transduction system.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Lomovskaya O, Lewis K, Matin A. EmrR is a negative regulator of the Escherichia coli multidrug resistance pump EmrAB. J Bacteriol. 1995;177:2328–2334. doi: 10.1128/jb.177.9.2328-2334.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma D, Alberti M, Lynch C, Nikaido H, Hearst JE. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol Microbiol. 1996;19:101–112. doi: 10.1046/j.1365-2958.1996.357881.x. [DOI] [PubMed] [Google Scholar]
- 3.Okusu H, Ma D, Nikaido H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol. 1996;178:306–308. doi: 10.1128/jb.178.1.306-308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanaka T, Horii T, Shibayama K, Sato K, Ohsuka S, Arakawa Y, Yamaki K, Takagi K, Ohta M. RobA-induced multiple antibiotic resistance largely depends on the activation of the AcrAB efflux. Microbiol Immunol. 1997;41:697–702. doi: 10.1111/j.1348-0421.1997.tb01913.x. [DOI] [PubMed] [Google Scholar]
- 5.White DG, Goldman JD, Demple B, Levy SB. Role of the acrAB locus in organic solvent tolerance mediated by expression of marA, soxS, or robA in Escherichia coli. J Bacteriol. 1997;179:6122–6126. doi: 10.1128/jb.179.19.6122-6126.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiong A, Gottman A, Park C, Baetens M, Pandza S, Matin A. The EmrR protein represses the Escherichia coli emrRAB multidrug resistance operon by directly binding to its promoter region. Antimicrob Agents Chemother. 2000;44:2905–2907. doi: 10.1128/aac.44.10.2905-2907.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 8.Nagakubo S, Nishino K, Hirata T, Yamaguchi A. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J Bacteriol. 2002;184:4161–4167. doi: 10.1128/JB.184.15.4161-4167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leblanc SK, Oates CW, Raivio TL. Characterization of the induction and cellular role of the BaeSR two-component envelope stress response of Escherichia coli. J Bacteriol. 2011;193:3367–3375. doi: 10.1128/JB.01534-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baranova N, Nikaido H. The baeSR two-component regulatory system activates transcription of the yegMNOB (mdtABCD) transporter gene cluster in Escherichia coli and increases its resistance to novobiocin and deoxycholate. J Bacteriol. 2002;184:4168–4176. doi: 10.1128/JB.184.15.4168-4176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirakawa H, Inazumi Y, Masaki T, Hirata T, Yamaguchi A. Indole induces the expression of multidrug exporter genes in Escherichia coli. Mol Microbiol. 2005;55:1113–1126. doi: 10.1111/j.1365-2958.2004.04449.x. [DOI] [PubMed] [Google Scholar]
- 12.Appia-Ayme C, Patrick EJ, Sullivan M, Alston MJ, Field SJ, Abuoun M, Anjum MF, Rowley G. Novel inducers of the envelope stress response BaeSR in Salmonella typhimurium: BaeR is critically required for tungstate waste disposal. PLoS One. 2011;6:e23713. doi: 10.1371/journal.pone.0023713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishino K, Nikaido E, Yamaguchi A. Regulation of multidrug efflux systems involved in multidrug and metal resistance of Salmonella enterica serovar Typhimurium. J Bacteriol. 2007;189:9066–9075. doi: 10.1128/JB.01045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu WS, Chen HW, Zhang RY, Huang CY, Shen CF. The expression levels of outer membrane proteins STM1530 and OmpD, which are influenced by the CpxAR and BaeSR two-component systems, play important roles in the ceftriaxone resistance of Salmonella enterica serovar Typhimurium. Antimicrob Agents Chemother. 2011;55:3829–3837. doi: 10.1128/AAC.00216-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto K, Ogasawara H, Ishihama A. Involvement of multiple transcription factors for metal-induced spy gene expression in Escherichia coli. J Biotechnol. 2008;133:196–200. doi: 10.1016/j.jbiotec.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Martinez-Hackert E, Stock AM. Structural relationships in the OmpR family of winged-helix transcription factors. J Mol Biol. 1997;269:301–312. doi: 10.1006/jmbi.1997.1065. [DOI] [PubMed] [Google Scholar]
- 17.Buckler DR, Zhou Y, Stock AM. Evidence of intradomain and interdomain flexibility in an OmpR/PhoB homolog from Thermotoga maritima. Structure. 2002;10:153–164. doi: 10.1016/s0969-2126(01)00706-7. [DOI] [PubMed] [Google Scholar]
- 18.Robinson VL, Wu T, Stock AM. Structural analysis of the domain interface in DrrB, a response regulator of the OmpR/PhoB subfamily. J Bacteriol. 2003;185:4186–4194. doi: 10.1128/JB.185.14.4186-4194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nowak E, Panjikar S, Konarev P, Svergun DI, Tucker PA. The structural basis of signal transduction for the response regulator PrrA from Mycobacterium tuberculosis. J Biol Chem. 2006;281:9659–9666. doi: 10.1074/jbc.M512004200. [DOI] [PubMed] [Google Scholar]
- 20.Friedland N, Mack TR, Yu M, Hung LW, Terwilliger TC, Waldo GS, Stock AM. Domain orientation in the inactive response regulator Mycobacterium tuberculosis MtrA provides a barrier to activation. Biochemistry. 2007;46:6733–6743. doi: 10.1021/bi602546q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King-Scott J, Nowak E, Mylonas E, Panjikar S, Roessle M, Svergun DI, Tucker PA. The structure of a full-length response regulator from Mycobacterium tuberculosis in a stabilized three-dimensional domain-swapped, activated state. J Biol Chem. 2007;282:37717–37729. doi: 10.1074/jbc.M705081200. [DOI] [PubMed] [Google Scholar]
- 22.Menon S, Wang S. Structure of the response regulator PhoP from Mycobacterium tuberculosis reveals a dimer through the receiver domain. Biochemistry. 2011;50:5948–5957. doi: 10.1021/bi2005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bachhawat P, Swapna GV, Montelione GT, Stock AM. Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure. 2005;13:1353–1363. doi: 10.1016/j.str.2005.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toro-Roman A, Wu T, Stock AM. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci. 2005;14:3077–3088. doi: 10.1110/ps.051722805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao R, Stock AM. Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr Opin Microbiol. 2010;13:160–167. doi: 10.1016/j.mib.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milani M, Leoni L, Rampioni G, Zennaro E, Ascenzi P, Bolognesi M. An active-like structure in the unphosphorylated StyR response regulator suggests a phosphorylation-dependent allosteric activation mechanism. Structure. 2005;13:1289–1297. doi: 10.1016/j.str.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 27.Blanco AG, Sola M, Gomis-Ruth FX, Coll M. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure. 2002;10:701–713. doi: 10.1016/s0969-2126(02)00761-x. [DOI] [PubMed] [Google Scholar]
- 28.Rhee JE, Sheng W, Morgan LK, Nolet R, Liao X, Kenney LJ. Amino acids important for DNA recognition by the response regulator OmpR. J Biol Chem. 2008;283:8664–8677. doi: 10.1074/jbc.M705550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winter G. xia2: an expert system for macromolecular crystallography data reduction. J Appl Cryst. 2010;43:186–190. [Google Scholar]
- 30.Sheldrick GM. A short history of SHELX. Acta Cryst. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 31.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 32.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams PD, Afonine PV, Bunkœczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.