

SnoN directly binds ALK1 on the plasma membrane, enhancing Smad1/5 activation, and is required for normal angiogenesis.
Abstract
In endothelial cells, two type I receptors of the transforming growth factor β (TGF-β) family, ALK1 and ALK5, coordinate to regulate embryonic angiogenesis in response to BMP9/10 and TGF-β. Whereas TGF-β binds to and activates ALK5, leading to Smad2/3 phosphorylation and inhibition of endothelial cell proliferation and migration, BMP9/10 and TGF-β also bind to ALK1, resulting in the activation of Smad1/5. SnoN is a negative regulator of ALK5 signaling through the binding and repression of Smad2/3. Here we uncover a positive role of SnoN in enhancing Smad1/5 activation in endothelial cells to promote angiogenesis. Upon ligand binding, SnoN directly bound to ALK1 on the plasma membrane and facilitated the interaction between ALK1 and Smad1/5, enhancing Smad1/5 phosphorylation. Disruption of this SnoN–Smad interaction impaired Smad1/5 activation and up-regulated Smad2/3 activity. This resulted in defective angiogenesis and arteriovenous malformations, leading to embryonic lethality at E12.5. Thus, SnoN is essential for TGF-β/BMP9-dependent biological processes by its ability to both positively and negatively modulate the activities of Smad-dependent pathways.
Introduction
Angiogenesis, the formation of new blood vessels from preexisting vessels, is essential for many physiological processes including embryonic development, wound healing, tissue repair, and regeneration (Carmeliet, 2003; Chung and Ferrara, 2011). During angiogenesis, the primary vascular plexus formed during the initial vasculogenesis undergoes a remodeling process in which the initial honeycomb-like pattern of vasculature is remodeled into a branched complex vascular network. Angiogenesis is composed of an activation phase when endothelial cells (ECs) are activated to undergo proliferation, migration, branching, and tube formation followed by a resolution phase. During resolution, EC proliferation stops while pericytes or vascular smooth muscle cells are induced to differentiate and recruited to the newly formed vessels to establish the vessel wall (Pepper, 1997). The complex process of vascular development is regulated by many growth factors, among which members of the transforming growth factor β (TGF-β) family, including TGF-β and BMP9/10, play a central and indispensable role (Goumans et al., 2003a; Pardali et al., 2010). More precisely, TGF-β is required for the formation and remodeling of the primary vascular plexus by regulating the proliferation, migration, and differentiation of vascular endothelial cells. Furthermore, TGF-β is required for the differentiation and recruitment of vascular smooth muscle cells and the interaction between endothelial cells and vascular smooth muscle cells. Inactivation of TGF-β ligand, its associated receptors, and downstream Smad proteins all lead to severe defects in yolk sac and embryonic angiogenesis, ultimately resulting in embryonic lethality (Mummery, 2001; Goumans et al., 2003a; Wu and Hill, 2009; Pardali et al., 2010).
TGF-β signals through the cell surface type II and I receptors and through downstream Smad proteins. Binding of TGF-β to its receptors results in activation of the ALK5 receptor kinase, which then phosphorylates and activates Smad2 and Smad3. The phosphorylated Smad2 and Smad3 form heterotrimeric complexes with Smad4 and translocate into the nucleus where they activate transcription of TGF-β target genes (Attisano and Wrana, 2002; Shi and Massagué, 2003; Derynck and Miyazono, 2008; Guo and Wang, 2009; Moustakas and Heldin, 2009). In endothelial cells, in addition to the conventional ALK5 type I receptor, an endothelial cell–specific type I receptor, ALK1, also mediates TGF-β signaling to activate the BMP R-Smads (Smad1, 5, and 8; Goumans et al., 2002). Recent advances suggest that in endothelial cells, BMP9/10 activates ALK1 and the downstream Smad1/5/8 to regulate endothelial cell proliferation and differentiation and might in fact be the bona fide ligand for ALK1 (David et al., 2007). It is known that the disruption of TGF-β or BMP9/10 signaling through the elimination of critical components of either pathway, including ALK1, ALK5, or Smad proteins, leads to defective maturation of the primary vascular plexus, resulting in dilated and fragile vessels and decreased integrity of vessel walls. These mice die in mid-gestation due to defective yolk sac and embryo angiogenesis (Pardali et al., 2010). In addition to these defects, ALK1-deficient embryos, but not ALK5-null embryos, also exhibit arteriovenous malformations (AVMs), due to the fusion of major arteries and veins, and a significantly reduced expression of Ephrin-B2, an artery marker (Urness et al., 2000; Park et al., 2008).
SnoN has been shown to be an important negative regulator of the Smad proteins (Luo, 2004). SnoN interacts with Smad2, Smad3, and Smad4 and represses their transactivation activity by disrupting the functional heteromeric Smad complexes, recruiting transcription corepressor complexes and blocking the binding of transcriptional coactivators to the Smads (Akiyoshi et al., 1999; Stroschein et al., 1999; Wu et al., 2002; Luo, 2004). Overexpression of SnoN can block TGF-β–induced growth arrest (Stroschein et al., 1999; Sun et al., 1999), a mechanism that is likely responsible for the pro-oncogenic activity of SnoN (He et al., 2003). To date, the majority of studies on SnoN have focused on its role in human cancer cells, whereas the function of SnoN in embryonic development is currently not well understood.
SnoN is ubiquitously expressed in all embryonic and adult tissues (Luo, 2004; Deheuninck and Luo, 2009; Jahchan and Luo, 2010; Zhu and Luo, 2012), and its expression is up-regulated during specific stages of development and tissue morphogenesis (Jahchan and Luo, 2010; Zhu and Luo, 2012), suggesting that it may play a role in embryonic development and normal tissue functions. Three snoN knockout mouse lines have been reported. Whereas one snoN-null line shows embryonic lethality before E3.5 (Shinagawa et al., 2000), the other two SnoN deletion mice are viable with only minor defects in T cell activation (Pearson-White and McDuffie, 2003). Thus, these knockout mice are not able to provide a definitive physiological role of SnoN in embryonic development.
In an effort to uncover the role of SnoN in regulating TGF-β signaling during embryonic development, we have generated a knock-in mouse (SnoNm/m) expressing a mutant SnoN defective in binding to the Smad proteins in the original snoN locus (Pan et al., 2009). In this paper, we report that SnoNm/m mice show partial embryonic lethality, mostly due to defects in angiogenesis in both the yolk sac and embryo body. Using mouse embryo endothelial cells (MEECs) derived from the knock-in embryos as well as human pulmonary artery endothelial cells (HPEACs), we have uncovered novel mechanisms by which SnoN enhances ALK1 signaling.
Results
SnoNm/m mice display partial embryonic lethality
To understand the functions of SnoN in regulation of Smad signaling during embryonic development, we generated a knock-in mouse by replacing the endogenous snoN gene with a mutant (mSnoN) that is defective in binding to both R-Smads and Co-Smad (Pan et al., 2009). In crossings of heterozygous SnoN knock-in mice (SnoN+/m) on either C57BL/6J or 129P2 background, the number of homozygous SnoNm/m pups was consistently lower than the expected 25% Mendelian ratio (unpublished data). Approximately 15.9% of the pups born from the SnoN+/m × SnoN+/m intercrossing were homozygous, 47.0% were heterozygous, and 37.1% were wild-type (WT), indicating that ∼36.4% of the homozygous embryos died before birth. This suggests that SnoNm/m mice display partial embryonic lethality. Interestingly, before E10.5–E11.5, 25% of embryos were homozygous for the knock-in allele while partial lethality could be observed after E11.5 (Table 1), indicating that embryonic lethality occurred largely between E11.5 and E12.5.
Table 1.
Partial embryonic lethality in SnoNm/m embryos
| Stage | SnoNm/m | SnoN+/m | SnoN+/+ | % |
| Postnatal | 43 | 127 | 100 | 15.9: 47.0: 37.1 |
| E16.5–E18.5 | 10 | 31 | 20 | 16.4: 50.8: 32.8 |
| E12.5 | 6 | 22 | 10 | 15.8: 57.9: 26.3 |
| E10.5–E11.5 | 15 | 28 | 17 | 25.0: 46.7: 28.3 |
Partial lethality of the SnoNm/m embryos occurs between E11.5 and E12.5. Offspring resulting from heterozygous intercrosses were genotyped at various embryonic developmental stages. Significant deviation from the Mendelian ratio is detected after E11.5.
Aberrant developmental phenotypes were observed as early as E8.75 in some of the SnoNm/m embryos (unpublished data). At E9.5, ∼22.7% of SnoNm/m embryos were retarded in growth, some of which failed to turn (Fig. 1 B; unpublished data). By E10.75, these mutants were much smaller than their littermate controls, and many of them exhibited cardiac abnormalities including pericardial effusion, a common manifestation of disrupted blood flow and osmotic imbalance (Fig. 1 D; Copp, 1995). From E11.5 to E12.75, some mutant embryos were resorbed, while the remaining embryos displayed dilated vessels and hemorrhage at various body locations (Fig. 1, F and H). These data suggest that SnoNm/m embryos may exhibit defects in the development of their vascular system.
Figure 1.
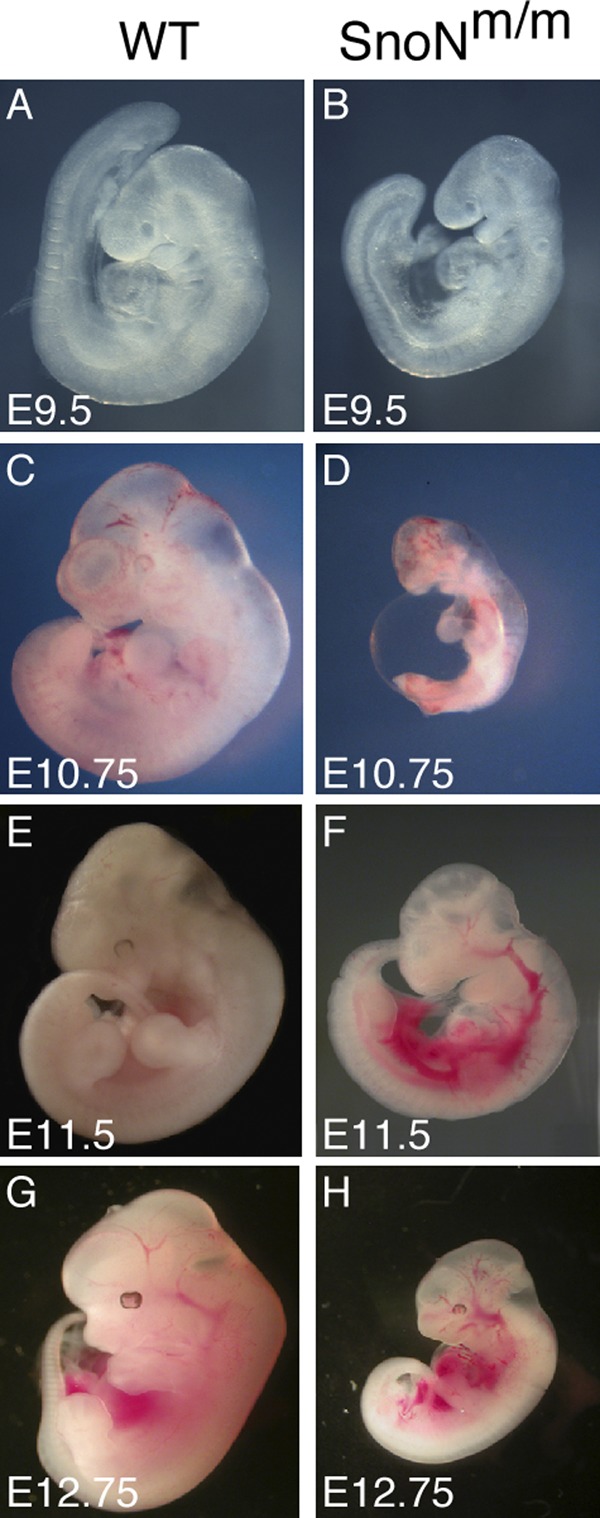
SnoNm/m embryos display growth retardation and cardiovascular defects. Lateral view of WT (A, C, E, and G) and mutant (B, D, F, and H) embryos at E9.5 (A and B), E10.75 (C and D), E11.5 (E and F) and E12.75 (G and H).
SnoNm/m embryos display defects in yolk sac angiogenesis
The yolk sac has long been recognized as the first site of vasculogenesis and hematopoiesis during embryonic development (Flamme et al., 1997). In WT embryos, beginning around E9.0, the primitive vascular plexus of the yolk sac undergoes a remodeling process in which the initial honeycomb-like pattern of vasculature is remodeled into a more branched pattern of mature vitelline vessels (Fig. 2 A, boxed area; Risau, 1997). At E10.5, the large, well-organized, tree-like vascular network was clearly visible in WT yolk sacs (Fig. 2 B). In contrast, more than 20% of the SnoNm/m yolk sacs looked pale, and the surface of these yolk sacs was dimpled and wrinkled with blood cells scattered in some regions (Fig. 2 A, boxed area). No distinct blood vessels were evident (Fig. 2 B). Whole-mount immunostaining of PECAM-1 (platelet endothelial cell adhesion molecule 1), an endothelial cell marker, showed that, at E10.5, the WT yolk sacs displayed an organized vascular network of branching vessels that were lined with PECAM-1–positive endothelial cells, indicating that the primitive vascular plexus had been fully remodeled (Fig. 2 C). In contrast, although the initial primitive vascular plexus had formed in SnoNm/m yolk sacs, this remodeling process did not take place and the vessels remained in the honeycomb-like pattern (Fig. 2 C). Histological analysis of SnoNm/m yolk sacs revealed that the vasculature was dilated, and the endoderm and mesoderm layers were barely connected (Fig. 2 D, boxed area). Immunohistochemical staining of α-smooth muscle actin (SMA), one of the earliest markers of pericytes in the developing mouse embryos (Takahashi et al., 1996), also confirmed that fewer vascular smooth muscle cells (VSMCs) were found surrounding the vessels in the mutant yolk sacs (Fig. 2 E). Therefore, the vasculature in SnoNm/m yolk sacs was not properly supported by the VSMCs.
Figure 2.

SnoNm/m embryos exhibit defects in yolk sac angiogenesis. Gross morphology of yolk sac of WT (top) and mutant (bottom) embryos at E9.5 (A) and E10.5 (B). The boxed areas in A are shown at a higher amplification to reveal the difference in vasculature between WT and mutant yolk sacs. (C) Whole-mount α-PECAM-1 staining of E10.5 yolk sac from WT (left) or SnoNm/m (right) mice. (D) H&E staining of cross sections of WT and SnoNm/m yolk sacs. The black arrowheads indicate blood vessels. The red arrow in the boxed area shows the area where the endoderm and mesoderm failed to connect properly. (E) α-SMA staining of cross sections of both WT and SnoNm/m yolk sacs shows a reduced number of smooth muscle cells surrounding the endothelium of the mutant yolk sac. The boxed area highlights the α-SMA staining in a single vessel in WT or SnoNm/m yolk sacs. (F) Semi-quantitative RT-PCR analysis of angiogenesis regulators from WT or SnoNm/m yolk sacs. The mRNA levels of these genes were normalized to that of β-actin. The data shown are from a single representative experiment out of three repeats. The relative fold changes in the SnoNm/m yolk sacs over that of WT yolk sacs were quantified and shown in the bar graphs below. Bars: (A, C, D, and E) 100 µm.
Consistent with the aberrant morphological and histological features in angiogenesis, the expression patterns of some of the molecules known to be involved in regulation of angiogenesis, including Angiopoietin 1 and 2 (Ang1 and Ang2), Angiopoietin 1 receptor (Tie2), and vascular endothelial growth factor receptor 1–3 (Flt1, Flk1, and Flt4), were all altered in SnoNm/m yolk sacs (Fig. 2 F). In addition, the expression of several Notch target genes, Hey1, Hey2, and Hes1, was decreased (Fig. 2 F), consistent with the idea that Notch signaling may be regulated by Flt4 and possibly BMP signaling (Larrivée et al., 2012). These changes confirmed that the normal angiogenesis program has been disrupted by the mutations of SnoN.
Taken together, our results suggest that a loss of the SnoN–Smads interaction impairs yolk sac angiogenesis, but not vasculogenesis.
SnoNm/m embryos also display angiogenesis defects
To determine if the vasculature of the embryo proper was also affected by the mutations of SnoN, we performed α-PECAM-1 whole-mount immunohistochemistry to visualize the vasculature of whole embryo bodies. At E9.5, the vascular system in WT embryos had begun the remodeling process. The branched cephalic large vessels and capillary networks were readily distinguishable in the dorsal midbrain (Fig. 3 A). In contrast, the cephalic plexus in SnoNm/m embryos failed to remodel into a branched vascular network and instead remained as a plexus with minor sprouting (Fig. 3 A). Although WT embryos displayed a continuous dorsal aorta along the length of the embryo and a well-defined cardinal vein from head to heart (Fig. 3 B), in SnoNm/m embryos the anterior cardinal vein appeared deformed and unusually close to the dorsal aorta, while the edges of the dorsal aorta were not clearly delineated (Fig. 3 B), suggesting a defect in embryonic angiogenesis. Indeed, sectioning of these α-PECAM-1–stained embryos at the level depicted in Fig. 3 B revealed that the dorsal aortas of the SnoNm/m embryos were enlarged and irregular in shape on both sides (Fig. 3 C). The relative position of the dorsal aortas was much closer to the anterior cardinal veins in the SnoNm/m embryos than that of the corresponding dorsal aortas in WT embryos, and one of the anterior cardinal veins was severely deformed (Fig. 3 C). The deformed vessels observed in E9.5 SnoNm/m embryos may be partially due to the defect in the differentiation and/or recruitment of vascular smooth muscle cells to the endothelial cells because in situ hybridization staining of E9.5 embryos with α-SMA detected a significant decrease in α-SMA expression around the dorsal aorta in these mutant embryos (Fig. 3 D). By E11.5, WT embryos have completed the remodeling process and exhibited extensive branching in the vasculature (Fig. 3 E). In contrast, a number of SnoNm/m embryos with pronounced angiogenesis defects still displayed the honeycomb-like pattern of primary capillary plexus, indicating that the vascular remodeling failed to take place. These embryos exhibited severe developmental delay and died around E11.75 to E12.5. The less severely affected SnoNm/m embryos survived through the vasculature remodeling process, but often displayed dilated vessels, developed severe hemorrhage at gut, brain, or heart regions, and died at a later stage (around E13.5; unpublished data).
Figure 3.

SnoNm/m mice display angiogenesis defects in the embryo proper. (A) α-PECAM-1 whole-mount immunostaining of the head region in E9.5 embryos shows a remodeled vascular plexus in the WT embryos but not in the SnoNm/m embryos. (B) α-PECAM-1 whole-mount immunostaining of the heart region reveals a deformed anterior cardinal vein in E9.5 SnoNm/m embryo. acv, anterior cardinal vein (indicated by white arrows); da, dorsal aorta (indicated by yellow arrowheads). The dashed black lines depict the level of the cross sections shown in C. (C) α-PECAM-1 immunohistochemistry of comparable paraffin sections in E9.5 WT and SnoNm/m embryos shows enlarged dorsal aortas (da) and deformed left anterior cardinal vein (acv) in the SnoNm/m embryo. (D) Whole-mount in situ α-SMA staining of E9.5 embryos. (E) Vasculature in the head area of E11.5 embryos as visualized by α-PECAM-1 whole-mount staining. Bars: (A–C) 100 µm; (D) 0.5 mm; (E) 0.25 mm.
SnoNm/m mice display arteriovenous malformations
Although mutations in both ALK1 and ALK5 signaling affect yolk sac and embryonic angiogenesis, ALK1- or Smad1/5-deficient embryos exhibit unique features that are not observed in mice defective in ALK5 signaling (Yang et al., 1999; Oh et al., 2000; Urness et al., 2000; Larsson et al., 2001). In particular, ALK1 is expressed at high levels in the arteries and is crucial for maintaining the arterial–venous identity because mice lacking ALK1 showed lethal AVM in multiple locations. Interestingly, immunohistochemistry staining of SnoN in E9.5 embryos showed that in addition to its expression in the epithelial and mesenchymal compartments, SnoN was expressed at a higher level in the arteries than in the cardinal veins (Fig. 4 A), similar to the expression pattern of ALK1 (Seki et al., 2003). Consistent with this, the expression of the artery marker ephrinB2 was significantly reduced in SnoNm/m embryos (Fig. 4 B).
Figure 4.

SnoN is involved in the regulation of arterial identity through modulating ALK1 signaling. (A) Immunofluorescence staining of SnoN on paraffin sections from E9.5 WT or SnoNm/m embryos. ov, otic vesicle; cv, cardinal vein; da, dorsal aorta. Bar, 50 µm. (B) RT-PCR analysis of ephrinB2 transcript from WT or SnoNm/m yolk sacs. β-Actin was used as a loading control. Data represent mean ± SEM from three independent experiments. (C) α-PECAM-1 immunohistochemistry staining of paraffin sections from E11.5 WT or SnoNm/m embryos at the level of the heart. An AVM (red arrow) formed by the fusion of the right dorsal aorta (da, blue arrowheads) with the right common cardinal vein (ca, blue arrows) is readily seen in the SnoNm/m embryo. Bar, 50 µm. (D) India ink injection in E11.5 embryos. The boxed areas highlight the paths of blood outflow from the heart in WT and SnoNm/m embryos. In WT embryos, the heart ejected the ink through the bronchial arches into the entire length of the descending dorsal aorta (black arrows), whereas in the SnoNm/m embryos, the ink flew directly into both anterior cardinal veins and dilated dorsal aorta, and shunted back to the heart (red arrows). Bar. 1 mm. (E) No apparent AVMs in subdermal blood vessels surrounding the wounds of SnoNm/m mutants. Latex dye–injected vascular images in the wound areas (*) of dorsal skin and ear of control (a–c), SnoN mutants (d–f), and ALK1-inducible knockout (iKO; g–i) are shown. Bar, 2 mm.
At E11.5, cross sectioning at the caudal heart position further revealed that ∼8.9% of the SnoNm/m embryos exhibited dilated cardinal vein and AVM resulting from fusion of the dorsal aorta with the common cardinal vein (Fig. 4 C, red arrows). Consistent with the histological analysis, the ink injection assay also suggested AVM (Fig. 4 D). In WT control embryos, after ink was injected into the proximal outflow tract of the heart, the ink exited through the paired bronchial arch arteries to enter the dorsal aorta and traversed the entire length of the embryo (Fig. 4 D, top, black arrows). In contrast, in some of the SnoNm/m embryos, after exiting the distal outflow tract, the injected ink directly entered the venous circulation via the anterior cardinal vein in the anterior part of the embryos. The ink injection simultaneously entered the descending dorsal aorta and was shunted back into the heart via fusion of the dorsal aorta with the common cardinal vein (Fig. 4 D, bottom, red arrows; and unpublished data). Thus, SnoNm/m embryos displayed AVM, a phenotype similarly found in mice lacking ALK1 (Yang et al., 1999; Urness et al., 2000; Seki et al., 2003), suggesting that SnoN can affect ALK1 signaling in endothelial cells.
It has been shown that endothelial-specific deletion of the ALK1 allele in adult mice (ALK1-iKO) results in the development of AVM in the lung, brain, and GI tract and dorsal skin after wounding (Park et al., 2009). We therefore wondered whether the surviving adult SnoNm/m mice might also develop AVM, similar to mice with a deletion of ALK1. To do this, we performed latex dye injection in four pairs of 2-mo-old WT and SnoNm/m mice after wounding as well as in two pairs of older 12-mo-old mice without wounding. Because the latex dye does not transverse the capillary bed, it only visualizes arterial branches unless there are AV shunts. When AVMs were present as shown in ALK1-iKO skin and ear (Fig. 4 E, g–i), arteries and veins appeared tortuous and excessive, and the latex dye could be found in venous branches. Unlike these, in the SnoNm/m mice, the latex dye was mostly found in arterial branches and no abnormal vessel tortuosity was found in the wound areas (Fig. 4 E, d–f), suggesting the absence of de novo AVM in these adult mice. However, in one of the two aged SnoNm/m mice examined, a clear sign of AVM was found in the blood vessels running the surface of the left cystic ovary of the mouse (Fig. S1). Because this is the only incidence of AVM, it is unclear whether this phenomenon is reproducible or whether it is specific to certain pathological conditions such as cystic ovary.
Taken together, our results suggest that a loss of the SnoN–Smads interaction impairs both extra-embryonic and embryonic angiogenesis, and that the defective angiogenesis may be responsible for the prenatal death of SnoNm/m embryos.
SnoNm/m embryos display aberrant ALK5 and ALK1 signaling
To determine the molecular mechanism responsible for the defective angiogenesis of the SnoNm/m embryos, we isolated MEECs from E11.5 SnoNm/m embryos and WT littermates. In the conventional 2D culture, the SnoNm/m MEECs displayed similar morphology and similar levels of apoptosis as WT MEECs (unpublished data), but showed markedly decreased proliferation (Fig. 5, A and B) and migration rates (Fig. 5 C). This reduced proliferation was due, at least partially, to inhibition by autocrine TGF-β through the ALK5 branch because treatment of cells with SB-431542, the ALK5 inhibitor, greatly increased proliferation of SnoNm/m MEECs (Fig. 5 A). The decreased proliferation was not due to autocrine BMP9 because addition of BMP9 blocking antibody did not enhance cell proliferation (Fig. 5 B). Interestingly, while WT MEECs underwent cell cycle arrest in response to BMP9, the proliferation of SnoNm/m MEECs was not as markedly affected (Fig. 5 B), suggesting a potential defect in ALK1–Smad1/5 signaling. Similarly, BMP9 was able to inhibit the migration of WT but not SnoNm/m MEECs (Fig. 5 C). Consistent with the defective angiogenesis observed in vivo, SnoNm/m MEECs also displayed impaired capillary formation ability in vitro. In a spheroid sprouting assay, VEGF induced endothelial sprouting in WT MEECs and BMP9 further enhanced this activity, whereas in the SnoNm/m MEECs sprouting in response to either VEGF alone or VEGF plus BMP9 was impaired (Fig. 5 D).
Figure 5.

SnoNm/m endothelial cells exhibit impaired ALK1 and ALK5 signaling. (A and B) SnoNm/m MEECs display reduced proliferation. Equal numbers of WT or SnoNm/m MEECs were treated either with or without 10 µM SB-431542 in A or in the absence or presence of 10 ng/ml of BMP9 or 5 ng/ml of α-BMP9 antibody in B and cultured for 4 d. Cells were counted at the indicated days. Data represent mean ± SEM from three independent experiments. (C) Transwell migration assays. 105 WT or SnoNm/m MEECs were placed in the upper chamber in media containing 1% FBS with or without 10 ng/ml BMP9 for 16 h. Migrated cells were stained, photographed (top panels), and quantified (shown in the graph below). Data represent mean ± SEM from three independent experiments. ns, not significant. (D) SnoNm/m MEECs show defective sprouting activity. MEECs spheroids were seeded in collagen gels with or without VEGF together with or without BMP9, and sprouting was evaluated after 24 h. Representative pictures are shown on the top. The lengths of the endothelial sprouts were quantified and shown in the graph below. At least 10 spheroids were measured per experiment, and data representing mean ± SEM from three independent experiments are shown. ns, not significant. Bar, 400 µm. (E) Semi-quantitative RT-PCR analysis of PAI-1 and Id-1 mRNA in yolk sacs (left) and MEECs (right). For MEECs, cells were serum starved for 16 h and stimulated with 100 pM TGF-β1 for 3 h. β-Actin was used as a loading control. (F) SnoNm/m MEECs show increased ALK5-dependent transcription and reduced ALK1-dependent transcription in luciferase assays. p3TP-Lux or (BRE)2lux was transfected into the WT and SnoNm/m MEECs, and luciferase activity was measured 16 h after TGF-β or BMP9 treatment.
Previous studies have suggested that two separate TGF-β receptor pathways (ALK1 and ALK5) in vascular endothelial cells may coordinate to regulate proliferation, maturation, and migration of cells during angiogenesis (Goumans et al., 2003a). We next compared the activities of these two signaling branches in WT and SnoNm/m cells. In SnoNm/m yolk sac and its derived MEECs, the expression of PAI-1, an ALK5 target gene and a negative regulator of endothelial migration (Goumans et al., 2003b), was highly elevated (Fig. 5 E). In SnoNm/m MEECs, this up-regulation of PAI-1 expression was further enhanced by TGF-β, possibly due to the lack of repression of Smad transcription by mSnoN. Interestingly, the expression of Id-1, a target gene of the ALK1 branch (Goumans et al., 2002), was significantly reduced in SnoNm/m yolk sacs and in SnoNm/m MEECs either in the presence or absence of TGF-β (Fig. 5 E) or BMP9 (not depicted). Similarly, in luciferase reporter assays the activity of p3TP-lux, the ALK5-dependent reporter, was elevated, whereas that of (BRE)2lux, the ALK1-dependent reporter, was significantly decreased upon TGF-β or BMP9 treatment in SnoNm/m MEECs (Fig. 5 F). Thus, the activity of ALK5 signaling is increased whereas that of the ALK1 branch is decreased in SnoNm/m MEECs.
Impaired ALK1 signaling in SnoN-deficient endothelial cells
We next examined upstream signaling events in both ALK5 and ALK1 pathways to determine how SnoN affected the activities of the two branches by using MEEC lines isolated from different embryos (Fig. 6 A and Fig. S2 A). Multiple lines of WT and SnoNm/m MEECs were treated with TGF-β. The phosphorylation of Smad proteins in these cell lines was examined by Western blot. As shown in Fig. 6 A and Fig. S2 A, although the total levels of R-Smad proteins (Smad1-5) remained the same, a marked decrease in the phosphorylation of Smad1/5 (pi-Smad1/5) after TGF-β stimulation was detected in SnoNm/m cells. Interestingly, the level of phospho-Smad2 (pi-Smad2) in SnoNm/m MEECs was indistinguishable from that in WT MEECs (Fig. 6 A). A time-course experiment indicated that the kinetics of Smad1/5 phosphorylation in response to TGF-β in SnoNm/m MEECs was similar to that in WT MEECs (Fig. 6 B). Because Smad1/5 can also be activated by BMP9 through ALK1 or BMP4 through ALK3 or ALK6, we asked whether the decrease in Smad1/5 phosphorylation also occurred in response to BMP signaling. WT or SnoNm/m MEECs were stimulated with either BMP9 or BMP4, and phospho-Smad1/5 levels were examined by Western blotting. Although no difference in BMP4-induced phosphorylation of Smad1/5 was detected between WT and SnoNm/m MEECs (Fig. S2 B), a decrease in BMP9-dependent phosphorylation of Smad1/5 was observed in SnoNm/m MEECs (Fig. 6 C), indicating that the reduced phosphorylation of Smad1/5 is specific to ALK1 signaling and independent of ALK2/3/6 pathways.
Figure 6.

Activation of Smad1/5 by ALK1 is impaired in SnoNm/m MEECs. (A) Reduced phosphorylation of Smad1/5, but not Smad2/3 in SnoNm/m MEECs. Cells were treated with 100 pM TGF-β1 for 1 h, and cell lysates were analyzed by Western blotting using the indicated antibodies on the left. α-Tubulin was included as a loading control. Black lines indicate the removal of intervening lanes for presentation purposes. (B) The kinetics of Smad1/5 phosphorylation is similar between WT and SnoNm/m MEECs. Cells were treated with 100 pM TGF-β1 for the indicated time periods, and phosphorylation of Smads was measured as described in A. (C) BMP9-induced phosphorylation of Smad1/5 is also decreased in SnoNm/m MEECs. Cells were treated with 10 ng/ml BMP9 for 1 h, and Smad1/5 phosphorylation was measured as described in A. (D) Endogenous interaction between Smad1/5 and ALK1 is impaired in SnoNm/m MEECs. Endogenous ALK1 was isolated by immunoprecipitation (IP) from WT and SnoNm/m MEECs stimulated or not with TGF-β, and ALK1-associated proteins were analyzed by Western blotting with the indicated antibodies (top panels). Controls of protein levels in whole-cell lysates are shown on the bottom. The black line in the SnoN cell lysate panel indicates the removal of intervening lanes for presentation purposes. (E) No difference in Smad2–ALK5 interaction is detected between WT and SnoNm/m MEECs. Interaction between endogenous ALK5 and Smad2 was isolated by immunoprecipitation with anti-ALK5 followed by Western blotting with anti-Smad2. (F) The GS domain in ALK1 is required for the SnoN–ALK1 interaction. Top: schematic drawing of WT ALK1, including the extracellular domain (ECD), transmembrane domain (TMD), glycine/serine-rich (GS) domain, and S/T kinase domains. Position of the N181-186 mutation is indicated. Bottom: association of HA-SnoN with various Flag-tagged ALK1 (F-ALK1) mutants was measured by immunoprecipitation with anti-Flag followed by Western blotting with anti-HA. Black lines indicate the removal of intervening lanes for presentation purposes.
A decrease in Smad1/5 phosphorylation could be due to the reduced receptor expression or an impaired ALK1–Smad1/5 interaction. As shown in Fig. 6 D, the expression level of ALK1 was the same in WT and SnoNm/m MEECs. Although the expression of Endoglin showed a slight decrease and that of TβRII displayed a moderate increase in mutant MEECs (Fig. S2 C), these changes are unlikely to be responsible for the dramatic decrease in Smad1/5 phosphorylation observed in mutant cells. Interestingly, upon TGF-β or BMP9 treatment, the ability of ALK1 to form a complex with its substrate, Smad1/5, was significantly weakened in SnoNm/m MEECs (Fig. 6 D and Fig. S2 D). In contrast, there was no change in the formation of the ALK5 and Smad2/3 complex between these two cell lines (Fig. 6 E). Therefore, the mutations in SnoN weakened the binding of Smad1/5 to the ALK1 receptor, leading to the decreased phospho-Smad1/5 in SnoNm/m MEECs.
SnoN facilitates activation of Smad1/5 by ALK1
We next investigated how SnoN specifically affected the interaction of ALK1 with Smad1/5, given that SnoN has always been thought to function in the nucleus or in the cytoplasm, but never been reported to act at the cell surface. In a coimmunoprecipitation assay to examine the interaction between endogenous ALK1 and Smad1/5, we were surprised to detect SnoN in the ALK1 immunoprecipitates in both WT and SnoNm/m MEECs in the presence of TGF-β (Fig. 6 D). This association appeared to require the GS domain of ALK1 (Fig. 6 F). In addition, the amount of mSnoN that associated with ALK1 was similar to that of WT SnoN even though mSnoN was present in the cells at a higher level than WT SnoN because it could not be degraded upon TGF-β stimulation (Fig. 6 D). Consistent with this finding, immunostaining results showed that in both WT and SnoNm/m MEECs, SnoN was expressed not only in the nucleus and cytoplasm as previously reported (Luo, 2004; Deheuninck and Luo, 2009; Jahchan and Luo, 2010), but a fraction of SnoN was also found in close proximity to the plasma membrane (Fig. S2 E). Because ALK1 binds to Smad1/5 to phosphorylate them, we asked whether SnoN could also associate with Smad1/5 and affect its interaction with ALK1. Indeed, WT SnoN could bind to Smad1/5 in a BMP9- or TGF-β–dependent manner in MEECs (Fig. 7 A). However, mSnoN was severely impaired in fostering this interaction. Thus, SnoN may play a direct role in facilitating Smad1/5 phosphorylation by ALK1 by forming a complex with both ALK1 and Smad1/5 to enhance their interactions. Consistent with this model, SnoN−/− MEECs also exhibited dramatically reduced Smad1/5 phosphorylation in response to either TGF-β (Fig. 7 B) or BMP9 (Fig. S3 A). Similar to SnoNm/m cells, SnoN−/− MEECs displayed significantly impaired Id-1, Smad7, and Smad6 expression and were refractory to BMP9 in proliferation, migration, and sprouting assays (Fig. S3, B–E). To further confirm that the reduced Smad1/5 phosphorylation and ALK1–Smad1/5 interaction in SnoNm/m and SnoN−/− MEECs were due to lack of SnoN activity, we used shRNA to knock down SnoN in WT MEECs and examined its effect on Smad1/5 phosphorylation and its interaction with ALK1. As shown in Fig. 7 C, reducing SnoN expression in WT MEECs caused a significant decrease in TGF-β–induced Smad1/5 phosphorylation as well as its interaction with ALK1.
Figure 7.

SnoN facilitates the interaction of Smad1/5 with ALK1. (A) WT but not mutant SnoN associates with Smad1/5 in endothelial cells. Flag-tagged WT or mutant SnoN was transiently transfected into WT MEECs. 48 h after transfection, the cells were stimulated with or without either 100 pM TGF-β or 10 ng/ml BMP9 for 30 min. Interaction between Flag-SnoN (F-SnoN) and endogenous Smad1/5 was detected by Western blotting analysis of anti-Flag IP (top panels). The levels of these proteins in cell lysates are shown in the bottom panels. (B) SnoN−/− MEECs also display a reduced Smad1/5 phosphorylation. MEECs isolated from E11.5 WT or SnoN−/− embryos were treated with 100 pM TGF-β1 for 1 h, and phosphorylation of Smad1/5 detected by Western blotting. (C and D) Knocking down SnoN by shRNA reduces the binding of Smad1/5 to ALK1. WT MEECs (C) or WT HPAECs (D) infected with a retrovirus expressing shSnoN or vector control were treated with 100 pM TGF-β or 10 ng/ml BMP9 for 1 h and subjected to coimmunoprecipitation assay with anti-ALK1. Black lines in the bottom two cell lysate panels indicate the removal of intervening lanes for presentation purposes. (E) SnoN enhances the Smad1–5-ALK1 interaction. 293T cells were transfected with a fixed amount of HA-ALK1 and Flag-Smad1 (F-Smad1) and increasing amounts (0, 0.5, 1, 2 µg) of Flag-SnoN (F-SnoN). Smad1 and SnoN that associated with ALK1 were isolated by coimmunoprecipitation with anti-HA (ALK1) and detected by Western blotting with anti-Flag. (F) Mutant SnoN fails to enhance the Smad1–ALK1 interaction. HA-ALK1 and Flag-Smad1 were cotransfected with Flag-tagged WT SnoN or mSnoN into 293T cells. The Smad1–ALK1 interaction was measured as described in E. (G) Model of SnoN regulation of TGF-β and BMP9 signaling in endothelial cells. SnoN enhances activation of Smad1/5 by ALK1 while repressing the activity of the ALK5 pathway. By regulating the activity of the two pathways, SnoN maintains the proper level of endothelial cell proliferation, migration, and maturation.
The ability of SnoN to promote Smad1/5 activation is not unique to mouse endothelial cells, but also true for human endothelial cells. Human pulmonary artery endothelial cells (HPAECs) expressing shSnoN showed a significant reduction in ALK1–Smad1/5 interaction and Smad1/5 phosphorylation in response to either BMP9 or TGF-β (Fig. 7 D). Consistent with this, these cells displayed decreased Id-1 transcription (Fig. S3 F) and sprouting activity (Fig. S3 G) in the presence of BMP9.
Finally, to confirm that the ALK1–Smad1/5 interaction can be strengthened by SnoN in a concentration-dependent manner, we coexpressed a fixed amount of HA-ALK1 and Flag-Smad1/5 in the absence or presence of increasing amounts of SnoN, and the effects of SnoN on the ALK1–Smad1/5 interaction were examined by a coimmunoprecipitation assay. As shown in Fig. 7 E, SnoN enhanced the interaction between ALK1 and Smad1/5 in a concentration-dependent manner. Because the mutant SnoN did not interact with Smad1/5 very well, it failed to facilitate the Smad1/5–ALK1 interaction (Fig. 7 F). Interestingly, SnoN did not affect the interaction between ALK5 and Smad2 (Fig. S4 A), possibly due to the much weaker interaction of SnoN with ALK5 (Fig. S4 B). Furthermore, no interaction between SnoN and ALK3 or ALK6 was detected (Fig. S4 B). Taken together, these data suggest that SnoN may bind to both ALK1 and Smad1/5 to enhance their interaction and phosphorylation in both mouse and human endothelial cells.
Based on both in vivo and in vitro observations, we propose that SnoN regulates angiogenesis through promoting ALK1 signaling while repressing ALK5 activity. By regulating the activity of the two pathways, SnoN maintains the proper level of endothelial cell proliferation, migration, and maturation (Fig. 7 G). Inactivation of this modulating capability either through deleting SnoN expression entirely or through disruption of the SnoN–Smads interaction disrupts both extra-embryonic and embryonic angiogenesis, specifically leading to angiogenesis defects observed in the SnoNm/m embryos.
Discussion
In endothelial cells, both ALK1 and ALK5 signaling pathways are critical for yolk sac and embryonic angiogenesis. We have uncovered a novel positive role of SnoN in enhancing ALK1-dependent Smad1/5 activation in endothelial cells during embryonic angiogenesis. Upon binding of BMP9 or TGF-β, SnoN directly binds to the ALK1 receptor on plasma membrane and facilitates the interaction between ALK1 and Smad1/5 and subsequently Smad1/5 phosphorylation. Disruption of the SnoN–Smad interaction significantly impairs Smad1/5 activation while simultaneously up-regulating Smad2/3 activity, resulting in a decrease in ALK1 signaling and an increase in the ALK5 activity (Fig. 7 G). This results in disrupted endothelial proliferation, migration, and differentiation as well as defective VSMC recruitment and differentiation, eventually leading to defective yolk sac and embryonic angiogenesis, AVM, and embryonic lethality at E12.5. Thus, SnoN can modulate Smad activities in both positive and negative manners depending on the tissue context. More importantly, our studies suggest that the overall impact of SnoN on TGF-β signaling in endothelial cells is not merely as a negative regulator as one might expect, but rather as an integral part of the TGF-β signaling circuit. SnoN promotes BMP9- or TGF-β–dependent biological processes by coordinating the actions of multiple TGF-β signaling branches or modulating the robustness of the signaling circuit.
Our discovery that SnoN can directly promote Smad1/5 phosphorylation by facilitating the interaction of ALK1 and Smad1/5 in endothelial cells is different from the recent report by Kawamura et al. (2012), whose findings show that SnoN can suppress BMP2-induced chondrocyte maturation at a step downstream of Smad1/5 phosphorylation, although the exact mechanism of this repression and whether this repression by SnoN is direct or through another molecule is not clear. It is possible that in different cell types and in the presence of different BMP ligands, SnoN may act on the signaling pathways differently. This highlights the unique tissue-specific nature of TGF-β family signaling activities.
Interestingly, although all MEEC cell lines isolated from the SnoN−/− mice displayed defects in Smad1/5 activation similar to the SnoNm/m MEECs, we did not detect similar vascular defects in the SnoN−/− embryos (unpublished data). The mechanism underlying this phenotype difference between the SnoNm/m and SnoN−/− embryos is not clear. We speculate that the lack of SnoN protein in the SnoN−/− embryos may trigger a compensatory program to unregulate the expression of proteins of similar function in order to ensure survival, whereas the presence of a SnoN protein, albeit a functionally inactive one, in the SnoNm/m embryos may prevent the activation of such a compensatory event, leading to the severe defects and embryonic lethality observed. Alternatively, the mutant SnoN protein may act in a “gain-of-function” manner. For example, the lack of Smad antagonism may alter the dynamics of the interaction between the mutant SnoN protein and other TGF-β–dependent or –independent pathways, leading to unexpected outcomes.
The ability of SnoN to act as a positive regulator of Smads appears to be specific for the ALK1-dependent Smad1/5 pathway in endothelial cells, but not for the ALK5–Smad2/3 pathway, nor for Smad1/5 activation by other BMP type I receptors (ALK3, 6). This specificity is likely due to the difference in binding affinities between SnoN and different ALK molecules. In endothelial cells, SnoN interacts strongly with ALK1, but not with ALK2–ALK5, in a TGF-β– or BMP9-dependent manner. Through its ability to bind to the Smads, SnoN can facilitate the ALK1–Smad1/5 interaction and subsequently Smad1/5 activation. mSnoN still binds to ALK1, but fails to bind to Smad1/5, and is therefore defective in promoting Smad1/5 activation. This activation mechanism of SnoN on Smad1/5 is entirely different from its repression on Smad2/3 in two aspects. First, SnoN activates Smad1/5 signaling at the plasma membrane, where SnoN physically binds to and colocalizes with ALK1 on the cell surface. Although SnoN is largely known as a nuclear and cytoplasmic protein, we found that a fraction of SnoN exists on the plasma membrane in endothelial cells, but not in other cell types. A recent report suggests that Ski may associate with ALK5 and enforce sequestration of the Smad2/4 complex by the receptor, leading to reduced transcriptional activation of TGF-β target genes (Ferrand et al., 2010). However, we failed to detect a strong interaction between SnoN and ALK5 in endothelial cells or other cancer cell lines (Zhu et al., 2007). Second, SnoN binds strongly to Smad2/3 in the nucleus to repress their transcription activity. In contrast, the interaction of SnoN with Smad1/5 is not as strong as that with Smad2/3. It is possible that this interaction is strengthened by ALK1 upon BMP9 or TGF-β stimulation. Consistent with this, binding of SnoN with Smad1/5 was not detected to a similar extent in a nonendothelial cell line (unpublished data). Perhaps due to this weaker affinity for Smad1/5, SnoN may not inhibit their transcription activity as effectively as it does for Smad2/3.
Disruption of the activities of ALK1 and ALK5 signaling not only directly affects the proliferation, differentiation, and migration of the endothelial cells, but also the recruitment and differentiation of vascular smooth muscle cells or pericytes. The proper activities of the two branches are also crucial for the expression of many cytokines including TGF-β1 itself, BMP9, and VEGF (unpublished data), which are also important regulators of yolk sac and embryonic angiogenesis. Interestingly, the SnoN knock-in mice also show markedly reduced expression of TGF-β1, which may further contribute to the defective angiogenesis (Goumans and Mummery, 2000). Consistent with its activity in facilitating Smad1/5 activation by ALK1, the SnoNm/m embryos display AVM and dramatically reduced expression of Ephrin-B2, a feature found in mice deficient in ALK1–Smad1/5 signaling, but not in mice lacking ALK5 branch signaling. Although similar, the defects in SnoNm/m mice are less severe than those found in mice lacking ALK1 or Smad5. In particular, among the limited number of surviving SnoNm/m mice examined, only one aged female mouse developed a noticeable AVM in the cystic ovary (Fig. S1), while all other adult SnoNm/m mice appeared to have normal vascular networks in the lungs and GI tracts. This differs from the phenotypes of mice with endothelial-specific ALK1 knockout, which exhibited vascular malformation and AVM in the yolk sacs and embryonic lungs by E17.5 (Park et al., 2008) as well as in the postnatal organs, such as lung, brain, and GI tracts (Park et al., 2009). Furthermore, the SnoNm/m mice display only a partial embryonic lethality instead of full penetrance as found in the knockout mice lacking any of the TGF-β receptors or Smads. The SnoNm/m embryos also died 1–2 days later than the ALK1–Smad5 KO embryos. This partial embryonic lethality cannot be due to the influence of genetic background because a similar percentage of embryonic lethality was observed in both 129P2 and C57BL/6J background (unpublished data). Interestingly, 100% of SnoN mutant MEEC cell lines show significantly decreased Smad1/5 activation, suggesting that all the mice display molecular defects in Smad1/5 activation in the endothelium, yet some of them are able to overcome this defect and develop well enough to survive. One possible explanation for this is that unlike deletion of the TGF-β receptors or Smad proteins, inactivation of SnoN does not lead to pathway elimination. Instead, SnoN mainly functions as a modulator of the activity of ALK1 and ALK5 signaling. In early embryos, these alterations in signaling activities could conceivably be compensated by other feedback loops or signaling pathways. Although we fail to detect any changes in the protein expression levels of known negative regulators of the pathway including Ski and Smad7 (unpublished data), we cannot exclude the involvement of other yet-to-be-identified feedback regulators of the pathway. The marked alteration in the expression of many cytokines involved in angiogenesis may also contribute to the milder phenotypes. Future studies may identify these important regulatory mechanisms.
Materials and methods
Mice, cells, antibodies, and constructs
The SnoNm/m mice expressing a mutant snoN gene have been described previously (Pan et al., 2009). In brief, this knock-in mouse expresses a snoN mutant containing point mutations that alter amino acid residues 88–92 and 267–277 to alanine and was generated through homologous recombination with the E14 mouse embryonic stem (ES) cells derived from 129P2/OlaHsd mice in C57BL/6 background. The SnoNm/m mice used in this study have been backcrossed to C57BL/6 for additional six generations. The SnoN−/− mice were obtained from S. Pearson-White (The Biomarker Consortium, Washington, DC). 293T cells were maintained in DMEM with 10% fetal bovine serum. MEECs were maintained in DMEM with 10% fetal bovine serum and 10 µM SB431542 (Tocris Bioscience). HPAECs were purchased from Invitrogen and cultured in Medium 200 plus low serum growth supplement (Invitrogen). Antibodies against Flag, HA, and α-smooth muscle actin (α-SMA) were purchased from Sigma-Aldrich; Smad2/3 from BD; SnoN (H317), Smad1/5/8 (sc-6031), ALK1 (RM0015-1B03), ALK1 (H150), ALK5 (v-22), and tubulin from Santa Cruz Biotechnology, Inc.; ALK1 (AF770) and BMP9 (MAB3209) from R&D Systems; phospho-Smad1/5/8 (#9511) from Cell Signaling Technology; and the Alexa fluorophore–conjugated secondary antibodies from Molecular Probes. Anti-phosphoSmad2 antisera (rabbit polyclonal antibody against a peptide [KKK-SSpMSp] containing two phosphoserine residues at the C terminus of Smad2) was a gift from A. Moustakas (Ludwig Institute for Cancer Research, Uppsala, Sweden). The ALK1- and BMP-responsive luciferase reporter construct (BRE)2lux (two copies of the BMP-responsive elements [BREs] from the Id-1 promoter fused to the major late minimal promoter from adenovirus and cloned into the pGL3basic luciferase vector [Promega]) was provided by P. ten Dijke (Leiden University Medical Center, Leiden, Netherlands).
Isolation of MEECs
MEECs were isolated from E11.5 embryos as described previously (Larsson et al., 2001; Goumans et al., 2002). In brief, the embryos were trypsinized at 37°C for 5 min and sheared to make a single cell suspension. The cells were seeded in a 0.1% gelatin-coated well in DMEM + 10% FCS at 37°C overnight, and then infected with viruses produced by PSI cells (expressing the Polyoma middle T antigen) for 16 h. After two rounds of infection, cells were cultured in DMEM with 10% FCS, 2 ng/ml bFGF, 25 ng/ml VEGF, 10 µM SB431542, and 400 ng/ml G418 for 10–15 d. After 12 passages, the cells were considered immortalized.
Transfection, retroviral infection, luciferase assay, immunoprecipitation, and Western blotting
293T cells, MEECs, and HPAECs were transiently transfected by using Lipofectamine Plus (Invitrogen) according to the manufacturer’s protocol. For retroviral infection of MEECs and HPAECs, cDNAs or shRNAs in retroviral vectors were cotransfected with packaging plasmids (pCMV-gag-pol and pBS-VSVG) into 293T cells to generated high-titer viral supernatant, which was used subsequently to infect the MEECs and HPAECs. After 48 h, infected cells were selected with 1.5 µg/ml puromycin. Pools of puromycin-resistant cells were analyzed in various experiments. For luciferase assays, a total of 2.5 µg of DNA (0.5 µg of either ALK5- or ALK1-responsive luciferase reporter construct [p3TP-lux or (BRE)2lux] was transiently transfected into MEECs or infected HPAECs. At 24 h after transfection, cells were serum starved for 8 h and stimulated with either 50 pM TGF-β1 or 10 ng/ml BMP9, and the luciferase activity was measured 16 h later according to the manufacturer’s protocol (Zhu et al., 2005).
Endogenous proteins were isolated by immunoprecipitation as described previously (Zhu et al., 2007). In brief, 6 µg of specific antibodies were bound to 40 µl of protein A–Sepharose beads (Sigma-Aldrich) and incubated with 1–2 mg whole-cell lysates in low salt lysis buffer (150 mM NaCl, 50 mM Hepes-KOH, pH 7.8, 5 mM EDTA, 0.1% NP-40, 3 mM dithiothreitol, and 0.5 mM PMSF) at 4°C for at least 2 h. Samples were eluted off the beads by boiling in 1× SDS buffer followed by standard SDS-PAGE and Western blot analysis.
Cell proliferation assay
2 × 104 WT or SnoNm/m or SnoN−/− MEECs were cultured either with or without 10 µM SB-431542 or 10 ng/ml BMP9 or 5 ng/ml α-BMP9 antibodies for 4 d. Relative cell growth was determined by counting cell numbers from day 1 to day 4.
Immunofluorescence
MEECs were grown on glass coverslips, fixed with ice-cold methanol for 20 min, permeabilized with 0.1% Triton X-100 for 5 min, and stained with the primary antibody α-ALK1 (1:50; R&D Systems) or α-SnoN (rabbit polyclonal antibody against a peptide located at the C terminus of human SnoN [KELKLQILKSSKTAKE], 1:500; Krakowski et al., 2005), and the appropriate Alexa fluorophore–conjugated secondary antibodies (Molecular Probes). Nuclei were detected by DAPI staining. Cells were visualized by a fluorescence microscope (AxioImager M2; Carl Zeiss) with a 20×/0.5 EC Plan-Neofluar objective (Carl Zeiss) at room temperature, and the images were captured with a camera (5 Mpix MicroPublisher; QImaging).
Whole-mount staining, in situ hybridization, and immunohistochemistry
Embryos were fixed in PBS containing 4% paraformaldehyde (PFA), and whole-mount α-PECAM staining was performed according to a standard protocol using anti–PECAM-1 (clone MEC13.3, 1:500; BD) and peroxidase-conjugated secondary antibody (BD Biosciences; Hogan et al., 1994). Whole-mount in situ hybridization with SMA probe was performed as described previously (Mavrakis et al., 2007). Sense or antisense RNA probes were generated using the Roche DIG RNA labeling kit. Pictures of whole-mount embryo stainings were taken under a fluorescence dissecting microscope (SterREO Lumar; Carl Zeiss) at room temperature with a color digital camera (3Mpix; QImaging) and iVision image analysis software (BioVision Technologies).
Immunohistochemistry was performed as described previously (Jahchan et al., 2010). In brief, paraffin-embedded mouse embryo transverse sections were deparaffinized in xylenes, rehydrated in a series of ethanol gradients, and permeabilized with 20 µg/ml proteinase K for 10 min at room temperature. Anti-SnoN, SMA, and PECAM-1 stainings were performed using the Tyramide Signal Amplification Biotin System kit (PerkinElmer) with anti-SnoN (1:200; Krakowski et al., 2005), anti-SMA at 1:200 (#017k4755; Sigma-Aldrich), and anti-PECAM-1 at 1:25 (clone MEC13.3; BD). For visualization, DAB was used as the peroxidase substrate (SK-4105; Vector Laboratories). All images were visualized under a fluorescence microscope (AxioImager M2; Carl Zeiss) with a 10×/0.3 EC Plan-Neofluar objective at room temperature and captured with a camera (5 Mpix MicroPublisher; QImaging) and iVision image analysis software.
India ink and latex dye injection
Ink injection assay was performed as described previously (Urness et al., 2000) in E9.5–11.5 embryos. In brief, embryos were dissected in PBS-Tween (0.1%) and immediately injected in the outflow tract of the heart with India ink diluted in PBS-Tween to 20% with a finely drawn Pasteur pipette. The embryos were either photographed directly or fixed in 4% PFA for later paraffin-embedded sectioning.
Latex dye injection was performed as described previously (Park et al., 2009). In brief, latex dye was perfused via left heart of anesthetized mice following sequential perfusion of heparin, vasodilators, and formalin. For generation of the skin wounds, full-thickness excisional wounds of 4 mm in diameter were inflicted into the dorsal skin and ear areas using a biopsy punch 8 d before the latex dye infusion. After overnight fixation with formalin, the stained organs or tissues were dehydrated by methanol series, cleared in benzyl alcohol/benzyl benzoate (1:1; Sigma-Aldrich), and observed under a microscope (model MZ8; Leica) with 0.63×, 1×, and 2× objectives for different organs and tissues. Images were captured by a camera (model DFC 320; Leica) and Application Suite version 2.4.0 R1 software (Leica).
RNA isolation and RT-PCR
Total RNA from yolk sacs or MEECs was extracted using the RNeasy Mini kit (QIAGEN). Complementary DNA (cDNA) was produced using reverse transcription (SuperScript II; Invitrogen), and was amplified with the following sets of primers: β-actin: 5′-GGACCTGGCTGGCCGGGACC-3′ (forward) and 5′-GCGGTGCACGATGGAGGGGC-3′ (reverse); PAI-1: 5′-GGGGCCGTGGAACAAGAATGAGAT-3′ (forward) and 5′-AGATGTTGGTGAGGGCGGAGAGGT-3′ (reverse); Id-1: 5′-TCCTGCAGCATGTAATCGAC-3′ (forward) and 5′-GAGAGGGTGAGGCTCTGTTG-3′ (reverse); ephrinB2: 5′-CAGACAAGAGCCATGAAGATCC-3′ (forward) and 5′-TCTCCCATTTGTACCAGCTTCT-3′ (reverse); Ang1: 5′-GGCACGGAAGGCAAGCGCTG-3′ (forward) and 5′-CAAGCATGGTGGCCGTGTGG-3′ (reverse); Ang2: 5′-GAACCCTCTGGGAGAGTACTG-3′ (forward) and 5′-GCTGCTATGCACTGGTGTCTC-3′ (reverse); Tie2: 5′-CCTTCCTACCTGCTACTTTA-3′ (forward) and 5′-CCACTACACCTTTCTTTACA-3′ (reverse); Flt1: 5′-TGTGGAGAAACTTGGTGACCT-3′ (forward) and 5′-TGGAGAACAGCAGGACTCCTT-3′ (reverse); Flk1: 5′-AGAACACCAAAAGAGAGGAACG-3′ (forward) and 5′-GCACACAGGCAGAAACCAGTAG-3′ (reverse); Flt4: 5′-CACCGAAGCAGACGCGCTGATGAT-3′ (forward) and 5′-AGCTGCTGTCTGCGAAGAAGGT-3′ (reverse); Hes1: 5′-TACCCCAGCCAGTGTCAACA-3′ (forward) and 5′-CCATGATAGGCTTTGATGACTTTCT-3′ (reverse); Hey-1: 5′-TGAGCTGAGAAGGCTGGTAC-3′ (forward) and 5′-ACCCCAAACTCCGATAGTCC-3′ (reverse); Hey-2: 5′-TGAGAAGACTAGTGCCAACAGC-3′ (forward) and 5′-TGGGCATCAAAGTAGCCTTTA-3′ (reverse); qSmad6: 5′-AGGAGAAACTCGCTCGCTCCAAGTGCAT-3′ (forward) and 5′-AGCTCCCTTCTTACTCCCTGCAAA-3′ (reverse); qSmad7: 5′-TCGGACAGCTCAATTCGGACAACA-3′ (forward) and 5′-AACACCTTGTGTACCAACAGCGTC-3′ (reverse); qId-1: 5′-ATCAGGGACCTTCAGTTGGAGC-3′ (forward) and 5′-AGACCCACAGAGCACGTAATTCC-3′ (reverse). For semi-quantitative PCR, the following PCR program was performed: 94°C for 5 min (initial denaturation), 94°C for 30 s, 55°C for 30 s, and 72°C for 45 s. Within the linear range of amplification, all PCR products were prepared under appropriate cycling conditions and separated on a 1% agarose gel.
The quantitative real-time PCR was performed in quadruplicates using the 7300 Real-Time PCR System (Applied Biosystems) and SYBRGreen PCR Master Mix (Applied Biosystems).
Transwell migration assay
The membrane of a transwell upper chamber (8 µm polycarbonate membrane; Corning) was precoated for 1 h with 0.05 mg/ml collagen I (BD). The lower chamber was covered with media containing 10% FBS. 105 cells in media containing 1% FBS were seeded in the upper chamber. 10 ng/ml BMP9 was added to both upper and lower chambers, and cell migration was allowed to proceed for 8–16 h at 37°C. Cells were stained with 1% Crystal Violet in 20% methanol for 5 min, and the number of migrated cells was scored by counting the stained cells from multiple randomly selected microscopic visual fields.
Endothelial spheroids sprouting assay
Sprouting assays were performed as described previously (Primo et al., 2010). In brief, MEECs or HPAECs in culture medium containing 0.4% methylcellulose were seeded in nonadhesive round-bottom 96-well plates (800 cells per well) overnight. The spheroids were collected (36 spheroids per assay), resuspended, and seeded in flat-bottom 96 well plates in medium containing 20% FBS, 0.66% methylcellulose, and 3 mg/ml collagen gel from rat tail (BD) in the presence or absence of growth factors (50 ng/ml VEGF and 10 ng/ml BMP9) for 24 h. Tubular structures were examined under an inverted phase-contrast microscope. The lengths of the sprout outgrowth were quantified using ImageJ software (National Institutes of Health).
Statistical analysis
Two-tailed unpaired Student’s t tests were used for statistical analysis. Quantitative data are presented as the mean ± SEM. Values of P ≤ 0.05 were considered statistically significant.
Online supplemental material
Fig. S1 shows an AVM detected in the blood vessels on the surface of a cystic ovary in an aged SnoNm/m mouse. Fig. S2 shows that SnoN is required for ALK1 phosphorylation of Smad1/5. Fig. S3 depicts how SnoN−/− MEECs and HPAECs expressing shSnoN show an impaired response to BMP9. Fig. S4 shows that SnoN is not required for the Smad2–ALK5 interaction and only binds strongly to ALK1, but not to ALK3, ALK6, or ALK5. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201208113/DC1.
Supplementary Material
Acknowledgments
We thank Marie-José Goumans and Peter ten Dijke for assistance and reagents with the isolation of MEECs, ALK1 constructs, and helpful discussions. Special thanks to Gerard C. Blobe for ALK1 truncation and deletion mutants and ALK3 and ALK6 constructs; Eric N. Olson for α-SMA cDNA probe; Vasso Episkopou for the help and advice on the isolation and analysis of mouse embryos; Hu Zhao and Yang Chai for assistance with in situ hybridization assay; Roberto Sessa and Federico Bussolino for advice on the endothelial spheroid sprouting assay; and Hitoshi Nishimura and Dragana Cado for generating the SnoNm/m mice. We also thank Christopher Hendrick, Angeline Protacio, and Fangjiu Zhang for mouse technical assistance; Steve Ruzin and Denise Schichnes at CNR Biological imaging facility at UC-Berkeley for assistance with microscopy; and the mouse pathology core at UCSF for histology services.
This study is supported by National Institutes of Health grants RO1 CA101891 and RO1 DK090347 to K. Luo.
The authors declare no conflicts of interest and no competing financial interests.
Author contributions: Q. Zhu and K. Luo designed research, Q. Zhu performed research, Q. Zhu and K. Luo analyzed data, Y.H. Kim performed latex dye injection, D. Wang performed wound generation, and S.P. Oh analyzed results from this experiment. Q. Zhu and K. Luo wrote the paper.
Footnotes
Abbreviations used in this paper:
- AVM
- arteriovenous malformation
- EC
- endothelial cell
- HPAEC
- human pulmonary artery endothelial cell
- MEEC
- mouse embryonic endothelial cell
- PECAM-1
- platelet endothelial cell adhesion molecule 1
- SMA
- smooth muscle actin
- TGF-β
- transforming growth factor β
- VSMC
- vascular smooth muscle cell
- WT
- wild type
References
- Akiyoshi S., Inoue H., Hanai J., Kusanagi K., Nemoto N., Miyazono K., Kawabata M. 1999. c-Ski acts as a transcriptional co-repressor in transforming growth factor-beta signaling through interaction with smads. J. Biol. Chem. 274:35269–35277 10.1074/jbc.274.49.35269 [DOI] [PubMed] [Google Scholar]
- Attisano L., Wrana J.L. 2002. Signal transduction by the TGF-beta superfamily. Science. 296:1646–1647 10.1126/science.1071809 [DOI] [PubMed] [Google Scholar]
- Carmeliet P. 2003. Angiogenesis in health and disease. Nat. Med. 9:653–660 10.1038/nm0603-653 [DOI] [PubMed] [Google Scholar]
- Chung A.S., Ferrara N. 2011. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 27:563–584 10.1146/annurev-cellbio-092910-154002 [DOI] [PubMed] [Google Scholar]
- Copp A.J. 1995. Death before birth: clues from gene knockouts and mutations. Trends Genet. 11:87–93 10.1016/S0168-9525(00)89008-3 [DOI] [PubMed] [Google Scholar]
- David L., Mallet C., Mazerbourg S., Feige J.J., Bailly S. 2007. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 109:1953–1961 10.1182/blood-2006-07-034124 [DOI] [PubMed] [Google Scholar]
- Deheuninck J., Luo K. 2009. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 19:47–57 10.1038/cr.2008.324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R., Miyazono K. 2008. TGF-β and the TGF-β family. The TGF-β Family. Vol. 50 Derynck R., Miyazono K., editor Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: 29-44 [Google Scholar]
- Ferrand N., Atfi A., Prunier C. 2010. The oncoprotein c-ski functions as a direct antagonist of the transforming growth factor-β type I receptor. Cancer Res. 70:8457–8466 10.1158/0008-5472.CAN-09-4088 [DOI] [PubMed] [Google Scholar]
- Flamme I., Frölich T., Risau W. 1997. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J. Cell. Physiol. 173:206–210 [DOI] [PubMed] [Google Scholar]
- Goumans M.J., Mummery C. 2000. Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int. J. Dev. Biol. 44:253–265 [PubMed] [Google Scholar]
- Goumans M.J., Valdimarsdottir G., Itoh S., Rosendahl A., Sideras P., ten Dijke P. 2002. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 21:1743–1753 10.1093/emboj/21.7.1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans M.J., Lebrin F., Valdimarsdottir G. 2003a. Controlling the angiogenic switch: a balance between two distinct TGF-b receptor signaling pathways. Trends Cardiovasc. Med. 13:301–307 10.1016/S1050-1738(03)00142-7 [DOI] [PubMed] [Google Scholar]
- Goumans M.J., Valdimarsdottir G., Itoh S., Lebrin F., Larsson J., Mummery C., Karlsson S., ten Dijke P. 2003b. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell. 12:817–828 10.1016/S1097-2765(03)00386-1 [DOI] [PubMed] [Google Scholar]
- Guo X., Wang X.F. 2009. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 19:71–88 10.1038/cr.2008.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Tegen S.B., Krawitz A.R., Martin G.S., Luo K. 2003. The transforming activity of Ski and SnoN is dependent on their ability to repress the activity of Smad proteins. J. Biol. Chem. 278:30540–30547 10.1074/jbc.M304016200 [DOI] [PubMed] [Google Scholar]
- Hogan B., Beddington R., Costantini F., Lacy E. 1994. Manipulating the Mouse Embryo: A Laboratory Manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Jahchan N.S., Luo K. 2010. SnoN in mammalian development, function and diseases. Curr. Opin. Pharmacol. 10:670–675 10.1016/j.coph.2010.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahchan N.S., You Y.H., Muller W.J., Luo K. 2010. Transforming growth factor-beta regulator SnoN modulates mammary gland branching morphogenesis, postlactational involution, and mammary tumorigenesis. Cancer Res. 70:4204–4213 10.1158/0008-5472.CAN-10-0135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura I., Maeda S., Imamura K., Setoguchi T., Yokouchi M., Ishidou Y., Komiya S. 2012. SnoN suppresses maturation of chondrocytes by mediating signal cross-talk between transforming growth factor-β and bone morphogenetic protein pathways. J. Biol. Chem. 287:29101–29113 10.1074/jbc.M112.349415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakowski A.R., Laboureau J., Mauviel A., Bissell M.J., Luo K. 2005. Cytoplasmic SnoN in normal tissues and nonmalignant cells antagonizes TGF-beta signaling by sequestration of the Smad proteins. Proc. Natl. Acad. Sci. USA. 102:12437–12442 10.1073/pnas.0504107102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrivée B., Prahst C., Gordon E., del Toro R., Mathivet T., Duarte A., Simons M., Eichmann A. 2012. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell. 22:489–500 10.1016/j.devcel.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson J., Goumans M.J., Sjöstrand L.J., van Rooijen M.A., Ward D., Levéen P., Xu X., ten Dijke P., Mummery C.L., Karlsson S. 2001. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 20:1663–1673 10.1093/emboj/20.7.1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K. 2004. Ski and SnoN: negative regulators of TGF-beta signaling. Curr. Opin. Genet. Dev. 14:65–70 10.1016/j.gde.2003.11.003 [DOI] [PubMed] [Google Scholar]
- Mavrakis K.J., Andrew R.L., Lee K.L., Petropoulou C., Dixon J.E., Navaratnam N., Norris D.P., Episkopou V. 2007. Arkadia enhances Nodal/TGF-beta signaling by coupling phospho-Smad2/3 activity and turnover. PLoS Biol. 5:e67 10.1371/journal.pbio.0050067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A., Heldin C.H. 2009. The regulation of TGFbeta signal transduction. Development. 136:3699–3714 10.1242/dev.030338 [DOI] [PubMed] [Google Scholar]
- Mummery C.L. 2001. Transforming growth factor beta and mouse development. Microsc. Res. Tech. 52:374–386 [DOI] [PubMed] [Google Scholar]
- Oh S.P., Seki T., Goss K.A., Imamura T., Yi Y., Donahoe P.K., Li L., Miyazono K., ten Dijke P., Kim S., Li E. 2000. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA. 97:2626–2631 10.1073/pnas.97.6.2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D., Zhu Q., Luo K. 2009. SnoN functions as a tumour suppressor by inducing premature senescence. EMBO J. 28:3500–3513 10.1038/emboj.2009.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardali E., Goumans M.J., ten Dijke P. 2010. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 20:556–567 10.1016/j.tcb.2010.06.006 [DOI] [PubMed] [Google Scholar]
- Park S.O., Lee Y.J., Seki T., Hong K.H., Fliess N., Jiang Z., Park A., Wu X., Kaartinen V., Roman B.L., Oh S.P. 2008. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 111:633–642 10.1182/blood-2007-08-107359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.O., Wankhede M., Lee Y.J., Choi E.J., Fliess N., Choe S.W., Oh S.H., Walter G., Raizada M.K., Sorg B.S., Oh S.P. 2009. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Invest. 119:3487–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson-White S., McDuffie M. 2003. Defective T-cell activation is associated with augmented transforming growth factor Beta sensitivity in mice with mutations in the Sno gene. Mol. Cell. Biol. 23:5446–5459 10.1128/MCB.23.15.5446-5459.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper M.S. 1997. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 8:21–43 10.1016/S1359-6101(96)00048-2 [DOI] [PubMed] [Google Scholar]
- Primo L., Seano G., Roca C., Maione F., Gagliardi P.A., Sessa R., Martinelli M., Giraudo E., di Blasio L., Bussolino F. 2010. Increased expression of alpha6 integrin in endothelial cells unveils a proangiogenic role for basement membrane. Cancer Res. 70:5759–5769 10.1158/0008-5472.CAN-10-0507 [DOI] [PubMed] [Google Scholar]
- Risau W. 1997. Mechanisms of angiogenesis. Nature. 386:671–674 10.1038/386671a0 [DOI] [PubMed] [Google Scholar]
- Seki T., Yun J., Oh S.P. 2003. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ. Res. 93:682–689 10.1161/01.RES.0000095246.40391.3B [DOI] [PubMed] [Google Scholar]
- Shi Y., Massagué J. 2003. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 113:685–700 10.1016/S0092-8674(03)00432-X [DOI] [PubMed] [Google Scholar]
- Shinagawa T., Dong H.-D., Xu M., Maekawa T., Ishii S. 2000. The sno gene, which encodes a component of the histone deacetylase complex, acts as a tumor suppressor in mice. EMBO J. 19:2280–2291 10.1093/emboj/19.10.2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroschein S.L., Wang W., Zhou S., Zhou Q., Luo K. 1999. Negative feedback regulation of TGF-beta signaling by the SnoN oncoprotein. Science. 286:771–774 10.1126/science.286.5440.771 [DOI] [PubMed] [Google Scholar]
- Sun Y., Liu X., Ng-Eaton E., Lodish H.F., Weinberg R.A. 1999. SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor beta signaling. Proc. Natl. Acad. Sci. USA. 96:12442–12447 10.1073/pnas.96.22.12442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Imanaka T., Takano T. 1996. Spatial and temporal pattern of smooth muscle cell differentiation during development of the vascular system in the mouse embryo. Anat. Embryol. (Berl.). 194:515–526 [DOI] [PubMed] [Google Scholar]
- Urness L.D., Sorensen L.K., Li D.Y. 2000. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 26:328–331 10.1038/81634 [DOI] [PubMed] [Google Scholar]
- Wu M.Y., Hill C.S. 2009. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell. 16:329–343 10.1016/j.devcel.2009.02.012 [DOI] [PubMed] [Google Scholar]
- Wu J.W., Krawitz A.R., Chai J., Li W., Zhang F., Luo K., Shi Y. 2002. Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF-beta signaling. Cell. 111:357–367 10.1016/S0092-8674(02)01006-1 [DOI] [PubMed] [Google Scholar]
- Yang X., Castilla L.H., Xu X., Li C., Gotay J., Weinstein M., Liu P.P., Deng C.X. 1999. Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development. 126:1571–1580 [DOI] [PubMed] [Google Scholar]
- Zhu Q., Luo K. 2012. SnoN in regulation of embryonic development and tissue morphogenesis. FEBS Lett. 586:1971–1976 10.1016/j.febslet.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q., Pearson-White S., Luo K. 2005. Requirement for the SnoN oncoprotein in transforming growth factor beta-induced oncogenic transformation of fibroblast cells. Mol. Cell. Biol. 25:10731–10744 10.1128/MCB.25.24.10731-10744.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q., Krakowski A.R., Dunham E.E., Wang L., Bandyopadhyay A., Berdeaux R., Martin G.S., Sun L., Luo K. 2007. Dual role of SnoN in mammalian tumorigenesis. Mol. Cell. Biol. 27:324–339 10.1128/MCB.01394-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.