

Abstract
There are a number of reports of interstitial deletions of the long arm of chromosome 6 that have developmental delay and obesity suggesting that this is a distinct phenotype almost like Prader-Willi syndrome. Here we report a patient with a similar deletion but a strikingly different phenotype, one more in keeping with Marfan syndrome, although he does not fulfil the criteria for that syndrome. Array comparative genomic hybridization was performed to investigate a patient with a striking phenotype. This revealed an interstitial deletion of 6q14.1q15. Parental FISH studies were normal, indicating that this is a de novo deletion. Our patient has a completely different phenotype compared to other patients reported to have similar deletions. The common feature is developmental delay, but the body features are quite different in that our patient is tall, strikingly thin with pectus excavatum, scoliosis, skin striae, arachnodactyly, pes planus, cataracts, and a high-arched palate. This contrasts with other patients who have a similar deletion but have short stature and obesity. 6q14.1q15 interstitial deletions can have a very variable phenotype and do not necessarily conform to a clinical recognizable microdeletion syndrome caused by haploinsufficiency of dosage-sensitive genes in that region as proposed by others.
Key Words: 6q deletion, Developmental delay, Marfanoid phenotype, Mental retardation, Tall stature
The usefulness of comparative genomic array in the investigation of previously unknown causes of developmental delay and/or learning disability has been shown many times with the result that new distinctive phenotypes are being recognized and reported. Unfortunately, there frequently is a wide variation in the phenotype which is typically attributed to differences in gene content due to the size and location of the imbalance. A recent screening of 100 patients with Marfanoid syndromes and intellectual disability [Callier et al., 2013] found 16 with genomic imbalances, none of which included the imbalance found in our patient.
An emerging syndrome associated with interstitial deletions of chromosome 6 is a Prader-Willi-like phenotype associated with developmental delay, early-onset obesity, hypotonia, and eye/vision anomalies [Bonaglia et al., 2008]. Haploinsufficiency of the SIM1 locus at 6q16.3 has been implicated in the profound obesity feature [Holder et al., 2000].
Recently, Wentzel et al. [2010] reported 2 cases with a phenotype strikingly similar to those with haploinsufficiency of SIM1 but have an interstitial deletion at 6q14.1q15, which did not involve that locus and, therefore, loss of SIM1 did not account for the obesity.
Our patient has a very similar deletion to patient 1, described by Wenzel et al. [2010], and overlaps all but the distal 35-90 kb of the smaller deletion in patient 2, but presents with a distinctive Marfanoid phenotype. The only features in common with cases reported by Wenzel et al. [2010] are developmental delay, hypotonia and feeding problems in infancy. Our patient does not have any of the facial features described in their patients and, in particular, is thin rather than obese. Therefore, the phenotypic variation observed cannot be explained by deletion of an apparently similar number of common genes.
Materials and Methods
Technical Details
Array comparative genomic hybridization (aCGH) was performed on DNA extracted from peripheral blood and hybridized with same-sex normal reference DNA (Promega), using the CytoChip™ ISCA 8×60K v2.0 Oligonucleotide array, NCB136/hg18.137 (BlueGnome Ltd, Cambridge, UK). This chip evaluates genomic gains or losses at 498 targeted regions across the entire genome with an effective backbone resolution of 170 kb; OMIM genes are presented with average oligo spacing of 3.5 kb and at centromeres and subtelomeres of 4.4 kb. Data were analyzed using Bluefuse Multi software v2.2 (8100), Cytochip v2 algorithm.
Array CGH (Microarray) analysis identified a copy number loss in 112 probes spanning a 7.4-Mb region of the long arm of chromosome 6, from nucleotide 80,867,031-88,301,985 (NCB136/hg18). The nearest oligonucleotide probes at each end that are not deleted are at positions 80,824,058, proximally, and 88,356,002, distally (fig. 1).
Fig. 1.

A Gene content in the 6q14.1q15 from Genomic Variants in the Human Genome (Build 36: March 2006; hg18). B Deletion in our patient (red bar) compared with similar, overlapping deletions in cases reported by Wenzel et al. [2010] (blue bars).
Verification of the copy number loss and parental analysis was performed by FISH using a BAC probe, RP11-122C7 mapping within the region of interest at band 6q14.1, and a chromosome 6 control probe, RP11-791F23 (RPC1-11 Human BAC library – The Centre for Applied Genomics). Metaphase FISH showed the loss of a signal for the target locus on one chromosome 6 and a normal hybridization pattern for the control locus. Parental FISH testing revealed no deletions or rearrangement of the region, indicating a de novo deletion in the patient.
Case Report
The male proband was born after an emergency Caesarean section for fetal distress, but he did not require resuscitation. The birth weight was 3 kg and the length 54 cm. At the time of birth, his father was age 31 and his mother was 27. Their heights are 188 and 175 cm, respectively. There is no consanguinity. The proband has an older half sister and an older full sister who have no health or developmental problems. Feeding was a problem throughout the first year, and he was very hypotonic with poor musculature. There was concern about possible hydrocephaly, but a CT of his head was normal. Both parents have head circumferences >98th centile. Poor weight gain has always been an issue with the proband.
His milestones were extremely delayed. He sat unsupported at 2 years, stood alone between 4 and 5 years and walked independently at 7 years. He is aged 19 and has no speech, but makes garbled sounds and has a developmental level of about a 9-year-old. Exotropia was detected at 3 years and cataracts at 6 years. A recent eye examination showed small angle exotropia and small posterior cortical cataracts bilaterally. There is no subluxation of lenses and very mild refractive errors of −1.25 right and −1.50 left. The discs, vessels, macula, and peripheral retina are all normal.
As an infant and young child, he had many episodes of middle ear infections and bronchitis, often requiring antibiotics and tympanic tubes on 2 occasions, but with no residual hearing loss. Examination aged 17 revealed a height of 188.6 cm (>97th centile), weight 55.9 kg (10-25th centile), OFC of 60.2 cm (>98th centile), upper segment/lower segment ratio 0.82, and arm span 188 cm. He is tall and very thin with marked pectus excavatum. Clinical features include a long narrow face, downslanting palpebral fissures, prominent nasal bridge and nose, and prominent ears (figs. 2, 3). Oral cavity examination showed a highly arched palate with a short soft palate and severe malocclusion, but no obvious nasal escape. There is mild kyphothoracolumbar scoliosis and skin striae. His hands show long fingers and thumbs with mild contractures of the proximal interphalangeal joints of both 5th digits, single palmar crease (right), hypoplastic nails on 1st and 2nd digits, very long, large toes, and contractures of toes 4 and 5 bilaterally (figs. 4, 5). Thumb and wrist signs were positive. There are no elbow, shoulder, hip or knee contractures. Deep tendon reflexes in upper and lower limbs are normal and plantars are downgoing. He has poor balance and is unable to ride a bicycle or jump and does not run. Recent heart examination showed normal heart sounds, blood pressure 90/60, normal first and second heart sounds, and normal carotid impulse with no bruit and no added sounds or murmurs. ECG showed a right bundle branch block and a non-specific repolarizing abnormality. Echocardiogram showed normal aortic dimensions and a structurally normal heart. Genitalia are normal post-pubertal male with both testes descended and normal masculinization.
Fig. 2.
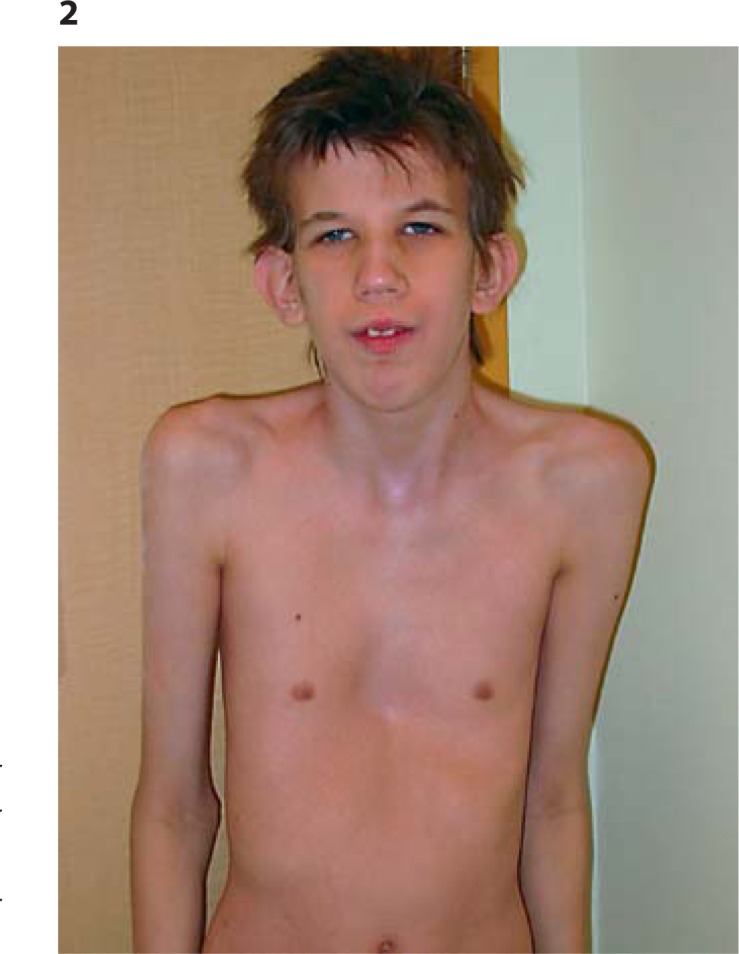
Patient at age 17. Thin habitus, pectus excavatum, downslanted palpebral fissures, and prominent ears.
Fig. 3.
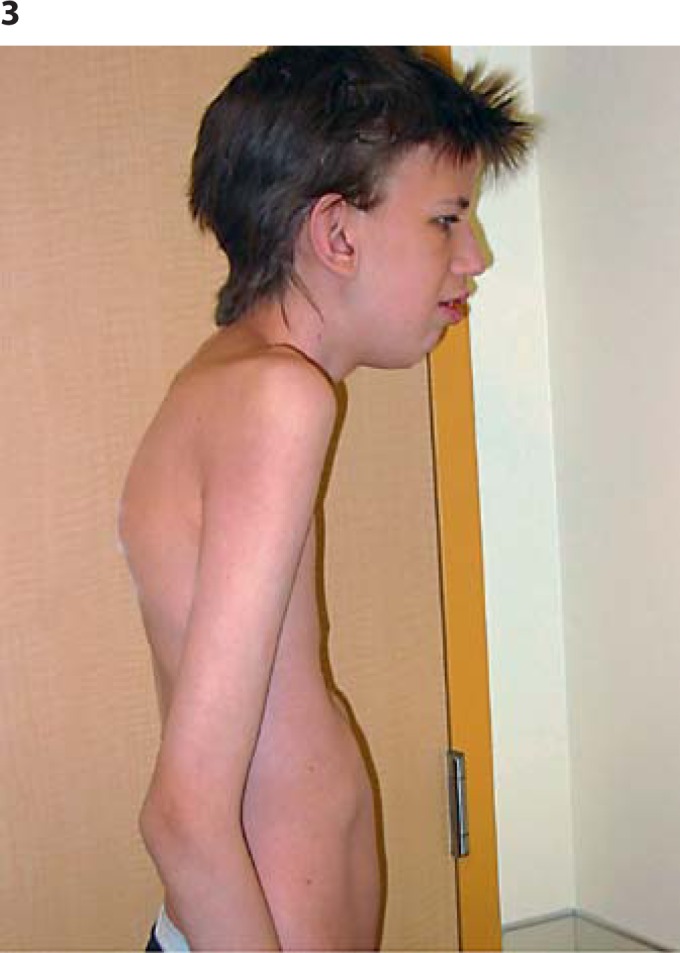
Patient at age 17. Thin habitus, kyphosis and prominent nose.
Fig. 4.
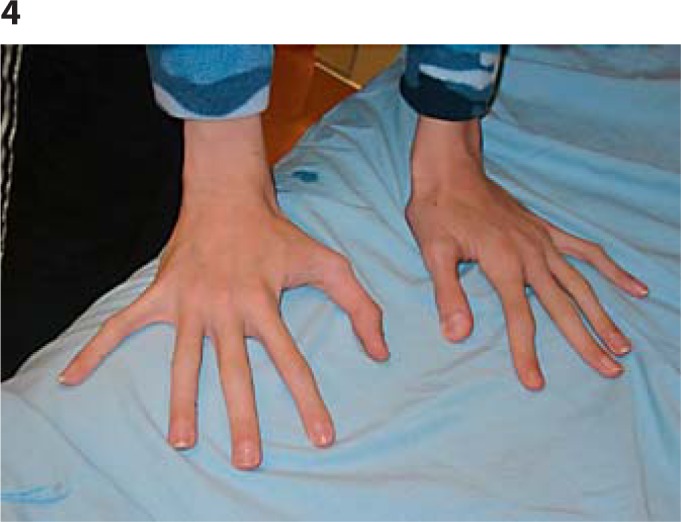
Hands: long fingers and thumbs, mild contracture of proximal interphalangeal joints of 5th digits.
Fig. 5.
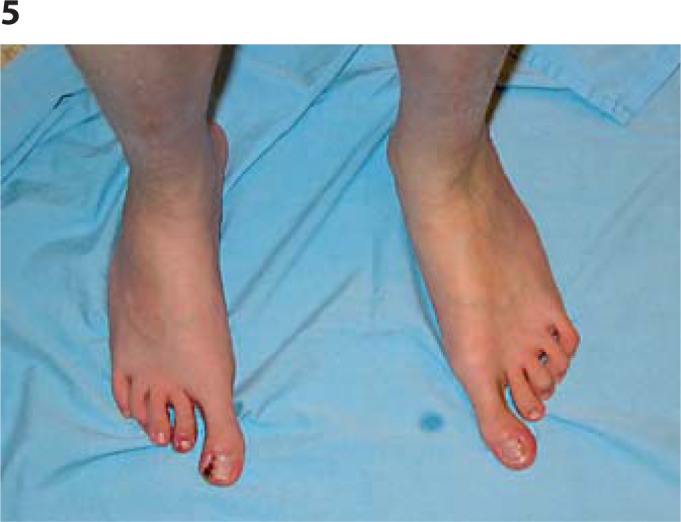
Feet: very long, large toes, contractures of toes 4 and 5, wide gap between 1 and 2.
There is no history of seizures, and there are no aggressive or disturbing behaviours. He is able to feed himself and do most of his toileting. Recently, he has developed nocturnal enuresis but has normal urinalysis and renal ultrasound. Past investigations, which were normal, have included karyotype, Fragile X testing, plasma amino acids, and 22q11.2 deletion testing.
Discussion
We report a unique patient with developmental delay and a Marfanoid habitus. Klinefelter syndrome and homocystinuria were excluded by appropriate tests, but other differential diagnoses included Marfan syndrome, Lujan-Fryns and Fragoso-Cantú syndromes. Marfan syndrome is clearly excluded based on the heart and eye signs and failure to fulfil the Ghent criteria. Lujan-Fryns was considered, but the behavioural or autistic components were lacking. In addition, sequencing for 7 exons of the MED12 gene, associated with Lujan-Fryns syndrome, was normal. Although Fragoso and Cantú [1984] syndrome was considered because there are many overlapping features, there are enough differences, particularly facial, to render that unlikely. At the age of 19 years, aCGH analysis revealed a 7.43-Mb deletion of the long arm of chromosome 6q14.1q15. This deletion encompasses 35 HGNC genes (17 OMIM), but does not include the SIM1 locus (BCKDHB, RPL17P25, FAM46A, IBTK, TPBG, UBE2CBP, DOPEY1, PGM3, RWDD2A, ME1, PRSS35, SNAP91, C6orf59, RIPPLY2, CYB5R4, MRAP2, KIAA1009, TBX18, KRT18P30, NT5E, SNX14, SYNCRIP, SNORD50A, SNORD50B, FAM165A, HTR1E, CGA, ZNF292, GJB7, C6orf162, C6orf163, C6orf164, C6orf165, SLC35A1, and RARS2).
Cytogenetically identified deletions of 6q are a rare but documented finding in patients with intellectual disability and congenital anomalies [McLeod et al., 1990; Pandya et al., 1995; Hopkin et al., 1997]. Hopkin et al. [1997] reviewed 60 patients which they grouped into 3 distinct phenotypes corresponding to proximal (6q11q16), middle (6q15q25) and terminal (6q256qter) deletions. Based on the breakpoints, our patient would fall into Group A (proximal deletions) characterized by upslanting palpebral fissures, thin lips, umbilical and inguinal hernias and an increase in whorls on finger tips. However, he shares none of these features. Prior to analysis by aCGH, genotype-phenotype correlation was complicated by the difficulty of precisely assigning breakpoints and the possible presence of confounding genomic imbalances that are due to cryptic karyotypic complexity.
Our patient's deletion 6q14.1q15 (chromosome 6:80867030..88301984) most closely matches that of patient 1 and overlaps all but the distal 35 (maximum size deletion) – 90 (minimum size) kb of the deletion of patient 2, reported by Wentzel et al. [2010]. Despite having a deletion which overlaps the common region deleted in these 2 patients, our patient has a completely different phenotype. Wentzel et al. [2010] postulate that ME1 (MIM 154250) is a candidate gene in the region for the obesity phenotype, supported by rodent models of altered ME1 enzyme activity. In contrast, Yang et al. [2009] demonstrated that knock-out Me1 mice showed decreased body weight, but not specifically adiposity, more in keeping with our patient who has a tall and very thin habitus.
A review of Decipher entries with overlapping deletions demonstrated none with a ‘Marfanoid’ habitus, and of those cases including a loss of the ME1 gene, besides the 2 Wenzel et al. [2010] cases (1,878 and 248,173); only one other case listed obesity as a phenotypic feature (249,539). Within our patient's deletion are genes that are likely to be haploinsufficient, including FAM46A, PGM3, SNAP91, SYNCRIP, ZNF292, and RARS2.
Therefore, the distinct clinical features in our patient cannot be attributable to loss of a different set of dosage-sensitive genes, and the obesity-developmental delay phenotype shared by the 2 patients of Wentzel et al. [2010] cannot be solely due to haploinsufficiency of genes in the common overlapping region because our patient does not share the obesity phenotype. Possible unmasking of a recessive allele in the region or altered long-range gene expression may account for the phenotypic differences. However, the phenotypic spectrum for many well-known microdeletion syndromes is quite broad and expanding with the use of genomic technologies in patients not previously suspected to have a diagnosis. Therefore, this may represent another variable microdeletion syndrome where genomic imbalance has an epigenetic and unpredictable effect on development.
A systematic investigation of 100 patients with Marfanoid phenotype and intellectual disability by Callier et al. [2013] came to the conclusion that aCGH should be the initial investigation. Subsequent steps depended on the presence or absence of thoracic aortic aneurysm. If present, sequence screening for FBN1 ± TGFBR1/2 should be undertaken. Since our patient does not have a thoracic aortic aneurysm, we have followed their suggested flow chart and proceeded no further.
The array diagnosis and negative testing for thoracic aortic aneurysm is extremely helpful in terms of management, since he will not need the extensive cardiovascular and ophthalmic follow-up examinations which would be required for a patient with Marfan syndrome. The diagnosis also provides definitive risk counselling for his sisters. Without this diagnosis, one could not exclude a rare X-linked recessive gene and the possibility that they could be carriers. Now they can be reassured that they are highly unlikely to have an offspring similar to their brother.
Acknowledgements
We thank the parents and patient for their cooperation, and Brenda McInnes and Judy Anderson for their assistance.
References
- 1.Bonaglia MC, Cicoone R, Gemelli G, Gemelli S, Marelli S, et al. Detailed phenotype-genotype study in five patients with chromosomes 6q16 deletion: narrowing the critical region for Prader-Willi-like phenotype. Eur J Hum Genet. 2008;16:1443–1449. doi: 10.1038/ejhg.2008.119. [DOI] [PubMed] [Google Scholar]
- 2.Callier P, Aral B, Hanna N, Lambert S, Dindy H, et al. : Systematic molecular and cytogenetic screening of 100 patients with marfanoid syndromes and intellectual disability. Clin Genet (2013), E-pub ahead of print. [DOI] [PubMed]
- 3.Fragoso R, Cantú JM. A new psychomotor retardation syndrome with peculiar facies and marfanoid habitus. Clin Genet. 1984;25:187–190. doi: 10.1111/j.1399-0004.1984.tb00483.x. [DOI] [PubMed] [Google Scholar]
- 4.Holder JLO, Jr, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 5.Hopkin RJ, Schorry E, Bofinger M, Milatovich A, Stern HJ, et al. New insights into the phenotypes of 6q deletions. Am J Med Genet. 1997;70:377–386. [PubMed] [Google Scholar]
- 6.McLeod DR, Fowlow SB, Robertson A, Samcoe D, Burgess I, Hoo JJ. Chromosome 6q deletions: a report of two additional cases and a review of the literature. Am J Med Genet. 1990;35:79–84. doi: 10.1002/ajmg.1320350115. [DOI] [PubMed] [Google Scholar]
- 7.Pandya A, Braverman N, Pyeritz RE, Juang-Lin Y, Kline AD, Falk RE. Interstitial deletion of the long arm of chromosome 6 associated with unusual limb anomalies: report of two new patients and review of the literature. Am J Med Genet. 1995;59:38–43. doi: 10.1002/ajmg.1320590109. [DOI] [PubMed] [Google Scholar]
- 8.Wentzel C, Lynch SA, Stattin EL, Sharkey FH, Annerén G, Thuresson AC. Interstitial deletions at 6q14.1-q15 associated with obesity, developmental delay and a distinct clinical phenotype. Mol Syndromol. 2010;1:75–81. doi: 10.1159/000314025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang X, Deignan JL, Hongxiu Q, Zhu J, Qian S, et al. Validation of candidate causal genes for abdominal obesity which affect shared metabolic pathways and networks. Nat Genet. 2009;41:415–423. doi: 10.1038/ng.325. [DOI] [PMC free article] [PubMed] [Google Scholar]