

Abstract
BACKGROUND
Environmental factors contribute to the etiology of cleft palate (CP). Identification of genes that are methylated during development of the secondary palate will contribute to a better understanding of the gene-environment link contributing to CP.
METHODS
Genomic DNA fragments from secondary palate tissue from gestational days (GDs) 12 to 14 were subjected to Selective Enrichment of Methylated DNA (SEMD) and used to probe NimbleGen 2.1M mouse promoter arrays. Input (control) and SEMD samples were labeled with Cy3 and Cy5, respectively, and used for array hybridization (three arrays per GD). Data were analyzed using the Bioconductor package Ringo. Gene methylation was verified by pyrosequencing analysis and expression by quantitative real-time PCR.
RESULTS
A total of 5577 methylated genes were identified during palate development: (1) 74% of genes were methylated on all three GDs; (2) CpG islands accounted for only 30% of methylated regions of interest (MRIs); (3) location of MRIs was more often observed in gene bodies (73%) than in promoters; (4) evaluation of MRIs on GDs 12–14 revealed no significant differentially methylated regions; (5) DAVID analysis of MRIs revealed that the cadherin and Wnt signaling pathways, as well as pathways involved in proteoglycan synthesis, were significantly enriched for methylated genes.
CONCLUSIONS
Our prior studies identified differentially expressed mRNAs and micro-RNAs in the developing palate. The current study complements these studies by identifying genes whose expression may be altered as a result of DNA methylation.
Keywords: cleft palate, CpG methylation, craniofacial, epigenetics, extra-cellular matrix, promoter microarray, pyrosequencing, secondary palate, Wnt signaling
INTRODUCTION
Orofacial clefts, such as cleft lip with or without cleft palate (CL/P), and isolated cleft palate (CP), affect about 6800 newborns in the US every year (Yazdy et al., 2007). Affected individuals experience a host of health complications, including ear infections and hearing loss, difficulty with feeding and speech, dental abnormalities, and psychological challenges such as social, emotional and behavioral problems because of their appearance. CP is primarily caused by the aberrant development of the secondary palate. The key phases of secondary palate development are the elevation of the paired palatal processes, oral projections from the embryonic maxillary processes, and their subsequent adhesion and fusion to each other forming the roof of the oral cavity (Bush and Jiang, 2012). This process is highly conserved in most mammals, and occurs, in mice, during gestational days (GD) 12 to 14. Genetic alterations are thought to contribute to ∼20 to 25% of CP cases (Marazita, 2002; Lidral and Moreno, 2005) while environmental factors represent contributing factors in the remaining cases. Analyses of mutations indicate that many of the genes responsible for palate development encode transcription factors, growth and signaling molecules and their receptors, and ECM (extracellular matrix) components (reviewed in Cobourne, 2004; Murray and Schutte, 2004; Stanier and Moore, 2004; Lidral and Moreno, 2005; Rice, 2005; Carinci et al., 2007; Gritli-Linde, 2007; Juriloff and Harris, 2008; Jugessur et al., 2009; Murthy and Bhaskar, 2009; Vieira, 2008; Yu et al., 2009; Bush and Jiang, 2012). Several of these genes are members of key signal transduction pathways required for palate development. These include the Wnt-, TGFβ-, PDGF-, FGF-, and Shh-signaling systems (Murray and Schutte, 2004; Gritli-Linde, 2007; Jugessur et al., 2009; Yu et al., 2009; Greene and Pisano, 2010; Rahimov et al., 2012).
Environmental factors such as exposure to alcohol, cigarette smoke, certain drugs, chemicals (e.g., pesticides), infection, and lack of nutrients (e.g., trace elements and folate) have been implicated in the etiology of orofacial malformations (Murray, 2002; Murray and Schutte, 2004; Lidral and Moreno, 2005; Rice, 2005; Juriloff and Harris, 2008; Vieira, 2008; Mossey et al., 2009; Murthy and Bhaskar, 2009; Zhu et al., 2009; Greene and Pisano, 2010). One mechanism that could account for many of these environmental effects is aberrant methylation of susceptible genes during embryonic or fetal development, resulting in the altered expression of critical genes necessary for normal palate development. Conventional screening techniques such as mutational analysis or Genome-Wide Association Studies (GWAS) do not identify such epigenetically altered genes. Moreover, few studies have attempted to clarify the role of DNA methylation in regulating secondary palate development, or to identify genes that are differentially methylated during this critical process. In our prior studies, differentially expressed mRNAs (Mukhopadhyay et al., 2006) and microRNAs (Mukhopadhyay et al., 2010) were identified in the developing murine secondary palate using microarray technology. The current study complements these studies by identifying genes whose expression may be altered as a result of (differential) DNA methylation in developing palatal tissue.
METHODS
Animals
Mice (ICR strain; Harlan, Indianapolis, IN) were used as per approved IACUC protocols and were housed in a climate-controlled room with a 12-hr alternating dark-light cycle. Mice were mated overnight and females were checked for vaginal plugs the following morning (GD0). On GD12, GD13, and GD14, pregnant dams were euthanized by CO2 asphyxiation and embryos collected in ice-cold calcium/magnesium-free PBS, and immediately processed.
Mouse Secondary Palate and Genomic DNA Isolation
Mouse secondary palates were isolated, dissected, and suspended in ice-cold PBS, as previously described (Mukhopadhyay et al., 2010). For each GD (GD12–14), three different litters (representing three biological replicates) were used for isolation of genomic DNA. The developmental state of the secondary palate at each GD was visually noted (GD12—palatal processes vertically oriented on either side of the tongue; GD13—palatal processes larger and either still vertically oriented, or horizontally oriented above the tongue but unfused; GD14—palatal processes fused to each other) to ensure sample consistency among the biological replicates. Genomic DNA was isolated from 20 to 40 mg of pooled tissues using the QIAamp DNA mini kit (QIAGEN Sciences, MD).
Microarray Experiments
Ten μg genomic DNA was suspended in 400 μl TE buffer and sonicated using a Microson Ultrasonic Cell Disruptor (Heat Systems Ultrasonics, Inc., Farmingdale, NY), set at 20% power output. Four 10-s pulses on ice, with a 1-min interval between pulses were sufficient to shear DNA to 300 to 1000 bp. Five microliters of the sonicate was examined for the desired DNA fragmentation size (∼300 to 1000 bp) by electrophoresis on 1% agarose gels. Samples were precipitated overnight at −20°C in 400 mM NaCl, 20 μg glycogen, and 2 vols of ethanol. DNA was pelleted, dissolved in 200 μl TE buffer and concentration determined using a NanoDrop ND1000 Spectrophotometer (Thermo Scientific, Wilmington, DE). Methylated DNA fragments were isolated using the MethylCollectorTM kit from Active Motif, Inc. (Carlsbad, CA). Briefly, 1 μg sonicated genomic DNA was incubated with nickel-coated magnetic beads bound to a protein that binds to methylated CpG residues (His-tagged MBD2b). Control DNA (provided with the MethylCollectorTM kit) consisted of genomic DNAs containing methylated and unmethylated versions of an unspecified gene, which allowed monitoring of the experimental protocol. Genomic DNA subjected to Selective Enrichment of Methylated DNA (SEMD) was recovered from the DNA-protein complexes, ethanol precipitated and subsequently amplified using a Whole Genome Amplification kit (Sigma; St. Louis, MO). As a control, an aliquot of the sonicated, genomic DNA not subjected to SEMD (i.e., input DNA) was similarly amplified.
Nine microarrays (mouse 2.1M Deluxe Promoter Arrays; Roche NimbleGen, Inc., Madison, WI) representing three biological replicates on each of GDs 12, 13, and 14, were probed with either SEMD DNA labeled with Cy5 or input control DNA labeled with Cy3. The NimbleGen 2.1M mouse promoter array contains 2.1 million oligonucleotide probes covering 10 kb of all annotated promoters, all known CpG islands, and all miRNA promoters. The arrays are tiled at 100 bp probe spacing and include positive, negative, and non CpG control regions to monitor the quality of array data. Signal intensity data, extracted from the scanned images of each array using NimbleScan, NimbleGen's data extraction software, was utilized for analysis. A schematic of the experimental steps taken to obtain the array data is illustrated in Supporting Information Figure S1.
Microarray Data Analysis
Data preprocessing
For each sample, the log (base 2) ratios, log2(Cy5/Cy3), for all reporters were computed. To adjust for systematic dye and labeling biases, we performed T-quantile normalization on all of the arrays. The T-quantile method uses quantile normalization in each of the Cy5 and Cy3 channels separately, and was recently found to outperform other normalization methods for DNA methylation arrays (Adriaens et al., 2012). The log2(Cy5/Cy3) values on each array were then recentered by subtracting the mode of the values on the array. The R package arrayQualityMetrics (Kauffmann et al., 2009) was used to assess array quality and identify any outliers. Plots of the distance between arrays, densities of the arrays, and comparisons of the average versus difference of the log2(Cy5/Cy3) values for each array relative to a “pseudo”-array consisting of the median values of all the arrays were used to check the expression levels of each chip after normalization. Outlier detection was performed by computing Hoeffding's statistic Da, which tests for the independence of the A and M distributions for each array (Hoeffding, 1948).
Smoothing of reporter intensities
A smoothing of individual reporter intensities was performed, prior to identifying SEMD regions. We chose a sliding window of 900 base pairs (bp), and the reporter level at genomic position x0 was replaced by the median over the intensities of those reporters mapped inside the window centered at x0. At any position x0 at which the window comprised less than three reporter-matched positions, the smoothed level was flagged as missing, as the data were insufficient to provide information about SEMD at such a position. The average spacing between reporter-matched positions in our data was roughly 100 bp.
Locating methylated regions of interest
To identify candidate methylated regions of interest (MRIs), the algorithm developed in the Bioconductor (Gentleman et al., 2004) package Ringo (Toedling et al., 2007) was applied. In this approach, a mixture model is fit to each array to determine an enrichment threshold y0 above which individual probes are considered methylated (Toedling and Huber, 2008). After fitting the mixture model to each array and identifying the methylated probes, a majority vote (2/3) was required to consider a probe to be methylated for a particular GD. Methylated regions corresponding to each GD were determined by the presence of six or more contiguous methylated probes within a 900 bp window. The requirement of six contiguous probes avoids identifying methylated regions supported by only a few reporters, and the window width was slightly less than the average fragment size to prevent calling disconnected methylated regions.
Relating MRIs to genomic features
MRIs were related to annotated genome elements, such as CpG islands and genes, using the Bioconductor package biomaRt (Durinck et al., 2009). Mouse genome annotation was obtained using Build 37 from the Ensembl database (mm9, July 2007), including Ensembl gene identifiers, MGI (mouse genome informatics) symbols, and descriptions of all genes annotated for the mm9 mouse assembly. An MRI was regarded as related to a gene if its center position was located less than 5 kb upstream of a gene's start coordinate, or between a gene's start and end coordinates. Genes associated with MRIs were determined separately for each GD, and genes uniquely associated with a particular GD were also identified. Associated genes were also segregated based on whether the MRI overlapped with the gene-body, promoter or regions further upstream (see Results for demarcation of these regions). CpG islands (Gardiner-Garden and Frommer, 1987) were determined using the UCSC Genome Browser (http://genome.ucsc.edu/), and MRIs were overlapped with CpG island regions using Galaxy (http://usegalaxy.org/) (Goecks et al., 2010).
Determining differentially methylated regions (DMRs)
Differentially methylated regions between GD12, 13, and 14 were determined using a statistical test based on a linear mixed-effects (LME) model (Everitt and Rabe-Hesketh, 2001). For each identified MRI on a particular GD, an LME model was fit to the cluster of probes belonging to the methylated region, to determine whether that region had statistically significant higher average log2(Cy5/Cy3) probe intensities for that GD versus the others. The LME model had the following form:
where yijk is the normalized log2(Cy5/Cy3) ratio for reporter k (k = 1,. . ., “# reporters in MRI”) on array j (j = 1,. . .,9) for gestational day i (i = 1,2,3). The are random effects representing array-specific variability, and the are residual errors. Note that the response yijk is the normalized, but unsmoothed log intensity ratios. The αi are main effects representing overall location shifts in intensity ratios within the MRI associated with gestational days. To test whether the intensity ratios were higher for one GD versus another, tests of H0 : αi = αj versus H1 : αi ≠ αj were performed for i = 1, 2, 3; j = 2, 3; i ≠ j (e.g., for each comparison between GDs) using t-tests based on the fitted model. Adjustments for multiple comparisons were done using the Benjamini-Hochberg approach (Benjamini and Hochberg, 1995) which controls the overall false discovery rate. Separate adjustments were made for each comparison between GDs, and for each set of MRIs identified on a particular GD. LME models were fit using the R (R Development Core Team, 2012) package nlme (Pinheiro et al., 2011), and adjustment for multiple comparisons was done using the R package multtest (Pollard et al., 2011).
Gene-set enrichment analysis
To determine the biological relevance of the genes associated with our MRIs, gene lists were uploaded to the DAVID software (Huang et al., 2007) for gene-set enrichment analysis (GSEA). Gene lists were identified based on the genes associated with the MRIs identified for each GD and further stratified by methylation status (gene-body/promoter). GSEA was performed using relevant databases in the broad areas of “GO terms,” “Pathways,” “Functional Categories,” and “Protein Domains.” The EASE p-value threshold was set to 0.001 for “GO terms,” “Functional Categories,” and “Protein Domains,” but for “Pathways” was set to a higher value of 0.1 due to the fewer overall number of genes annotated in these databases.
Pyrosequencing
Methylation levels of representative CpG residues found within the highly methylated regions of MRIs in eight selected genes—Aph1a, Bmp8b/Oxct2b, Ctnnb1, Dkk4, Fat1, Olfr66, Siah1b, and Wnt5a—were evaluated by pyro-sequencing. These genes were selected based on the location of MRIs in different gene compartments—e.g., the gene-body (Bmp8b/Oxct2b, Wnt5a, Aph1a, and Fat1), the putative promoter (Olfr66 and Siah1b) and the 5′ upstream region (Dkk4 and Ctnnb1). The gene-body was defined as a region extending from the TSS (transcription start site) to the end of the 3′ UTR; the putative promoter, as a region spanning ± 500 bp of the TSS; and the 5′ upstream region as a DNA region within 5 kb upstream of the TSS excluding the promoter region. Genomic DNA was bisulfite-modified using the EpiTect Bisulfite kit (Qiagen). Primers for the amplification of genes from bisulfite-modified DNA were either procured from Qiagen's PyroMark CpG assay or custom designed using the PyroMark Assay Design 2.0 software. The various primers used and the sequences subjected to pyrosequencing are depicted in Supporting Information Table S1. Samples were amplified using the PyroMark PCR kit (Qiagen) and CpG methylation levels were analyzed using the PyroMark Q24 system (Qiagen). PCR samples were pyrosequenced a minimum of three times using at least two independently amplified PCR templates. For each CpG site, the mean ± SD (standard deviation) was calculated and p values (unpaired t-test) for each comparison between GDs was performed. A CpG residue was considered differentially methylated if it exhibited both a p value <0.05 and an arbitrary ≥5% change in methylation levels.
mRNA Expression Analysis
mRNA expression levels were determined by quantitative real-time PCR (qRT-PCR) using cDNA samples prepared from GD12, GD13, and GD14 secondary palatal tissue. cDNAs were synthesized, using the SuperScript III First Strand Synthesis kit (Life Technologies), from 1 μg of total mRNA, isolated using the RNeasy Protect Mini kit (Qiagen). qRT-PCR was undertaken on cDNA samples using the Fast SYBR Green Mastermix (Applied Biosystems) in a Viia7 Real-time PCR System (Applied Biosystems). Primers for PCR amplification were selected using the Primer3 v 0.4.0 software (http://frodo.wi.mit.edu/primer3/) and synthesized by IDT (Coralville, Iowa). Primers were first validated by melt curve analysis of the amplified product before quantification by the ΔΔCt method (Livak and Schmittgen, 2001). Effects of GD on normalized Ct values (ΔCt) from qRT-PCR for mRNA expression were analyzed using a one-way ANOVA model. Post hoc comparisons to GD12 were presented with Dunnett's adjustment for multiple comparisons. Means and standard deviations from two independent experiments, followed by differences or fold-changes relative to GD12 levels were presented with simultaneous 95% confidence limits and statistical significance after multiple comparison adjustment. p Values of <0.05 were considered significant. Statistical analyses were performed using SAS v9.3 (SAS Institute Inc., Cary, NC). Mean Ct values were used to arbitrarily categorize mRNA expression as low (Ct: >32), moderate (Ct: 25–32), or high (Ct: <25).
Genome-wide mRNA expression levels during the period of murine orofacial development were also obtained from previously published data (Affymetrix GeneChip Arrays; Mukhopadhyay et al., 2004). Data were pre-processed as previously described, and probes were cross-referenced with genes determined to be gene-body or promoter methylated. This enabled us to evaluate the genome-wide effect of promoter/gene-body methylation during the period of murine secondary palate development. Expression levels of gene-body/promoter methylated versus non-methylated genes were visually compared using boxplots, and evaluated for statistically significant differences using a linear model with main effects for gestational day (GD 12, 13, and 14), and methylation status (gene-body methylated vs. non-methylated and promoter methylated vs. non-methylated).
RESULTS
Array Normalization and Determination of MRIs
After array normalization, methylation enrichment values appeared adequately adjusted (see Supporting Information Fig. S2–S3). Distributions of smoothed reporter levels were relatively homogeneous across arrays (Fig. 1), with the slight exception of arrays 1 (GD12-A) and 7 (GD14-A), which had more skewed right tails relative to the other arrays. The overall percent methylation of probes on each GD was not statistically different (4.83%, 2.89%, and 4.97% for GDs 12, 13, and 14, respectively, p = 0.69 based on one-way analysis of variance (ANOVA)). Percent methylation of probes also did not differ significantly when comparing methylation of individual chromosomes (p = 0.61, one-way ANOVA). Based on our algorithm, we identified a total of 16,357 MRIs across all GDs: 5590 on GD12, 4819 on GD13, and 5948 on GD14. A full listing of all MRIs is given in Supporting Information Table S2. The overall mean and standard deviation for the MRI length was 969 ± 524 base pairs, with a mean span of 10.4 ± 5.1 probes. MRI length was consistent across GDs (p = 0.15 for differences, one-way ANOVA), though significant differences existed across chromosomes (p < 0.001, see Supporting Information Table S3). Figure 2 displays the number of MRIs per chromosome as a function of chromosome length (Y chromosome excluded), color-coded by GD. As expected, there is a clear trend in increasing number of MRIs with chromosome size, though there is considerable variability about the trend lines.
Figure 1.
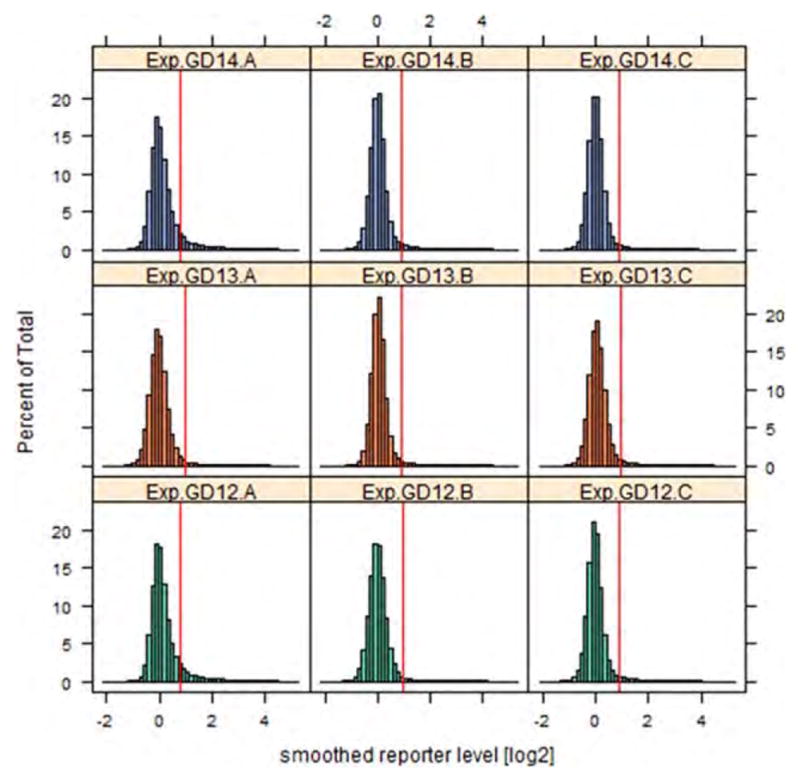
Histograms of smoothed reporter intensities. Histograms of smoothed log2(Cy5/Cy3) levels for each array, color-coded according to gestational day (GD). Vertical red lines in each case represent the cut-off above which individual reporters were declared to be methylated. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 2.
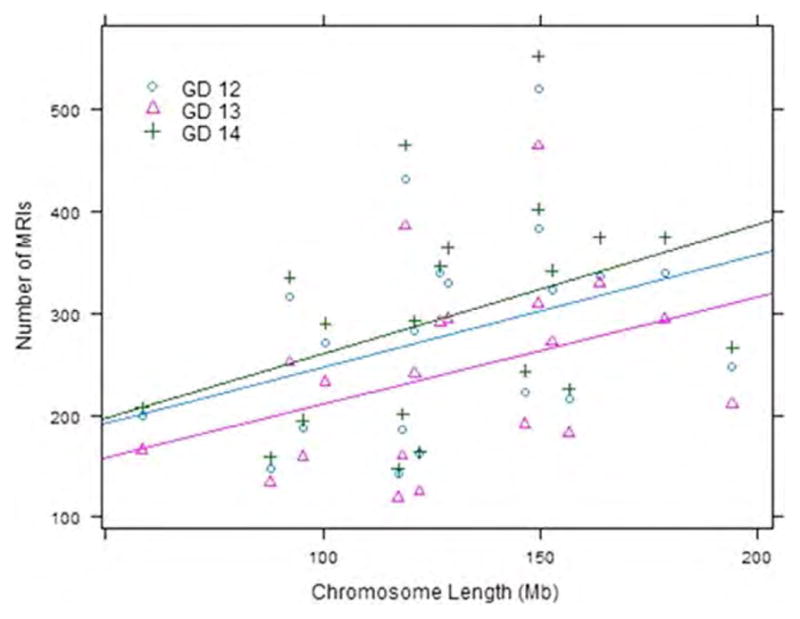
Methylated regions of interest (MRIs) per chromosome. Scatterplot showing the relationship between chromosome length and number of MRIs, color-coded by gestational day (GD). Straight lines indicate best linear regression fit of number of MRIs onto chromosome length, for each GD.
After associating each MRI with its cognate gene, the genes were sorted into those that were associated with methylated regions on GD12, 13, or 14. These data are depicted using a Venn diagram (Fig. 3). A majority of genes (74%; 4150 of 5577) were associated with MRIs on all 3 gestational days, with only a small fraction of genes associated with a methylated region on a single GD (4.45% on GD12, 1% on GD13, and 8.5% on GD14). An equally small fraction of genes were associated with MRIs on two of the three GDs (1.4% on GD12 and GD13, 2.3% on GD13 and GD14). It is stressed that Figure 3 does not statistically identify differentially methylated genes. This is accomplished using the LME model and the results are described in the following subsection Evaluation of DMRs.
Figure 3.
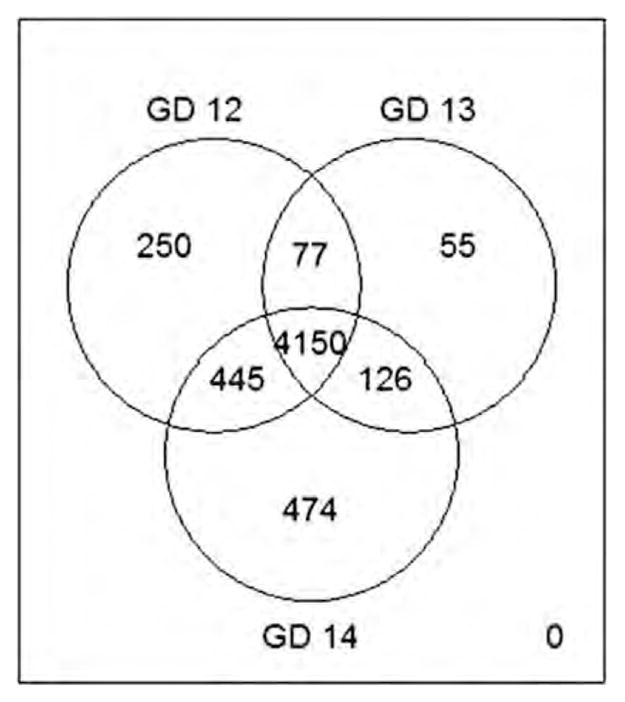
Genome-wide promoter-specific methylation in mouse secondary palate. Venn diagram depicting the number of methylated genes on GD12, GD13, and GD14. Note that a predominant number of genes (4150/5577; 74%) are methylated on all three gestational days. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com]
Location of the MRIs occurred predominantly within the gene-body (4071 of 5577, 73%), while 2095 genes (37.6%) were associated with MRIs in the 5′ upstream region including the putative promoter region (up to 5 kb from the TSS) and 589 (10.5%) of genes were associated with MRIs in both the gene-body and upstream/promoter regions. A total of 821 genes (14.7%) were associated with MRIs specifically in the promoter region (±500 bp of the TSS) of the gene. Out of a total of 16,027 CpG islands, the number of methylated CpG islands was roughly constant at 10% for each GD (1634 (10.2%) on GD12, 1598 (10.0%) on GD13, and 1633 (10.2%) on GD14). Most CpG islands were associated with only one MRI, with a small number (between 6 and 10) associated with two MRIs on each GD. The methylation status of each CpG island was also highly conserved across GDs 12 through 14, with 84.5% of methylated CpG islands identical between the GDs. Only 4.4%, 1.9%, and 3.8% of methylated CpG islands were unique to GDs 12, 13, and 14, respectively. Of the 16,357 total MRIs identified for all GDs, 11,468 (70%) were in non-CpG island regions while only 4889 (30%) were associated with CpG island regions. The percentage of MRIs in CpG island regions was similar for each GD (29% on GD12, 32% on GD13, and 27% on GD14).
Evaluation of DMRs
After determining the MRIs for each GD, a linear mixed-effects (LME) model (Everitt and Rabe-Hesketh, 2001) was fitted to each MRI to evaluate whether there was differential methylation between the GDs. After adjustment for multiple comparisons, no significant differentially methylated regions were identified. This is consistent with the high level of overlap found between methylated regions indicated in Figure 3. However, to identify potential candidate regions on which to perform follow-up experiments, Table 1 reports the DMRs with an unadjusted p value <0.05. This identified some potentially interesting regions, including an MRI on chromosome 7 that had ≈fourfold increase in methylation intensity for both GD12 and 13 relative to GD14 (Table 1; see Olfr66 in Fig. 4). This region was upstream of the gene Olfr66, an olfactory receptor, and additionally spanned the gene bodies of two relatively uncharacterized Ensembl Gene IDs. Another region upstream of the genes Poln and BC023882 was ≈2.8 fold higher in methylation intensity on GD13 relative to GD14 (Table 1). Lastly, several methylated regions on GD14 were found to have between 1.6- and 2.8-fold higher methylation intensity relative to GD12 and GD13 (Table 1), including a region upstream of Itih5 on chromosome 7 and a region in the gene-body of Map3k1 on chromosome 8 (not shown).
Table 1.
Differentially Methylated Regions in the Murine Secondary Palate Development.
| MRI | Chromosome: Start-End | No. Probes | Genes Methylated | p-Valuea | Fold Changeb | ||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Upstream | Inside | ||||||||
| GD 12 MRIsc | |||||||||
| GD12 vs. 13 | MRI-225 | 3: 96164030-544 | 6 | ENSMUSG00000065308 | 0.032 | 1.72 | |||
| MRI-272 | 3: 142224032-532 | 6 | Gbp3 | 0.037 | 1.41 | ||||
| MRI-128 | 10: 85418246-9200 | 7 | D10Wsu52e | 0.049 | 1.67 | ||||
| MRI-26 | 10: 117237033-514 | 6 | 0.05 | 1.75 | |||||
| GD12 vs. 14 | MRI-381 | 7: 111030780-2184 | 15 | Olfr66 | ENSMUSG00000057330. | 0.001 | 4.00 | ||
| ENSMUSG00000083854 | |||||||||
| MRI-196 | 16: 18622844-3430 | 7 | Gb1bb | 5-Sep | 0.03 | 0.52 | |||
| MRI-129 | 13: 112645638-6819 | 13 | 0.032 | 1.49 | |||||
| GD 13 MRIsc | |||||||||
| GD13 vs. 14 | MRI-345 | 7: 111030780-2184 | 15 | Olfr66 | ENSMUSG00000057330. | 0.001 | 4.35 | ||
| ENSMUSG00000083854 | |||||||||
| MRI-34 | 5: 34514088-5210 | 12 | Poln BC023882 | 0.021 | 2.78 | ||||
| MRI-9 | 18: 17119863-20371 | 6 | 0.022 | 1.69 | |||||
| MRI-281 | 8: 125844173-5004 | 9 | Fanca | 0.031 | 0.54 | ||||
| MRI-2 | 19: 4066470-7274 | 6 | 0.045 | 1.61 | |||||
| MRI-229 | 7: 51032468-968 | 6 | Klk10 | Klk11 | 0.048 | 0.74 | |||
| GD 14 MRIsc | p-Valuea | Fold Changeb | |||||||
|
|
|
||||||||
| 14 vs. 12 | 14 vs. 13 | 14 vs. 12 | 14 vs. 13 | ||||||
| GD14 vs. 12 & GD14 vs. 13 | MRI-126 | 13: 112555392-6084 | 8 | Map3k1 | 0.01 | 0.05 | 2.09 | 1.61 | |
| MRI-131 | 13: 112645718-6618 | 10 | 0.03 | 0.05 | 0.63 | 0.68 | |||
| MRI-390 | 5: 149697150-650 | 6 | Katnal1 | 0.05 | 0.03 | 1.43 | 1.51 | ||
| MRI-121 | 17: 29458944-9564 | 7 | Pi16 | 0.05 | 0.05 | 1.36 | 1.35 | ||
| GD14 vs. 12 only | MRI-70 | 13: 56405158-6048 | 10 | 0.03 | 0.06 | 2.2 | 1.85 | ||
| MRI-29 | 5: 24680486-1096 | 7 | 0.03 | 0.11 | 2.22 | 1.63 | |||
| MRI-436 | 11: 118942392-3028 | 7 | Cbx4 | 0.03 | 0.06 | 1.63 | 1.5 | ||
| MRI-26 | 16: 18622524-3544 | 11 | Gp1bb | 5-Sep | 0.03 | 0.06 | 1.71 | 1.56 | |
| MRI-132 | 13: 112661540-2225 | 8 | 0.04 | 0.75 | 0.62 | 0.94 | |||
| MRI-351 | 5: 144016414-914 | 6 | Zfp316 | 0.04 | 0.1 | 1.75 | 1.52 | ||
| MRI-219 | 17: 45682380-876 | 6 | Tmem151b | 0.05 | 0.13 | 1.86 | 1.53 | ||
| GD14 vs. 13 only | MRI-8 | 2: 10073117-704 | 7 | Itih5 | 0.15 | 0.01 | 1.59 | 2.78 | |
| MRI-7 | 5: 5381920-2538 | 7 | Pftk1 | 0.2 | 0.02 | 1.32 | 1.86 | ||
| MRI-260 | 4: 141404848-5474 | 7 | Ctrc | 0.58 | 0.02 | 1.12 | 1.82 | ||
| MRI-293 | 5: 136293196-778 | 7 | 2900083l11Rik | 0.27 | 0.02 | 1.34 | 2.07 | ||
| MRI-117 | 16: 91182850-3440 | 7 | 0.54 | 0.03 | 1.12 | 1.58 | |||
| MRI-345 | 8: 125843980-5204 | 13 | Fanca | 0.27 | 0.04 | 1.34 | 1.87 | ||
| MRI-76 | 18: 37897160-644 | 6 | Pcdhga2 Pcdhga9 | 0.32 | 0.04 | 1.3 | 1.87 | ||
| MRI-55 | 12: 74651881-2661 | 6 | Tmem30b | 0.25 | 0.04 | 1.26 | 1.58 | ||
| MRI-307 | X: 136144506-998 | 6 | Rnf128 | 0.16 | 0.04 | 1.32 | 1.54 | ||
| MRI-246 | 7: 31743166-670 | 6 | Usf2 | Lsr | 0.8 | 0.04 | 1.07 | 1.86 | |
| MRI-208 | 3: 142224032-532 | 6 | Gbp3 | 0.85 | 0.04 | 0.98 | 1.38 | ||
| MRI-22 | 6: 18792144-836 | 8 | 0.25 | 0.05 | 1.34 | 1.76 | |||
Differentially methylated regions identified during murine secondary palate development, based on a linear mixed-effects model (see Methods). Reported p values are unadjusted for multiple comparisons. No comparisons were significant after controlling for the overall false-discovery rate. Results are listed separately for the MRIs identified for each GD.
p-Value unadjusted for multiple comparisons.
Fold-change based on average fold-change for all replicates and all probes identified in MRI.
Differential methylation was evaluated separately for MRIs identified on GDs 12, 13, and 14; e.g., GD 12 versus 14 lists MRIs identified on GD 12 that were differentially methylated relative to GD 14, while GD 14 versus 12 lists MRIs identified on GD 14 that were differentially methylated relative to GD 12.
Figure 4.



Pyrosequencing analysis of CpG residues localized within selected MRIs. Eight genes with MRIs localized in gene bodies, putative promoter or 5′ upstream regions (within 5kb of TSS; see text for definition) were selected for validation by pyrosequencing analysis. The most highly methylated segment of each MRI (black horizontal bar) was selected for pyrosequencing of CpG residues in a PyroMark Q24 (Qiagen) instrument. MRIs are in triplicates for each of the three GDs (GD12–GD14) and are depicted in the left panel. Methylation levels of CpG residues are indicated in the right panel. The MRI profiles and the bar graphs are color-coded for each GD. The choice and number of CpGs to be analyzed were determined by the PyroMark Design 2.0 software (Qiagen). Mean methylation levels for each CpG residue were deduced from at least three pyrosequencing runs using two independent PCR templates. Statistical significance was computed using a Student's t-test for changes in methylation levels relative to GD12 or GD13. Significant changes are indicated only for CpG residues that exhibit ≥5% change in methylation levels and p values of: *<0.05; **<0.01; ***<0.005. Significant changes relative to GD12 are depicted by asterisks without (or below) a bar, while significant changes relative to GD13 are indicated above a bar (see details in Supporting Information Table S10).
In light of the overall null findings for differential methylation, we added a post-hoc power analysis to characterize what differences in methylation between GDs we were powered to detect. Calculations determined the power to detect a given difference in log2 R/G intensity levels between two GDs, using a two-way repeated measures ANOVA with one between factor (GD) and one within factor (multiple probes constituting a methylated region, here set to six). Parameters were representative of the actual array data, including standard deviation of replicates of 0.52 and correlation between measurements at subsequent probes of 0.65. Power analysis was conducted using Power and Sample Size (PASS; Hintze, 2008). To account for multiple comparisons, an adjusted alpha level was determined by controlling the false discovery rate (FDR) at 0.05. Assuming a total number of 5500 tested methylated regions (the mean number of MRIs found on each GD), with 50 true DMRs and a true positive rate of 80%, then an approximate alpha which controls the FDR at 0.05 is (50 × 0.8 × 0.05)/(5450 × 0.95) ≈ 0.0004 (Lin et al., 2010). With our sample size of three per group, we had 11% power to detect a mean difference of log2 R/G values of two (four-fold increase) and 62% power to detect a mean difference of 4 (16-fold increase). With the given variability, correlation between probes and number of methylated regions evaluated, a sample size of five per group provides 82% power to detect a fourfold increase in methylation levels, while a sample size of 11 per group is needed for 83% power to detect twofold increases in methylation. The maximum fold-change increase in log2 R/G values we observed was approximately 4 (GD 12 MRI-381 and GD 13 MRI-345, see Table 1), though these were not validated using pyrosequencing (Fig. 4, gene Olfr66).
Methylation of Genes of Potential Interest in Secondary Palate Development
MRIs identified for each GD were subjected to DAVID analysis (Huang et al., 2007) to identify affected pathways in palate development. Full results from the DAVID analysis for MRIs identified in GD12–14 are given in Supporting Information Tables S4 to S6 for gene-body methylated genes and Supporting Information Tables S7 to S9 for promoter methylated genes. Some of the pathways, identified using KEGG (Kanehisa and Goto, 2000; Kanehisa et al., 2012) and PANTHER (Thomas et al., 2003), harbored genes that are methylated and relevant to secondary palate development (see Table 2). These include Wnt signaling, apoptosis, Notch signaling, calcium signaling, and cadherin signaling. Of these, the cadherin and Wnt signaling PANTHER pathways were significantly enriched (adjusted p value <0.05) for methylated genes. Particularly noteworthy is a preponderance of pathways involved in proteoglycan synthesis. These include categories associated with the biosynthesis of chondtroitin sulfate, heparan sulfate, galactose metabolism and glycosphingolipid (Table 2; see Discussion).
Table 2.
Pathways Enriched for Methylated Genes
| GD12 | GD13 | GD14 | ||||
|---|---|---|---|---|---|---|
|
|
|
|
||||
| N | Fold Enrichment | N | Fold Enrichment | N | Fold Enrichment | |
| Gene-body methylated pathways | ||||||
| Acute myeloid leukemia | 16 | 1.77 | 13 | 1.62 | 18 | 1.90 |
| Aldosterone-regulated sodium reabsorption | 13 | 1.95 | 13 | 2.20 | 15 | 2.15 |
| Alzheimer disease-amyloid secretase pathwaya | 23 | 1.57 | 19 | 1.43 | 22 | 1.47 |
| Alzheimer disease-presenilin pathwaya | 38 | 1.43 | 34 | 1.42 | 37 | 1.37 |
| Apoptosis | 22 | 1.59 | 18 | 1.47 | 23 | 1.59 |
| Arrhythmogenic right ventricular cardiomyopathy | 19 | 1.60 | 17 | 1.61 | 22 | 1.76 |
| Axon guidance | 31 | 1.49 | 28 | 1.52 | 33 | 1.52 |
| Basal cell carcinoma | 17 | 1.95 | 15 | 1.94 | 15 | 1.64 |
| Cadherin signaling pathwaya | 56 | 1.79c | 54 | 1.91c | 55 | 1.72c |
| Caffeine metabolism | 5 | 3.50 | 5 | 3.96 | 5 | 3.34 |
| Calcium signaling pathway | 45 | 1.49 | 44 | 1.64 | 45 | 1.42 |
| Chondroitin sulfate biosynthesis | 9 | 2.58 | 9 | 2.91 | 8 | 2.19 |
| Dilated cardiomyopathy | 25 | 1.71 | 23 | 1.78 | 27 | 1.77 |
| Endothelin signaling pathwaya | 28 | 1.45 | 24 | 1.38 | 27 | 1.37 |
| Extrinsic prothrombin activation pathwayb | 5 | 2.76 | 5 | 3.21 | 6 | 3.13 |
| Gap junction | 24 | 1.76 | 20 | 1.66 | 24 | 1.68 |
| Glioma | 18 | 1.77 | 18 | 2.00 | 23 | 2.16d |
| Glycosphingolipid biosynthesis | 11 | 2.89 | 8 | 2.37 | 10 | 2.51 |
| GnRH signaling pathway | 23 | 1.50 | 20 | 1.47 | 25 | 1.55 |
| Hedgehog signaling pathwaya | 13 | 1.66 | 13 | 1.84 | 13 | 1.63 |
| Heparan sulfate biosynthesis | 10 | 2.34 | 9 | 2.37 | 10 | 2.23 |
| Hypertrophic cardiomyopathy | 22 | 1.65 | 19 | 1.61 | 23 | 1.65 |
| Inositol phosphate metabolism | 14 | 1.63 | 14 | 1.85 | 17 | 1.89 |
| Insulin signaling pathway | 35 | 1.60 | 32 | 1.65 | 38 | 1.66 |
| Leloir pathway of galactose metabolismb | 3 | 6.07 | 3 | 7.05 | 3 | 5.74 |
| MAPK signaling pathway | 51 | 1.21 | 49 | 1.32 | 56 | 1.27 |
| Melanogenesis | 29 | 1.83 | 26 | 1.85 | 28 | 1.68 |
| Metabotropic glutamate receptor group 1 pathwaya | 15 | 1.92 | 13 | 1.84 | 13 | 1.63 |
| Neurotrophin signaling pathway | 33 | 1.60 | 31 | 1.70 | 40 | 1.85d |
| Spliceosome | 32 | 1.63 | 30 | 1.72 | 33 | 1.60 |
| Tight junction | 33 | 1.54 | 27 | 1.42 | 34 | 1.51 |
| Transcriptional activation of dbpb from mRNAb | 4 | 6.07 | 4 | 7.05 | 4 | 5.74 |
| Type II diabetes mellitus | 15 | 1.93 | 15 | 2.18 | 15 | 1.84 |
| Wnt signaling pathway | 34 | 1.44 | 32 | 1.53 | 34 | 1.37 |
| Wnt signaling pathwaya | 99 | 1.47c | 92 | 1.51c | 95 | 1.38d |
| Promoter methylated pathways | ||||||
| Ribosome | 12 | 6.45c | 9 | 5.69d | 13 | 6.55c |
| Systemic lupus erythematosus | 6 | 2.79 | 7 | 3.82 | 9 | 3.92 |
Pathways relevant to secondary palate development and enriched for methylated genes, as identified using gene-set enrichment analysis in DAVID (Huang et al., 2007). Fold enrichment was determined by the ratio of the number of methylated genes associated with term divided by the number of methylated genes to the total number of genes associated with term divided by the total number of mouse genes.
Panther pathway.
BioCarta pathway.
Adjusted p value <0.01;
adjusted p value <0.05.
Confirmation of Gene Methylation by Pyrosequencing
Pyrosequencing was undertaken on a stretch of CpGs localized within the most methylated region of the MRIs for eight genes (Fig. 4). Figure 4 presents the region of the MRI targeted for analysis (black bar, left panel) and the methylation levels of contiguous CpG residues within that region for all three GDs (right panel). Six of the eight methylated genes were selected from the aforementioned DAVID categories pertinent to palate development (Table 2)—Ctnnb1 (Cadherin and Wnt signaling), Bmp8b/Oxct2b (Hedgehog signaling), Dkk4 (Wnt signaling), Fat1 (Wnt signaling), Siah1b (Wnt signaling), and Wnt5a (Cadherin, Hedgehog and Wnt signaling). These genes were also selected for the location of MRIs in different gene compartments (e.g., gene-body, promoter or 5′ upstream regions). In addition, the MRI for Aph1a (Notch signaling) and Olfr66 was chosen for pyrose-quencing analysis; the latter was one of the few genes that exhibited marked differential methylation - poor methylation on GD14 compared with GD12 or GD13 (Fig. 4).
Pyrosequencing indicated that MRIs associated with the 8 genes were indeed methylated, to varying degrees, on the three GDs (Fig. 4): (1) Aph1a—the single CpG site analyzed displayed high (>93%), but no differential, methylation on all three GDs. (2) Bmp8b/Oxct2b—methylation levels varied from 41 to 69% for the five CpGs analyzed. Significant changes in methylation levels of specific CpG residues was observed on GD13 and GD14, relative to GD12. Furthermore, methylation levels for CpGs 1, 2, and 3 were also significantly different on GD14, compared with GD13 (asterisks above bar; Fig. 4). (3) Ctnnb1—the 3 CpGs analyzed exhibited high methylation levels (84–93%) but were not significantly differentially methylated on any of the three GDs. (4) Dkk4—all three CpGs tested exhibited high methylation (88–100%). Methylation level of one CpG residue (CpG3) exhibited a significant 8% decrease on GD14 relative to GD12 (p < 0.005) and a 6% decrease relative to GD13 (p < 0.05). (5) Fat1—all five CpG residues analyzed exhibited high methylation levels (89–100%) but none were significantly differentially methylated during palate development. (6) Siah1b—the 6 CpGs analyzed showed variable levels of methylation. CpG5 (9–11%) and CpG6 (13–16%) were poorly methylated on all three GDs; the remaining CpGs exhibited modest levels of methylation (∼20–33%). Overall, there was a progressive increase in methylation from GD12 to GD14 for all CpG residues. Significant increases in methylation levels were observed for CpGs 1, 3, and 4 on GD14, relative to GD12, and for CpG3, relative to GD13. (7) Wnt5a—the 8 CpGs analyzed showed variable levels of methylation, ranging from 35 to 73%. Six of the eight CpGs (CpGs3–8) exhibited significant changes in methylation levels on GD14 relative to GD12 (8–14% change; p < 0.005); in addition, CpGs 5, 6, and 8 also exhibited significant differential methylation (∼7% change) on GD13 compared with GD12. Three CpG residues (CpGs 3, 6, and 7) also had significantly higher methylation levels on GD14 compared with GD13. (8) Olfr66—the seven CpGs analyzed exhibited methylation levels ranging from 58 to 83%. Only three CpGs had significant changes in methylation levels—CpG4 (>5% change on GD14 relative to GD13), CpG5 (>5% on GD14 relative to both GD12 and GD13) and CpG7 (>7% on GD14 relative to GD12). While the MRI data for Olfr66 were consistent with pyrosequencing analysis on GD12 and GD13, the poor methylation observed on GD14 (left panel, Fig. 4) was not reflected by the pyrosequencing data which showed 56 to 77% methylation on this GD (right panel, Fig. 4). Methylation levels for individual CpG sites determined for each gene by pyrosequencing is presented in Supporting Information Table S10.
Determination of mRNA Expression of Selected Methylated Genes
The mRNA expression profiles of all 8 genes selected for pyrosequencing analysis were determined by qRT-PCR (Table 3). High expression (Ct: <25) was observed for Ctnnb1, Fat1, and Siah1b on all three GDs. Wnt5a was also considered to be highly expressed on all three GDs (its Ct value, 25.2, on GD13 being at the threshold for high expression). Bmp8b/Oxct2b and Aph1a were moderately expressed (Ct: 25–32) on all three GDs. For Dkk4 and Olfr66, we could not detect expression. Differential expression was observed in 5 out of 6 genes that were expressed (Bmp8b/Oxct2b, Ctnnb1, Fat1, Siah1b, and Wnt5a) and expressed as fold-change relative to GD12 (Table 3). Significant differential expression was based on the Dunnett's adjusted p values for multiple comparisons. Expression of Bmp8b/Oxct2b, Ctnnb1, and Wnt5a were up-regulated on GD14 in comparison with GD12; while Siah1b was down-regulated on GD14. Expression of Fat1 was up-regulated on GD13 in comparison with GD12; while Wnt5a was down-regulated on GD13.
Table 3.
mRNA Expression Levels of Selected Genes Methylated from GD12 to GD14 in Mouse Secondary Palate
| Gene | Primers | GD | Mean Ct Valuesa | ΔCtb,c | ΔΔCtc | Dunnett's p Valuec | Fold Change (2−ΔΔ Ct)c | 95% Confidence Limits |
|---|---|---|---|---|---|---|---|---|
| Aph1a | F: GTGTTTTTCGGATGCACCTT | 12 | 27.58 | 8.21±0.38 | – | – | 1.00 | |
| R: CCAAAAATCAGGAGGCCATA | 13 | 27.67 | 8.33±0.25 | 0.12 | 0.922 | 0.92 | (0.37–2.30) | |
| 14 | 27.90 | 8.84±0.38 | 0.63 | 0.254 | 0.65 | (0.26–1.61) | ||
| Bmp8b/Oxct2b | F: CTATGCAGGCCCTGGTACAT | 12 | 31.79 | 12.39±0.65 | – | – | 1.00 | |
| R: AGGCCTGGACTACCATGTTG | 13 | 30.91 | 11.47±0.70 | −0.92 | 0.346 | 1.89 | (0.38–9.53) | |
| 14 | 28.41 | 9.24±0.42 | −3.15 | 0.023d | 8.86 | (1.76–44.65) | ||
| Ctnnb1 | F: GTGCAATTCCTGAGCTGACA | 12 | 21.12 | 1.75±0.06 | – | – | 1.00 | |
| R: CTTTAAAGATGGCCAGCAAGC | 13 | 21.14 | 1.80±0.07 | 0.05 | 0.575 | 0.97 | (0.84–1.11) | |
| 14 | 20.51 | 1.44±0.01 | −0.31 | 0.015d | 1.24 | (1.08–1.42) | ||
| Dkk4 | Not determined | |||||||
| Fat1 | F: TCAGGAGACAGCGAAAACCT | 12 | 24.69 | 5.21±0.08 | – | – | 1.00 | |
| R: GCTTTCTCCACTGCCTTGAC | 13 | 24.28 | 4.75±0.06 | −0.47 | 0.020d | 1.38 | (1.10–1.74) | |
| 14 | 24.73 | 5.45±0.11 | 0.23 | 0.117 | 0.85 | (0.67–1.07) | ||
| Olfr66 | Not determined | |||||||
| Siah1b | F: AAGTGTCCACCATCCCAGAG | 12 | 24.02 | 4.55±0.05 | – | – | 1.00 | |
| R: ATGTAAGTTTGGGGCGACAG | 13 | 24.20 | 4.66±0.02 | 0.11 | 0.067 | 0.93 | (0.85–1.01) | |
| 14 | 23.97 | 4.68±0.02 | 0.13 | 0.040d | 0.91 | (0.84–0.99) | ||
| Wnt5a | F: AGGAGTTCGTGGACGCTAGA | 12 | 24.37 | 4.89±0.12 | – | – | 1.00 | |
| R: GCCGCGCTATCATACTTCTC | 13 | 25.21 | 5.74±0.06 | 0.85 | 0.003e | 0.56 | (0.45–0.69) | |
| 14 | 23.75 | 4.51±0.03 | −0.38 | 0.028d | 1.30 | (1.05–1.61) | ||
| Gapdh | F: GAGCATCTCCCTCACAATTTCCAT | Ct values normalized to control gene | ||||||
| R: GTGCAGCGAACTTTATTGATGGTAT | ||||||||
Expression based on mean Ct values from two sets of experiments, each done in triplicate: high (Ct: <25), moderate (Ct: 25–32), or low (Ct: >32).
Data presented represent mean ΔCt ± standard deviation. All data were normalized to the amplification signal from the housekeeping gene, GAPDH; i.e., the Ct value for each gene was subtracted from the corresponding GAPDH Ct value for each sample type.
Expressed relative to GD12. Fold-change values were determined according to the relationship: fold change =2−ΔΔCt, where Ct represents the threshold value for real-time PCR amplification detection; ΔCt = Ct, sample − Ct, GAPDH, and ΔΔCt the difference in ΔCt for the same probe: primer pair (e.g., Aph1a, Bmp8b, etc.) on GD13 and 14 samples versus GD12 samples
p <0.05;
p <0.005.
mRNA Expression of Gene-Body/Promoter Methylated Versus Nonmethylated Genes
Of the 500 genes that were promoter methylated on every GD, 167 were uniquely associated with methylation in the promoter. Twenty-nine of these genes mapped to probes on the Affymetrix Murine Genome U74Av2 GeneChip arrays. Amongst the 3030 genes found to be methylated in the gene-body on every GD, 2697 were uniquely methylated in the gene-body region. Of these, 1236 genes mapped to probes on the Affymetrix U74Av2 array. There were 333 genes methylated on both promoter and gene-body regions, of which only 86 genes were mapped to probes on the Affymetrix U74Av2 array. Furthermore, probes not linked with either of these methylation status groups were labeled as “non-methylated.” Figure 5 displays boxplots of log2 expression levels of probes associated with gene-body/promoter methylated versus nonmethylated genes, for each GD. Overall, expression for gene-body specific methylated genes was approximately 1.21 fold higher compared with nonmethylated genes (95% CI 1.17- to 1.24-fold, p < 0.001) and 1.51 fold higher compared with promoter methylated genes (95% CI 1.27- to 1.79-fold, p < 0.001). Whereas expression for promoter specific methylated genes was approximately 0.80-fold compared with non-methylated genes (95% CI 0.68- to 0.95-fold, p = 0.010). Expression for both gene-body/promoter methylated genes was approximately 0.86 fold compared with non-methylated genes (95% CI 0.78- to 0.95-fold, p = 0.002).
Figure 5.

Expression of gene-body versus promoter methylated genes. Boxplots of log2 expression levels for gene-body only methylated genes, promoter only methylated genes, genes that have both gene-body and promoter methylation and nonmethylated genes on all three gestational days. Data were obtained from previously published Affymetrix Murine Genome U74Av2 GeneChip arrays (Mukhopadhyay et al., 2004).
DISCUSSION
Epigenetics refers to inherited changes in phenotype or gene expression caused by mechanisms other than changes in the underlying DNA sequence. Alterations in DNA methylation represent one such epigenetic effect that can stably alter gene expression. To better understand how such events may function during normal and aberrant palate development, it is first necessary to establish the DNA methylation profile of a normal developing palate. Here, we provide an overview of DNA methylation changes taking place during development of the murine secondary palate. While numerous genetic mutations that cause orofacial clefts have been identified (reviewed in Cobourne, 2004; Murray and Schutte, 2004; Stanier and Moore, 2004; Lidral and Moreno, 2005; Rice, 2005; Carinci et al., 2007; Gritli-Linde, 2007; Juriloff and Harris, 2008; Vieira, 2008; Jugessur et al., 2009; Murthy and Bhaskar, 2009; Yu et al., 2009; Bush and Jiang, 2012), far less is known about the susceptibility of genes to adverse environmental exposures. A gene-environment link affecting palatal ontogeny (Murray and Schutte, 2004; Lidral and Moreno, 2005; Rice, 2005; Juriloff and Harris, 2008; Vieira, 2008; Murthy and Bhaskar, 2009; Greene and Pisano, 2010) may be mediated by epigenetic mechanisms involving DNA methylation, the actions of miRNAs, and/or histone modifications. To date, few studies have attempted to link alterations in these processes with the etiology of cleft palate (CP). Significant changes in gene expression profiles were seen when B-lymphoblasts from CLP patients were cultured in folate-deficient and folate supplemented media (Bliek et al., 2008). Since folate is an important mediator in the transfer of methyl groups for DNA methylation, this implied that methylation may play important roles in palate development. Indeed, changes in the methylation status of CpG islands, as well as in global DNA methylation were observed in the secondary palates of mice exposed to all-trans retinoic acid at doses that induced CP (Kuriyama et al., 2008). Only one study, thus far, has attempted to clarify the epigenetic role of DNA methylation in regulating secondary palate development (Plamondon et al., 2011). In this particular study, the Clf2 gene was found to affect the methylation and silencing of IAP retrotransposons in the Wnt9b gene, thereby altering susceptibility to CLP. In the current paper, we describe the CpG methylation profile of the developing murine secondary palate and identify specific genes that are methylated during palate development and the major cellular processes that are likely affected as a result. We validate our observations by pyrosequencing of CpG residues located within the MRIs of eight selected genes and correlate their methylation status and their localization in different genomic compartments (particularly, gene bodies and putative promoters) to their mRNA expression levels.
Genome-wide methylation analysis of all 21 mouse chromosomes on each of GDs 12–14 indicated that a predominant number of genes (4150 of 5577 genes or 74%) were methylated on all three gestational days with only a smaller fraction appearing to be gestational-day specific. On average, CpG islands accounted for only 30% of MRIs during the gestational period analyzed, while a considerable number (70%) were found in non-CpG island regions. Interestingly, it has been reported that DMRs in CpG islands account for only a small fraction of the total DMRs in tissue-specifically expressed genes, with the bulk of the methylation changes occurring at CpG sites outside CpG islands (Irizarry et al., 2009; Maunakea et al., 2010). Irizarry et al. (2009) have observed that such DMRs are more likely to be located at ‘CpG island shores’ rather than at ‘CpG islands’, the former referring to regions located at the outer periphery of CpG islands. More recent observations by Maunakea et al. (2010) indicate that only 2–3% of CpG islands manifest tissue-specific methylation.
To identify some of the key signaling systems/pathways affected by gene methylation during GD12-GD14, Gene Ontology (GO) analysis was performed using DAVID on each of the 3 gestational days (Table 2). A considerable overlap of pathways was observed for all three GDs, consistent with the observation that ∼74% of MRIs were shared between all the 3 GDs. Some of the major pathways affected by gene methylation and associated with palate development include: proteoglycan synthesis, Wnt signaling, cadherin signaling, calcium signaling, hedgehog signaling, and apoptosis. The biosynthesis of proteoglycans, critical for ECM formation, is particularly interesting because of the preponderance of hits for this category when analyzed by DAVID (Table 2). The synthesis of proteoglycans by palatal mesenchymal cells is considered essential for proper elevation of the palatal shelves (Mansell et al., 2000; Morris-Wiman et al., 2000; de Oliveira Demarchi et al., 2010). Moreover, proteoglycans have a major role in cell signaling with some binding TGF-β (Border et al., 1992), FGF and other growth factors (Ruoslahti and Yamaguchi, 1991). The critical roles of the TGF-β and FGF signaling systems to the proper development of the secondary palate have been well documented (Rahimov et al., 2012).
Wnt signaling is another major category affected by gene methylation. This category includes several Wnt genes (Wnt 3, 3a, 4, 5a, 7a, 7b and 8a). Several studies have implicated the canonical Wnt/β-catenin signaling in the regulation of normal palate development (Murray and Schutte, 2004; Gritli-Linde, 2007; Jugessur et al., 2009; Yu et al., 2009; Greene and Pisano, 2010). Epithelial Wnt/β-catenin signaling likely controls the expression of Tgfβ3 in the medial edge epithelium during palate fusion (He et al., 2011). Indeed, the specific ablation of Wnt signaling in the oral epithelium blocks rugae formation and induces CP (Lin et al., 2011). Several genes encoding cadherins are also found to be methylated during palate development. Cadherins which depend on Ca2+ ions for activity are involved in cell adhesion, a process necessary for tissue organization during palatogenesis. Another interesting category affected by gene methylation during palatogenesis is caffeine metabolism. Our analyses indicate that genes encoding specific members of the cytochrome P450 family, xanthine dehydrogenase, and N-acetyl transferase 3 are normally methylated on all three gestational days during secondary palate development (Table 2). Several studies have examined the teratogenic potential of caffeine exposure during pregnancy on oral clefting (Johansen et al., 2009). Although no strong evidences link normal caffeine use during pregnancy to oral clefts, excessive caffeine use has been demonstrated in vitro to prevent palatal fusion in the mouse (Kosazuma and Kawauchi, 1994). Thus, the effect of high dose caffeine on the methylation of these genes in the palate could be of potential interest.
A predominant number of gene methylation peaks were observed in gene bodies (73%) rather than in promoter regions or in regions further upstream (up to 5kb of TSS). The array probes which cover ∼10 kb of all annotated promoters and CpG islands presumably overlap gene bodies of adjacent genes. Interestingly, methylation of gene bodies has been associated with tissue-specificity and has been found to positively correlate with gene expression (Ball et al., 2009; Maunakea et al., 2010; Jones, 2012). It can, therefore, be assumed that the majority of methylated genes in the palate are likely expressed. To validate this hypothesis, we examined the mRNA expression of four genes that were gene-body methylated by qRT-PCR analysis (see below).
A total of eight genes were chosen for validation by pyrosequencing, seven of which (Aph1a, Bmp8b/Oxct2b, Ctnnb1, Dkk4, Fat1, Siah1b and Wnt5a) were associated with pathways/signaling systems considered relevant for secondary palate development. Three of these genes are also associated with CP. Ctnnb1 and Wnt5a play prominent roles in palate development. Both canonical (β-catenin dependent) and non-canonical Wnt signaling pathways play distinct but essential roles in palate development (He and Chen, 2012). β-catenin (encoded by Ctnnb1) is abundantly expressed in the rugae epithelium of the secondary palate (Lin et al., 2011), and elimination of β-catenin expression in the palate epithelium results in loss of Shh expression and CP due to ablation of Wnt signaling (Lin et al., 2011). Deficiency of Wnt5a, which is expressed along the AP (anterior-posterior) axis of the palate and which regulates directional cell migration, results in a complete cleft of the secondary palate (He et al., 2008). The role for Fat1 (a member of the Fat family of tumor suppressors) in secondary palate development is less clear but removal of a chromosomal region harboring this gene often results in a phenotype that consists of dysmorphic facial features, CP, limb deformities, heart defects and mental retardation (Bendavid et al., 2007).
The eight genes were methylated on all 3 GDs. The selected genes exhibited MRIs in different genomic compartments—the gene-body, the putative promoter or in the 5′ upstream region within 5 kb of the TSS. Pyrose-quencing, undertaken on selected CpGs located within the highly methylated regions of the MRIs, confirmed that all genes were methylated to varying degrees on all three GDs. The four gene-body methylated genes (Aph1a, Bmp8b/Oxct2b, Fat1, and Wnt5a) had moderate to high gene expression as evidenced by qRT-PCR analysis (Table 4). These observations are consistent with the widely emerging view that gene-body methylation is reflective of active transcription (Ball et al., 2009; Maunakea et al., 2010; Hahn et al., 2011). Indeed, the CpG residues constituting the MRIs of Aph1a and Fat1 were not only highly methylated (89–100%) but both genes were highly expressed in secondary palate tissues on all three gestational days. Siah1b, one of the few promoter methylated genes, was found to be highly expressed on all three gestational days. This seemingly contradictory observation can be explained when one examines the methylation levels of the CpG residues spanning the Siah1b MRI. These CpG residues were not highly methylated—of the six CpGs analyzed, two were essentially unmethylated (∼9–11%) while the remaining four were <35% methylated on all three GDs. Thus, the high expression of Siah1b could be attributed to the low level of promoter methylation of this gene. The MRIs for Dkk4 and Ctnnb1 did not occur in the immediate vicinity of the TSS, or in the gene-body, but were observed far upstream of the gene. Unlike gene-body and promoter methylation which are, respectively, positively and negatively correlated to gene expression, it is not clear how methylation of far upstream regions impact gene expression. Nevertheless, Ctnnb1 mRNA was found to be highly expressed in palatal tissues and the analyzed CpG residues spanning the cognate MRI were highly methylated (84–93%) suggesting that the MRI could span a putative repressive element/s. Alternatively, this MRI may be unrelated to Ctnnb1 expression. For Dkk4, CpGs spanning the MRI were also highly methylated (88–100%) on all three GDs but mRNA expression could not be detected in palatal tissues on any of the three GDs. Overall, pyrosequencing of CpG residues associated with the MRIs of the seven selected genes, considered relevant for palate development, were consistent with the cognate MRI and mRNA expression profiles on all three GDs.
Table 4.
Relation Between CpG Methylation and mRNA Expression for Selected Genes Methylated During Palate Development
| Gene | Location of MRI | CpG Methylation Levels (%)a | mRNA Expressionb |
|---|---|---|---|
| Aph1a | Gene-body | 93–94 | Moderate |
| Bmp8b/Oxct2b | Gene-body | 41–69 | Moderate |
| Ctnnb1 | Upstream | 84–93 | High |
| Dkk4 | Upstream | 88–100 | ND |
| Fat1 | Gene-body | 89–100 | High |
| Olfr66 | Promoter | 58–83 | ND |
| Siah1b | Promoter | 9–33 | High |
| Wnt5a | Gene-body | 35–73 | High/moderate |
Represents mean values ranging from the lowest methylated CpG site to the highest methylated CpG site for all three gestational days analyzed by pyrosequencing analysis (see Supporting Information Fig. S3).
Based on Ct values arbitrarily defined as low (>32), moderate (25–32) or high (<25); ND, not detectable.
In terms of expression, a significant increase in Bmp8b mRNA levels was observed on GD14 compared with levels on GD12 or GD13 and a significant drop in expression in Wnt5a on GD13 with respect to levels on either GD12 or GD14 (Table 3). Expression of Bmp8b/Oxct2b is particularly noteworthy. Oxct2b is encoded within intron 3 of Bmp8b and both genes are transcribed in the same orientation. Whereas the methylation data (i.e., MRI location and pyrosequencing) apply to both genes, mRNA analysis was specifically targeted toward Bmp8b. Bmp8b expression progressively increased during palate development: approximately twofold on GD13 and ∼9-fold (p < 0.005) on GD14 (relative to GD12). The significance of this increase during palate development remains to be established. The high expression of Bmp8b on all three GDs is consistent with it being gene-body methylated. However, the pronounced increase in expression on GD14 does not appear to be related to an equally pronounced change in GD14 methylation levels (based either on MRI profiles or CpG methylation data; Fig. 4), suggesting that other factors, besides methylation, may be involved in regulating Bmp8b expression. It is also possible that critical CpG residues involved in Bmp8b regulation were not targeted by MRI profiling or pyrosequencing. Wnt5a, however, exhibited a transient decrease in expression on GD13 (Table 3). Wnt5a plays a critical role in directional cell migration and cell proliferation along the A-P axis during palatogenesis (He et al., 2008). Indeed, He et al. (2008) have shown that a deficiency of Wnt5a causes CP while numerous studies have suggested a strong association between Wnt5a expression and non-syndromic CLP (Blanton et al., 2004; Chiquet et al., 2008).
After adjusting for multiple comparisons, our analysis did not reveal any genomic regions that were significantly differentially methylated. This result may reflect the biology of a relatively stable methylome during this period of development, or indicate that changes in methylation status are more subtle than our array study was powered to detect. Our post hoc power analysis revealed that our current study did not have high power to detect (declare as statistically significant) fourfold differences in array intensities (the maximum observed difference). Given our observed variability between replicates, a sample size of 5 arrays per group would be needed to reliably detect a fourfold difference in intensity, while a sample size of 11 arrays per group would be needed to detect a twofold difference. However, as with many array studies, our use of the NimbleGen arrays was primarily intended as a screening tool, and thus we reported regions found to be differentially methylated based on unadjusted p values to provide potential areas for further study (Table 1). Given that our maximum observed difference was four fold (which was ultimately not validated by pyrosequencing of Olfr66), it appears safe to declare that very few, if any, regions undergo drastic changes in methylation status (e.g., from complete methylation to unmethylation, or vice-versa) during the period of palatogenesis. Pyrosequencing of the eight selected genes, described above, however, identified, individual CpG residues that were significantly differentially methylated (>5% change; p < 0.05). For example, while no differential methylation of CpG residues was observed during palate development for Aph1a, Ctnnb1 and Fat1, the remaining five genes harbored specific CpG residues that exhibited differential methylation (Fig. 4). It should be emphasized that the observed differential methylation is based on the methylation state of representative CpGs, whose direct impact on mRNA expression remains to be proven (using more focused gene-specific experimental approaches such as promoter analysis, site-directed mutation analysis, etc.). The association between location of the methylated region and gene expression varied (Table 4), though globally there was ≈ 1.2-fold increase in gene expression for gene-body specific methylated genes versus non-methylated genes and ≈ 0.8-fold decrease in expression for promoter specific methylated genes versus non-methylated genes (Fig. 5).
In conclusion, this study has identified a number of methylated genes during palatogenesis (GD12-GD14) whose expression/nonexpression may affect the functioning of major signaling pathways. A majority of them are gene-body methylated, indicating that they are likely expressed in the secondary palate, participating in processes such as Wnt and cadherin signaling, and ECM formation. Future work should evaluate if aberrant methylation (i.e., demethylation) of these genes can contribute to CP and CL/P.
Supplementary Material
Acknowledgments
Grant sponsor: Supported in part by grant from NIH (DE018215, HD053509, DE021460) and NIH (P20 RR017702) from the COBRE program of the NIGMS.
References
- Adriaens ME, Jaillard M, Eijssen LM, et al. An evaluation of two-channel ChIP-on-chip and DNA methylation microarray normalization strategies. BMC Genomics. 2012;13:42. doi: 10.1186/1471-2164-13-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendavid C, Pasquier L, Watrin T, et al. Phenotypic variability of a 4q34->qter inherited deletion: MRKH syndrome in the daughter, cardiac defect and Fallopian tube cancer in the mother. Eur J Med Genet. 2007;50:66–72. doi: 10.1016/j.ejmg.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- Blanton SH, Bertin T, Serna ME, et al. Association of chromosomal regions 3p21.2, 10p13, and 16p13.3 with nonsyndromic cleft lip and palate. Am J Med Genet A. 2004;125:23–27. doi: 10.1002/ajmg.a.20426. [DOI] [PubMed] [Google Scholar]
- Bliek BJ, Steegers-Theunissen RP, Blok LJ, et al. Genome-wide pathway analysis of folate-responsive genes to unravel the pathogenesis of orofacial clefting in man. Birth Defects Res A Clin Mol Teratol. 2008;82:627–635. doi: 10.1002/bdra.20488. [DOI] [PubMed] [Google Scholar]
- Border WA, Noble NA, Yamamoto T, et al. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature. 1992;360:361–364. doi: 10.1038/360361a0. [DOI] [PubMed] [Google Scholar]
- Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231–343. doi: 10.1242/dev.067082. Erratum: 139: 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carinci F, Scapoli L, Palmieri A, et al. Human genetic factors in nonsyndromic cleft lip and palate: an update. Int J Pediatr Otorhinolaryngol. 2007;71:1509–1519. doi: 10.1016/j.ijporl.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Chiquet BT, Blanton SH, Burt A, et al. Variation in WNT genes is associated with non-syndromic cleft lip with or without cleft palate. Hum Mol Genet. 2008;17:2212–2218. doi: 10.1093/hmg/ddn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobourne MT. The complex genetics of cleft lip and palate. Eur J Orthod. 2004;26:7–16. doi: 10.1093/ejo/26.1.7. [DOI] [PubMed] [Google Scholar]
- de Oliveira Demarchi AC, Zambuzzi WF, Paiva KB, et al. Development of secondary palate requires strict regulation of ECM remodeling: sequential distribution of RECK, MMP-2, MMP-3, and MMP-9. Cell Tissue Res. 2010;340:61–69. doi: 10.1007/s00441-010-0931-6. [DOI] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, et al. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt B, Rabe-Hesketh S. Analyzing medical data using S-PLUS. New York, NY: Springer-Verlag; 2001. pp. 243–268. [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Gentleman R, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J, et al. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene RM, Pisano MM. Palate morphogenesis: current understanding and future directions. Birth Defects Res C Embryo Today. 2010;90:133–154. doi: 10.1002/bdrc.20180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A. Molecular control of secondary palate development. Dev Biol. 2007;301:309–326. doi: 10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- Hahn MA, Wu X, Li AX, et al. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS One. 2011;6:e18844. doi: 10.1371/journal.pone.0018844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Chen Y. Wnt signaling in lip and palate development. Front Oral Biol. 2012;16:81–90. doi: 10.1159/000337619. [DOI] [PubMed] [Google Scholar]
- He F, Xiong W, Yu X, et al. Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development. 2008;135:3871–3879. doi: 10.1242/dev.025767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Xiong W, Wang Y, et al. Epithelial Wnt/β-catenin signaling regulates palatal shelf fusion through regulation of Tgfβ3 expression. Dev Biol. 2011;350:511–519. doi: 10.1016/j.ydbio.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintze J. PASS. Kaysville, Utah: NCSS, LLC; 2008. [Accessed July 1, 2012]. Available at: www.ncss.com. [Google Scholar]
- Hoeffding W. A non-parametric test of independence. Ann Math Stat. 1948;19:546–557. [Google Scholar]
- Huang DW, Sherman BT, Tan Q, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen AM, Wilcox AJ, Lie RT, et al. Maternal consumption of coffee and caffeine-containing beverages and oral clefts: a population-based case-control study in Norway. Am J Epidemiol. 2009;169:1216–1222. doi: 10.1093/aje/kwp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Jugessur A, Farlie PG, Kilpatrick N. The genetics of isolated orofacial clefts: from genotypes to subphenotypes. Oral Dis. 2009;15:437–453. doi: 10.1111/j.1601-0825.2009.01577.x. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ. Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res A Clin Mol Teratol. 2008;82:63–77. doi: 10.1002/bdra.20430. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, et al. KEGG for integration and interpretation of large-scale molecular datasets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics—a bio-conductor package for quality assessment of microarray data. Bioinformatics. 2009;25:415–416. doi: 10.1093/bioinformatics/btn647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosazuma T, Kawauchi S. Effects of caffeine and related methyl-xanthines on fetal mouse palates cultured in vitro. J Toxicol Sci. 1994;19:175–180. doi: 10.2131/jts.19.4_175. Erratum in: 20:73. [DOI] [PubMed] [Google Scholar]
- Kuriyama M, Udagawa A, Yoshimoto S, et al. DNA methylation changes during cleft palate formation induced by retinoic acid in mice. Cleft Palate Craniofac J. 2008;45:545–551. doi: 10.1597/07-134.1. [DOI] [PubMed] [Google Scholar]
- Lidral AC, Moreno LM. Progress toward discerning the genetics of cleft lip. Curr Opin Pediatr. 2005;17:731–739. doi: 10.1097/01.mop.0000185138.65820.7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WJ, Hsueh HM, Chen JJ. Power and sample size estimation in microarray studies. BMC Bioinformatics. 2010;11:48. doi: 10.1186/1471-2105-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Fisher AV, Yin Y, et al. The inductive role of Wnt-β-Catenin signaling in the formation of oral apparatus. Dev Biol. 2011;356:40–50. doi: 10.1016/j.ydbio.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–428. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mansell JP, Kerrigan J, McGill J, et al. Temporal changes in collagen composition and metabolism during rodent palatogenesis. Mech Ageing Dev. 2000;119:49–62. doi: 10.1016/s0047-6374(00)00168-8. [DOI] [PubMed] [Google Scholar]
- Marazita ML. Genetic etiologies of facial clefting. In: Mooney MP, Siegel MI, editors. Understanding craniofacial anomalies: the etiopathogenesis of craniosynostoses and facial clefting. New York: Wiley-Liss; 2002. pp. 147–161. [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris-Wiman J, Burch H, Basco E. Temporospatial distribution of matrix metalloproteinase and tissue inhibitors of matrix metalloproteinases during murine secondary palate morphogenesis. Anat Embryol. 2000;202:129–141. doi: 10.1007/s004290000098. [DOI] [PubMed] [Google Scholar]
- Mossey PA, Little J, Munger RG, et al. Cleft lip and palate. Lancet. 2009;374:1773–1785. doi: 10.1016/S0140-6736(09)60695-4. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Greene RM, Zacharias W, et al. Developmental gene expression profiling of mammalian, fetal orofacial tissue. Birth Defects Res A Clin Mol Teratol. 2004;70:912–926. doi: 10.1002/bdra.20095. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Greene RM, Pisano MM. Expression profiling of TGFß superfamily genes in developing orofacial tissue. Birth Defects Res A Clin Mol Teratol. 2006;76:528–543. doi: 10.1002/bdra.20276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Brock G, Pihur V, et al. Developmental micro-RNA expression profiling of murine embryonic orofacial tissue. Birth Defects Res A Clin Mol Teratol. 2010;88:511–534. doi: 10.1002/bdra.20684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JC. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002;61:248–256. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- Murray JC, Schutte BC. Cleft palate: players, pathways, and pursuits. J Clin Invest. 2004;113:1676–1678. doi: 10.1172/JCI22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy J, Bhaskar L. Current concepts in genetics of nonsyndromic clefts. Indian J Plast Surg. 2009;42:68–81. doi: 10.4103/0970-0358.53004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro J, Bates D, DebRoy S, et al. nlme: Linear and nonlinear mixed effects models. R package version 3.1–102. 2011 http://cran.r-pro-ject.org/web/packages/nlme/index.html.
- Plamondon JA, Harris MJ, Mager DL, et al. The clf2 gene has an epigenetic role in the multifactorial etiology of cleft lip and palate in the A/WySn mouse strain. Birth Defects Res A Clin Mol Teratol. 2011;91:716–727. doi: 10.1002/bdra.20788. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Gilbert HN, Ge Y, et al. multtest: Resampling-based multiple hypothesis testing R package version 2.10.0. 2011 http://www.bioconductor.org/packages/release/bioc/html/multtest.html.
- R Development Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2012. [Last accessed 7/1/12]. Available at: http://www.R-project.org/ [Google Scholar]
- Rahimov F, Jugessur A, Murray JC. Genetics of nonsyndromic orofacial clefts. Cleft Palate-Craniofac J. 2012;49:73–91. doi: 10.1597/10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DP. Craniofacial anomalies: from development to molecular pathogenesis. Curr Mol Med. 2005;5:699–722. doi: 10.2174/156652405774641043. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Yamaguchi Y. Proteoglycans as modulators of growth factor activities. Cell. 1991;64:867–869. doi: 10.1016/0092-8674(91)90308-l. [DOI] [PubMed] [Google Scholar]
- Stanier P, Moore GE. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet. 2004;13:R73–R81. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toedling J, Huber W. Analyzing ChIP-chip data using bioconductor. PLoS Comput Biol. 2008;4:e1000227. doi: 10.1371/journal.pcbi.1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toedling J, Sklyar O, Krueger T, et al. Ringo—an R/Bioconductor package for analyzing ChIP-chip readouts. BMC Bioinformatics. 2007;8:221. doi: 10.1186/1471-2105-8-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira AR. Unraveling human cleft lip and palate research. J Dent Res. 2008;87:119–125. doi: 10.1177/154405910808700202. [DOI] [PubMed] [Google Scholar]
- Yazdy MM, Honein MA, Rasmussen SA, et al. Priorities for future public health research in orofacial clefts. Cleft Palate Craniofac J. 2007;44:351–357. doi: 10.1597/06-233.1. [DOI] [PubMed] [Google Scholar]
- Yu W, Serrano M, Miguel SS, et al. Cleft lip and palate genetics and application in early embryological development. Indian J Plast Surg. 2009;42(Suppl):S35–S50. doi: 10.4103/0970-0358.57185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Kartiko S, Finnell RH. Importance of gene-environment interactions in the etiology of selected birth defects. Clin Genet. 2009;75:409–423. doi: 10.1111/j.1399-0004.2009.01174.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.