

Abstract
Objective:
To investigate default mode network (DMN) functional connectivity MRI (fcMRI) in a large cross-sectional cohort of subjects from families harboring pathogenic presenilin-1 (PSEN1), presenilin-2 (PSEN2), and amyloid precursor protein (APP) mutations participating in the Dominantly Inherited Alzheimer Network.
Methods:
Eighty-three mutation carriers and 37 asymptomatic noncarriers from the same families underwent fMRI during resting state at 8 centers in the United States, United Kingdom, and Australia. Using group-independent component analysis, fcMRI was compared using mutation status and Clinical Dementia Rating to stratify groups, and related to each participant's estimated years from expected symptom onset (eYO).
Results:
We observed significantly decreased DMN fcMRI in mutation carriers with increasing Clinical Dementia Rating, most evident in the precuneus/posterior cingulate and parietal cortices (p < 0.001). Comparison of asymptomatic mutation carriers with noncarriers demonstrated decreased fcMRI in the precuneus/posterior cingulate (p = 0.014) and right parietal cortex (p = 0.0016). We observed a significant interaction between mutation carrier status and eYO, with decreases in DMN fcMRI observed as mutation carriers approached and surpassed their eYO.
Conclusion:
Functional disruption of the DMN occurs early in the course of autosomal dominant Alzheimer disease, beginning before clinically evident symptoms, and worsening with increased impairment. These findings suggest that DMN fcMRI may prove useful as a biomarker across a wide spectrum of disease, and support the feasibility of DMN fcMRI as a secondary endpoint in upcoming multicenter clinical trials in Alzheimer disease.
Alzheimer disease (AD) is a neurodegenerative disorder characterized by progressive synaptic failure.1–4 Recent advances in functional neuroimaging techniques allow for the indirect assessment of polysynaptic connections in the human brain. Analyses of coordinated, spontaneous, blood oxygen level–dependent signal fluctuations during task-independent fMRI (termed resting-state functional connectivity MRI [fcMRI]) have demonstrated the presence of ubiquitous large-scale neural networks.5–7 Of particular relevance to AD is a set of cortical regions collectively known as the default mode network (DMN).4,8–11
Intact functional connectivity within the DMN at rest, as well as the ability to modulate DMN activity during memory encoding and retrieval tasks, is thought to be critical for successful memory function.12,13 The DMN includes the posterior cingulate, lateral parietal, and medial frontal cortices. The neocortical regions of the DMN are preferential (but not exclusive) sites for amyloid-β (Aβ) deposition in early AD.14 Studies in sporadic late-onset AD (LOAD) have demonstrated decreases in DMN fcMRI.2,4 Similar changes are also seen in prodromal and preclinical AD, as evidenced by decreased DMN fcMRI in subjects with mild cognitive impairment (MCI) who progress to AD,11 asymptomatic APOE ε4 carriers,15–17 and Aβ biomarker–positive older individuals.18–20
Together, these findings have prompted the development of DMN connectivity as a noninvasive biomarker for detection of early synaptic dysfunction in AD and for tracking potential therapeutic response in clinical trials. Despite being well studied in the symptomatic stages of LOAD,2,4 alterations in DMN fcMRI have been largely unstudied in cases of familial autosomal dominant AD (ADAD).
In the present report, we examine changes in DMN fcMRI across the continuum of impairment in a large cohort of subjects drawn from 8 international sites within the Dominantly Inherited Alzheimer Network (DIAN).21,22 This cohort of subjects presents a rare opportunity to examine fcMRI across the spectrum of AD in a young group of subjects carrying highly penetrant ADAD mutations. Taking advantage of the relatively conserved age of dementia onset within each family,23–25 we also model changes in DMN connectivity regarding each subject's estimated years from expected symptom onset (eYO) in their families to generate a temporal pattern for altered DMN fcMRI in ADAD.
METHODS
Participants, standard protocol approvals, registrations, and participant consents.
Persons with first-degree relatives known to carry ADAD mutations in presenilin-1 (PSEN1), presenilin-2 (PSEN2), or amyloid precursor protein (APP) were recruited into the DIAN study (www.dian-info.org; NIA-U19-AG032438, Clinical Trial Identifier NCT00869817) irrespective of ADAD mutation status. All subjects provided informed consent in accordance with the local institutional review boards of each participating site. A detailed protocol for the DIAN study has previously been published, and is available in the supplementary data of reference 22.
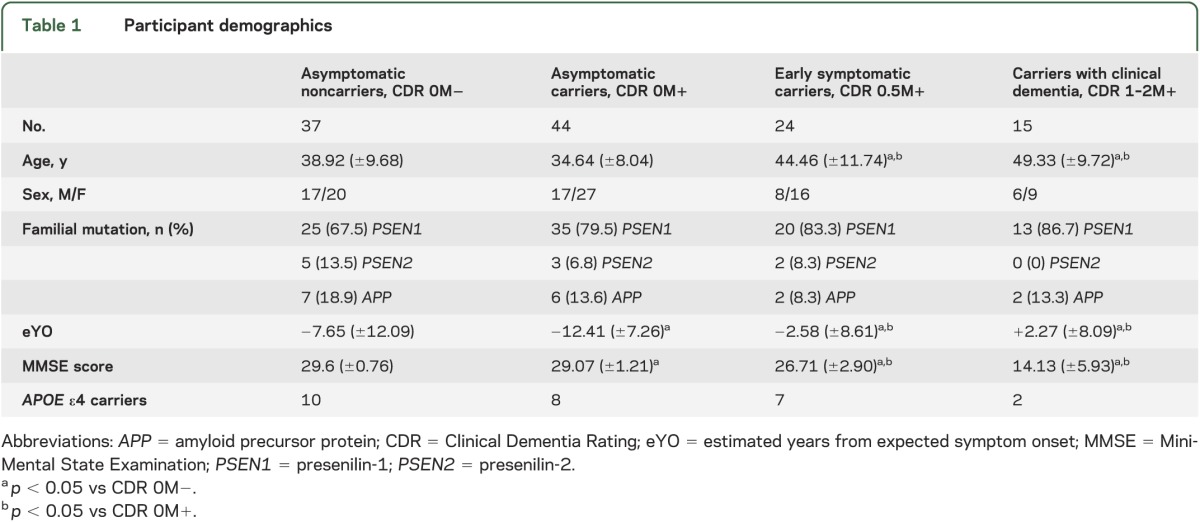
Cross-sectional clinical and imaging data from 120 people from 61 families, including 83 carriers of ADAD mutations (PSEN1: 68; PSEN2: 5; APP: 10) collected at 8 DIAN sites were analyzed. Comparisons were made to a control group of 37 asymptomatic, mutation-negative subjects from the same families. Four mutation-negative individuals with Clinical Dementia Rating (CDR) ≥0.5 were not included because of their low number and ambiguity regarding their underlying diagnosis. The remaining 120 subjects were classified into 4 groups on the basis of mutation and CDR status (table 1): asymptomatic mutation-negative participants (CDR 0M−), asymptomatic mutation carriers (CDR 0M+), early symptomatic mutation carriers (CDR 0.5M+), and mutation carriers with clinical dementia (CDR 1–2M+). Only baseline data available in the DIAN database as of February 2012 were included in this study.
Table 1.
Participant demographics
Clinical evaluation.
Participants underwent an extensive evaluation, including physical examination, cognitive testing, and CDR, in which a score of 0 indicates no dementia, 0.5 indicates questionable or very early dementia, and 1, 2, and 3 correspond to mild, moderate, and severe dementia, respectively. DIAN investigators were blinded to participant mutation status in asymptomatic individuals and received no confirmation of genetic status in symptomatic individuals. Outside of medical necessity, no research data (including genetic status) were provided to participants as part of the DIAN study. Participants wishing to know their mutation status were offered free-of-charge genetic counseling, separate from the conduct of the study.
Using semistructured interviews with cognitively intact family members, each participant's eYO was computed as the subject's age minus the age at which the subject's parent or sibling first showed symptoms of progressive cognitive decline22 (familial age at onset). In cases in which multiple first-degree relatives were available to calculate familial age at onset, an average value was used. Accordingly, negative eYO values indicate participants younger than the age at which his/her parent or sibling(s) developed the first signs of progressive cognitive decline.
Genetic analysis.
DNA was extracted from blood samples sent to the DIAN Genetics Core (DGC) at Washington University and the National Cell Repository for Alzheimer's Disease (NCRAD). For quality control purposes, sequencing for DIAN mutations and APOE genotyping (using rs7412 and rs429358 as markers) and DNA fingerprinting was performed by DGC personnel in parallel on DNA samples extracted by DGC and NCRAD. Only subjects with concordant data for DGC and NCRAD extractions were included in the analysis.
Imaging methods.
Data acquisition.
Participants underwent eyes-open resting-state fMRI using a 12-channel phased-array head coil. Earplugs and noise-reduction headphones were used to attenuate scanner noise, and head motion was minimized with foam padding. Only data from Siemens Trio TIM 3T (109 sessions) or Verio 3T (11 sessions) scanners (Siemens Medical Solutions, Erlangen, Germany) were used in this report. Trio TIM Data were acquired using a gradient-echo echo-planar pulse sequence sensitive to blood oxygen level–dependent contrast using the following parameters: repetition time = 2,200 milliseconds; echo time = 30 milliseconds; fractional anisotropy = 80°; 64 × 64 matrix; 3.125 × 3.125 mm in-plane resolution with 3.3 mm slice thickness, with equivalent parameters used on the Verio. Thirty-six interleaved axial slices covered a field of view of 119 mm. Images were acquired in a single run of 120 time points, lasting approximately 5 minutes.
Preprocessing.
Using SPM8 (http://www.fil.ion.ucl.ac.uk/spm/; version r4290) each run was slice-time corrected, realigned to the first volume of each run with INRIAlign,26,27 normalized to the Montreal Neurological Institute (MNI) ICMB152 EPI template, and smoothed with a 6-mm full width at half maximum Gaussian kernel. Realignment parameters were used to calculate the mean movement across the resting-state MRI, which was then used to screen out sessions with excessive movement using a threshold set at 0.15 mm/repetition time.28 This resulted in the exclusion of 14 subjects (8 CDR 0M−, 4 CDR 0M+, 1 CDR 0.5M+, and 1 CDR 1–2M+), leaving a total of 120 subjects.
Functional connectivity analysis.
Group spatiotemporal independent component analysis (ICA) was performed using the GIFT toolbox (http://mialab.mrn.org/software/gift/;v1.3h) for MATLAB. The SPM gray matter template with a probability threshold of 0.30 was used to mask out extra gray matter voxels. First-pass principal component analysis using 40 components per session was followed by use of the Infomax algorithm to generate 20 independent components. The GICA3 algorithm29 was used to back-construct spatial component maps and time courses for each subject. A goodness-of-fit approach4 was used to quantitatively identify the component that most accurately matched a DMN template created from an independent set of young subjects using 10-mm spherical precuneus/posterior cingulate (PPC) (MNI coordinates: 0, −53, 26) seed-based maps. Similar fcMRI analysis of the motor network was performed to determine whether fcMRI was universally altered across networks in mutation carriers.
Statistical analyses.
Given the lack of a sufficient group of individuals who were CDR >0M− to assess for a classic interaction between CDR and mutation status, we ran 3 separate general linear models (GLMs). The first GLM was a simple 1-way analysis of variance (ANOVA) across the 4 groups described above (CDR 0M−; CDR 0M+; CDR 0.5M+; CDR >0.5M+). The second GLM was an analysis of covariance modeled as eYO × mutation, which allowed for the assessment of an interaction between the eYO variable and the presence of an ADAD mutation. As a final check, we performed this second analysis while controlling for CDR to evaluate the additional explanatory contribution of the model beyond what could be explained by CDR status alone.
Importantly, we also investigated the influence of family membership, performance site, APOE, and nonlinear terms for eYO, as well as the appropriateness of the eYO construct relative to the inclusion of both age and familial age at onset as separate regressors (see supplemental data on the Neurology® Web site at www.neurology.org). We found no contribution of family membership (modeled as a random effect), APOE, sex, and nonlinear eYO terms to the models. Additionally, we visualized the relationship between eYO and PPC fcMRI using local regression (LOESS) to facilitate comparison with earlier reports from DIAN (figure e-1).
Lastly, we evaluated whether performance site was a substantial contributor to the observed findings (see e-Methods, table e-1). Given the similarity of results when site was included or excluded in the model, we did not include site as a covariate in the primary analyses (in keeping with prior reports from DIAN in which site was not used as a covariate22). Backward elimination was used to evaluate the utility of additional model terms and resulted in the following final model: connectivity = eYO + mutation + (eYO × mutation), which was used for the analyses presented below.
RESULTS
Demographics.
As expected, asymptomatic mutation carriers were significantly younger on average than symptomatic mutation carriers and further from their expected age of symptom onset (see table 1). In our cohort, asymptomatic non–mutation carriers were modestly but significantly older than asymptomatic mutation carriers (p = 0.033). No significant differences in APOE ε4 carrier status were observed across groups. Modest but statistically significant differences in Mini-Mental State Examination scores between CDR 0M− and CDR 0M+ groups were observed.
ICA analysis of task-free fMRI data.
ICA analysis including all subjects who were M+ and M− revealed a single, easily identifiable DMN, including the PPC cortices, medial prefrontal cortex (mPFC), lateral parietal regions (left lateral parietal cortex [LPC] and right lateral parietal cortex [RPC]), lateral temporal regions, and bilateral medial temporal regions (medial temporal lobe; figure 1). Although not examined in detail, several previously described neural networks appeared in our ICA analysis. Among these, the motor network was analyzed to determine whether general changes in fcMRI in mutation carriers were present. No significant changes in motor network fcMRI were seen at p < 0.005 (not shown), arguing against universally decreased fcMRI in mutation carriers.
Figure 1. DMN component derived from group ICA.

The DMN was readily identified in the study population in one component of the group ICA. Dorsal (A) and ventral (B) portions of this component are shown above. DMN = default mode network; ICA = independent component analysis.
ANOVA comparing DMN fcMRI across groups.
One-way ANOVA across groups defined by mutation and clinical status demonstrated decreased functional connectivity within much of the DMN in ADAD mutation carriers compared with noncarriers (figure 2, A–E, and figure 3A). Decreased connectivity was most apparent within the major posterior node of the DMN (PPC; F3,116 = 14.72, p < 0.001; figure 2, A and B), along with significantly decreased connectivity in the anterior node of the DMN (mPFC; F3,116 = 10.10, p < 0.001; figure 2, A and C). Significant differences were also observed in the lateral parietal cortices (RPC: F3,116 = 10.09, p < 0.001; LPC: F3,116 = 11.92, p < 0.001) as well as numerous other peaks within the DMN (table e-2).
Figure 2. Whole-brain analysis of variance.

(A) Whole-brain map depicting regions of significantly altered DMN fcMRI across groups (p < 0.001 threshold). Comparison across groups using a 10-mm sphere region of interest in the PPC (B), mPFC (C), LPC (D), and RPC (E). For post hoc analyses, error bars are SEM. a p < 0.05 compared with CDR 0M−; b p < 0.001 compared with CDR 0M−; c p < 0.001 compared with CDR 0M+; d p < 0.005 compared with CDR 0M−; e p < 0.05 compared with CDR 0M+. CDR = Clinical Dementia Rating; DMN = default mode network; fcMRI = functional connectivity MRI; LPC = left lateral parietal cortex; mPFC = medial prefrontal cortex; PPC = precuneus/posterior cingulate; RPC = right lateral parietal cortex.
Figure 3. Decreasing DMN connectivity as subjects approach and surpass their expected age of symptom onset.

(A) Illustration of the DMN component (a; red); regions within the DMN that show negative correlation with eYO (p < 0.001; b; blue); overlap between the left and middle panels with common regions in green (c). Decreased connectivity within the DMN as measured in a 10-mm sphere based in the PPC (B), mPFC (C), LPC (D), and RPC (E). Solid lines denote linear correlation (with M− in blue and M+ in red) and curved dotted boundaries represent 95% confidence intervals. DMN = default mode network; eYO = estimated years from expected symptom onset; fcMRI = functional connectivity MRI; LPC = left lateral parietal cortex; mPFC = medial prefrontal cortex; PPC = precuneus/posterior cingulate; RPC = right lateral parietal cortex.
Post hoc comparison of CDR 0M− with CDR 0.5M+ and CDR 1–2M+ demonstrated that DMN functional connectivity was decreased in the PPC, mPFC, LPC, and RPC in symptomatic mutation carriers as compared with asymptomatic non–mutation carriers (figure 2, A–E). Post hoc comparison of asymptomatic mutation carriers with noncarriers (i.e., CDR 0M+ with CDR 0M− groups) yielded significantly decreased functional connectivity in the PPC (t79 = 2.51; p = 0.014; figure 2, A and B) and RPC (t79 = 3.28; p = 0.0016; figure 2, A and E), but these effects were relatively modest in size.
Post hoc comparisons demonstrated decreases in functional connectivity with advancing CDR in the PPC (CDR 0M+ vs CDR 0.5M+, t66 = 3.88; p < 0.001; CDR 0M+ vs CDR 1–2M+, t57 = 4.04; p < 0.001), mPFC (CDR 0M+ vs CDR 0.5M+, t66 = 3.94; p < 0.001; CDR 0M+ vs CDR 1–2M+, t57 = 4.49; p < 0.001), and RPC (CDR 0M+ vs CDR 0.5M+, t66 = 1.06; p = 0.295; CDR 0M+ vs CDR 1–2M+, t57 = 2.47; p = 0.016) nodes of the DMN. A more complex pattern was seen in the LPC, where asymptomatic mutation carriers showed very similar levels of connectivity compared with asymptomatic noncarriers (CDR 0M+ vs CDR 0M−, t79 = 0.05; p = 0.96), but decreases in connectivity were seen at higher CDRs (CDR 0M− vs CDR 0.5M+, t66 = 4.50; p < 0.001; CDR 0M− vs CDR 1–2M+, t57 = 3.80; p < 0.001).
Decreased connectivity as a function of proximity to familial age at symptom onset.
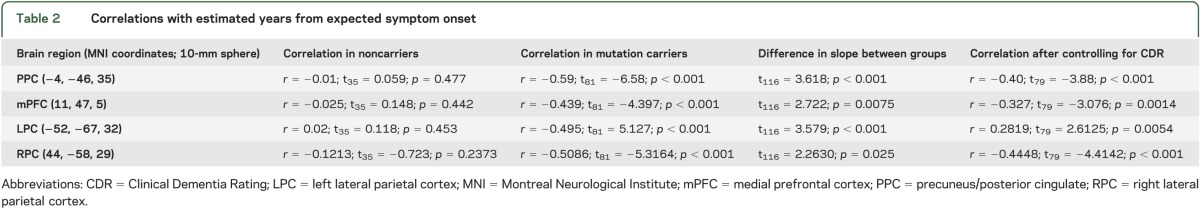
We next focused on examining the rate and pattern of degraded connectivity within the DMN by plotting changing DMN fcMRI against eYO. The strongest effect was seen in the PPC where we observed a significant interaction between mutation carrier status and eYO (t116 = 3.86, p = 0.017), with a significant negative correlation between fcMRI and eYO in mutation carriers (r = −0.59, t81 = −6.64; p < 0.001), but no significant relationship observed in noncarriers (r = −0.11, t35 = 0.06; p = 0.95; figure 3 and table 2). The relationship between eYO and fcMRI in mutation carriers remained significant after controlling for CDR status (PPC: r = −0.412, t79 = −2.39; p = 0.010). Similar patterns were observed in the mPFC, LPC, and RPC (figure 3, table 2).
Table 2.
Correlations with estimated years from expected symptom onset
DISCUSSION
In the present study, we observed alterations in DMN connectivity in both symptomatic and asymptomatic carriers of pathogenic mutations in PSEN1, PSEN2, and APP. Connectivity within the DMN is decreased in symptomatic carriers of ADAD mutations and this decrease is greater in magnitude in more advanced disease. DMN fcMRI differs in asymptomatic mutation carriers as compared with noncarriers, with subtle decreases in DMN connectivity observable before symptom onset. A negatively sloped, mutation status–dependent decrease in DMN fcMRI is observed as mutation carriers approach and surpass their expected age of symptom onset, even when controlling for CDR. Although further study is needed to assess the test-retest reliability of fcMRI and to compare its performance with other biomarkers in early AD, these results suggest that DMN fcMRI may prove useful as a noninvasive biomarker for tracking disease progression in the preclinical30,31 and early clinical phases of ADAD. Additionally, these data demonstrate the feasibility of performing multicenter clinical trials using resting-state fcMRI, and support the inclusion of fcMRI as an exploratory outcome measure in upcoming AD prevention trials.21,31
Our study benefits from the unique strengths of the large DIAN cohort. Specifically, the relative youth of these subjects (as compared with the older populations studied in LOAD) allowed us to decrease the influence of “normal” age-related decreases in DMN functional connectivity,3,32 allowing for the clearer interpretation of decreasing DMN connectivity related to progression of AD rather than age. This is especially true as mutation noncarriers in our cohort were members of the same families as mutation carriers, lessening the possibility that polygenic variation outside of PSEN1, PSEN2, and APP are contributing to decreased DMN connectivity. However, the present study is limited in its ability to assess mutation-specific effects, given that our results are largely based on individuals carrying PSEN1 mutations. Notably, the observation of decreased fcMRI before symptom onset is consistent with CSF-based biochemical markers in both ADAD22,33 and in LOAD,34 where both CSF Aβ and tau abnormalities are manifest years before the onset of clinical symptoms. While the present study focuses on an initial description of fcMRI in ADAD and its potential use as a biomarker in clinical trials, additional studies are under way to examine connectivity changes in other networks and subnetworks, to place changes in fcMRI in temporal relation with respect to other biomarkers (especially CSF tau, Aβ, and PET amyloid imaging) and to compare the variability in fcMRI measures with that seen with other biomarker measures in this unique cohort. Finally, follow-up reports on longitudinal changes in fcMRI in this cohort are needed to more robustly evaluate the nature of DMN dysfunction with disease progression.
Modeling of DMN connectivity as a function of eYO benefits from the extremely high penetrance of ADAD mutations, despite the approximate nature of the eYO measure. The relative certainty that mutation carriers (regardless of cognitive status at the time of enrollment) will develop AD contrasts with similar studies in LOAD, where progression to AD dementia in MCI populations is quite variable35 and it is possible that subjects with MCI will develop other types of dementia. In this context, the high penetrance of ADAD mutations allows us to more confidently model changes in connectivity across the entire disease spectrum in a manner that is considerably more difficult in nonlongitudinal studies of LOAD, given the uncertainty surrounding when and if sporadic subjects with MCI will develop AD.
The early decreases in DMN fcMRI in ADAD generally accord well with prior fcMRI studies in LOAD,2,4 in asymptomatic older individuals with biomarker evidence of Aβ deposition,19,20,36 and in asymptomatic APOE ε4 carriers.15,16,18 Additionally, the presence of observable changes in DMN fcMRI before symptom onset fits well with recent evidence that young, asymptomatic ADAD mutation carriers show changes in memory-task fMRI years before their estimated age of symptom onset.37 Although we did not directly compare ADAD with LOAD in our study, a subsequent analysis with the objective of comparing network disruption in ADAD and LOAD is presently in preparation by our colleagues at Washington University.38 That analysis includes fcMRI data from a subset of the DIAN subjects presented in this report, and uses a different analytic method to generate “seed-based” fcMRI network composite scores to compare ADAD subjects with a large LOAD cohort from Washington University. In contrast, we used ICA to identify whole-brain network maps of the DMN followed by region-of-interest analyses. By using this regional analytic approach, we were able to detect subtle regionally specific alterations in the DMN in presymptomatic mutation carriers. The ability to identify regionally specific changes in the DMN that occur early in the course of AD may be an important consideration for interpretation of data from upcoming secondary prevention clinical trials that plan to use DMN fcMRI as a secondary endpoint.
Lastly, our findings of decreased connectivity within specific regions of the DMN associated with increasing proximity to eYO and advancing CDR raise the intriguing question of how selective the pathobiologic process of AD is for the DMN, especially early in the course of the disease. Recent studies have elegantly demonstrated that other neurodegenerative diseases have distinctive patterns with which they affect distributed neural networks.39 The present findings of regional changes in DMN fcMRI early in ADAD, coupled with studies demonstrating that DMN is an early site for amyloid deposition14,36 and metabolic dysregulation in LOAD,14,40 suggest that specific regions within the DMN may be early, preferential targets of the AD pathophysiologic process. Multimodality imaging studies in ADAD, where AD pathology is relatively disentangled from the effects of advanced age and identification of those in the earliest stages of AD is more straightforward, may help us to understand the consequences of AD pathology on intrinsic functional networks and elucidate the neural basis for the complex cognitive and behavioral domains so adversely affected in AD.
Supplementary Material
ACKNOWLEDGMENT
The authors gratefully acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study. Additionally, the authors gratefully acknowledge the contributions of Drs. William Klunk, Chester Mathis, Michael Weiner, and Krista Moulder to the DIAN Imaging Core and data collection for this project. This manuscript has been reviewed by DIAN study investigators for scientific content and consistency of data interpretation with previous DIAN study publications.
GLOSSARY
- Aβ
amyloid-β
- AD
Alzheimer disease
- ADAD
autosomal dominant Alzheimer disease
- ANOVA
analysis of variance
- APP
amyloid precursor protein
- CDR
Clinical Dementia Rating
- DGC
DIAN Genetics Core
- DIAN
Dominantly Inherited Alzheimer Network
- DMN
default mode network
- eYO
estimated years from expected symptom onset
- fcMRI
functional connectivity MRI
- GLM
general linear model
- ICA
independent component analysis
- LOAD
late-onset Alzheimer disease
- LPC
left lateral parietal cortex
- MCI
mild cognitive impairment
- MNI
Montreal Neurological Institute
- mPFC
medial prefrontal cortex
- NCRAD
National Cell Repository for Alzheimer's Disease
- PPC
precuneus/posterior cingulate
- PSEN1
presenilin-1
- PSEN2
presenilin-2
- RPC
right lateral parietal cortex
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Drs. Chhatwal and Schultz: data analysis, writing of manuscript, statistical analysis and interpretation. Dr. Johnson: study design, study supervision, data analysis, writing of manuscript, statistical analysis and interpretation of results, critical revision of the manuscript for important intellectual content. Dr. Benzinger: study design, acquisition of data, critical revision of the manuscript for important intellectual content. Dr. Jack: study design, acquisition of data, critical revision of the manuscript for important intellectual content, interpretation of results. Dr. Ances: study design, acquisition of data, critical revision of the manuscript for important intellectual content. Ms. Sullivan: acquisition of data, critical revision of the manuscript for important intellectual content. Drs. Salloway, Ringman, Koeppe, Marcus, Thompson, Saykin, Correia, Schofield, Rowe, Fox, Brickman, Mayeux, McDade, Bateman, Fagan, Goate, Xiong, Buckles: study design, acquisition of data, critical revision of the manuscript for important intellectual content. Dr. Morris: study supervision, study design, critical revision of the manuscript for important intellectual content. Dr. Sperling: study design, study supervision, data analysis, writing of manuscript, statistical analysis and interpretation of results, critical revision of the manuscript for important intellectual content.
STUDY FUNDING
Data collection for this project was supported by the Dominantly Inherited Alzheimer Network (DIAN, U19AG032438 to J.C.M.) and data analyses by a K24 grant (AG035007 to R.A.S.), both funded by the National Institute on Aging (NIA).
DISCLOSURE
J. Chhatwal and A. Schultz report no disclosures. K. Johnson has served as paid consultant for Bayer, Bristol-Myers Squibb, GE Healthcare, Janssen Alzheimer's Immunotherapy, Siemens Medical Solutions, and Genzyme. He is a site coinvestigator for Lilly/Avid, Bristol-Myers Squibb, Pfizer, Janssen Immunotherapy, and Navidea. He has spoken at symposia sponsored by Janssen Alzheimer's Immunotherapy and Pfizer. T. Benzinger has served on an advisory board for Eli Lilly and has received research funding from Avid Radiopharmaceuticals. These relationships are not related to the content in the manuscript. C. Jack serves as a consultant for Janssen, Bristol-Myers Squibb, General Electric, Siemens, and Johnson & Johnson and is involved in clinical trials sponsored by Allon and Baxter, Inc. He receives research funding from the NIH and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation. B. Ances and C. Sullivan report no disclosures. S. Salloway has received research support from Avid-Lilly, GE, Bayer, and Merck for studies related to amyloid imaging. He is a paid consultant to Lilly, GE, and Bayer. These relationships are not related to the content in the manuscript. J. Ringman receives research support from the NIA and the Jim Easton Consortium for Alzheimer's Disease Drug Discovery and Biomarkers at UCLA, and has received compensation as an advisory board member for Takeda Pharmaceuticals and StemCells, Inc. These relationships are not related to the content of the manuscript. R. Koeppe reports no disclosures. D. Marcus is founder of Radiologics, Inc. This relationship is not related to the content of the manuscript. P. Thompson reports no disclosures. A. Saykin has received research support from Siemens Healthcare and Welch Allyn. He has served as a consultant to Eli Lilly and Siemens Healthcare in the past year. He serves as Editor-in-Chief of Brain Imaging and Behavior, a Springer journal. None of these relationships are relevant to the content in the manuscript. S. Correia reports no disclosures. P. Schofield has been a paid speaker for Janssen. This relationship is not related to the content in the manuscript. C. Rowe has been a consultant on brain PET imaging to GE Healthcare, Bayer, and Astra Zeneca, and received research grants from these companies and Avid Radiopharmaceuticals. These relationships are not related to the content of the manuscript. N. Fox reports that in the last 3 years his research group has received funding for image analysis services or consultancy from Bristol-Myers Squibb, Elan/Janssen, GE, Eli Lilly, Lundbeck, and Pfizer/Wyeth. These relationships are not related to the content in the manuscript. A. Brickman, R. Mayeux, and E. McDade report no disclosures. R. Bateman receives research support from the NIH, Alzheimer's Association, American Health Assistance Foundation, Ruth K. Broadman Biomedical Research Foundation, Anonymous Foundation, Eli Lilly research collaboration, Merck research collaboration, AstraZeneca research collaboration, and the DIAN Pharma Consortium: AIP, Biogen, Elan, Genentech, Lilly, Mithridion, Novartis, Pfizer, Roche, and Sanofi. He is a cofounder of C2N Diagnostics, and invited speaker at BMS, Lilly, Merck, Pfizer, Elan, Wyeth, Novartis, Abbott, Biogen, Takeda Foundation, and consulting relationships for the DZNE, Probiodrug AG, Medscape, En Vivo (SAB), and Merck. A. Fagan is a member of the Alzheimer's Disease CSF Biomarker Development Advisory Board for Roche and the US Alzheimer's Disease Advisory Board for Lilly USA. These relationships are not related to the content of the manuscript. A. Goate is a principal investigator on research grants from Pfizer, Genentech, and iPierian. She has served as a consultant for Finnogan, Henderson, Farabow, and Garret & Dunner, LLP. These relationships are not related to the content of the manuscript. C. Xiong has served as a paid consultant for Biogen-IDEC. This relationship is not related to the content in the manuscript. V. Buckles reports no disclosures. J. Morris has participated or is currently participating in clinical trials of antidementia drugs sponsored by the following companies: Janssen Immunotherapy, Eli Lilly and Company, and Pfizer. He has served as a consultant for the following companies: Avid Radiopharmaceuticals, Eisai, Esteve, Janssen Alzheimer Immunotherapy Program, Glaxo-Smith-Kline, Novartis, Otsuka Pharmaceutical, and Pfizer. R. Sperling has served as a paid consultant for Bayer, Biogen-IDEC, Bristol-Myers Squibb, Eisai, Janssen Alzheimer Immunotherapy, Pfizer, Merck, and Roche, and as an unpaid consultant to Avid. She is a site coinvestigator for Avid, Bristol-Myers Squibb, Pfizer, and Janssen Alzheimer Immunotherapy clinical trials. She has spoken at symposia sponsored by Eli Lilly, Pfizer, and Janssen Alzheimer Immunotherapy. These relationships are not related to the content in the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Hardy J, Selkoe D. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356 [DOI] [PubMed] [Google Scholar]
- 2.Celone KA, Calhoun VD, Dickerson BC, et al. Alterations in memory networks in mild cognitive impairment and Alzheimer's disease: an independent component analysis. J Neurosci 2006;26:10222–10231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones DT, Machulda MM, Vemuri P, et al. Age-related changes in the default mode network are more advanced in Alzheimer disease. Neurology 2011;77:1524–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci USA 2004;101:4637–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, Raichle ME. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci USA 2005;102:9673–9678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeo BT, Krienen FM, Sepulcre J, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol 2011;106:1125–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo CC, Kurth F, Zhou J, et al. One-year test–retest reliability of intrinsic connectivity network fMRI in older adults. Neuroimage 2012;61:1471–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greicius MD, Krasnow B, Reiss AL, Menon V. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc Natl Acad Sci USA 2003;100:253–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chhatwal JP, Sperling RA. Functional MRI of mnemonic networks across the spectrum of normal aging, mild cognitive impairment, and Alzheimer's disease. J Alzheimers Dis 2012;31(suppl 3):S155–S167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vannini P, Hedden T, Becker JA, et al. Age and amyloid-related alterations in default network habituation to stimulus repetition. Neurobiol Aging 2012;33:1237–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petrella JR, Sheldon FC, Prince SE, Calhoun VD, Doraiswamy PM. Default mode network connectivity in stable vs progressive mild cognitive impairment. Neurology 2011;76:511–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daselaar SM, Prince SE, Cabeza R. When less means more: deactivations during encoding that predict subsequent memory. Neuroimage 2004;23:921–927 [DOI] [PubMed] [Google Scholar]
- 13.Miller SL, Celone K, DePeau K, et al. Age-related memory impairment associated with loss of parietal deactivation but preserved hippocampal activation. Proc Natl Acad Sci USA 2008;105:2181–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vlassenko AG, Vaishnavi SN, Couture L, et al. Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc Natl Acad Sci USA 2010;107:17763–17767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Machulda MM, Jones DT, Vemuri P, et al. Effect of APOE 4 status on intrinsic network connectivity in cognitively normal elderly subjects. Arch Neurol 2011;68:1131–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Filippini N, Ebmeier KP, MacIntosh BJ, et al. Differential effects of the APOE genotype on brain function across the lifespan. Neuroimage 2011;54:602–610 [DOI] [PubMed] [Google Scholar]
- 17.Trachtenberg AJ, Filippini N, Ebmeier KP, Smith SM, Karpe F, Mackay CE. The effects of APOE on the functional architecture of the resting brain. Neuroimage 2012;59:565–572 [DOI] [PubMed] [Google Scholar]
- 18.Sheline YI, Morris JC, Snyder AZ, et al. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. J Neurosci 2010;30:17035–17040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedden T, Van Dijk KR, Becker JA, et al. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci 2009;29:12686–12694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mormino EC, Smiljic A, Hayenga AO, et al. Relationships between beta-amyloid and functional connectivity in different components of the default mode network in aging. Cereb Cortex 2011;21:2399–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bateman RJ, Aisen PS, de Strooper B, et al. Autosomal-dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res Ther 2011;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bateman RJ, Xiong C, Benzinger TLS, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bird TD. Familial Alzheimer's disease. Ann Neurol 1994;36:335–336 [DOI] [PubMed] [Google Scholar]
- 24.Lampe TH, Bird TD, Nochlin D, et al. Phenotype of chromosome 14-linked familial Alzheimer's disease in a large kindred. Ann Neurol 1994;36:368–378 [DOI] [PubMed] [Google Scholar]
- 25.Haltia M, Viitanen M, Sulkava R, et al. Chromosome 14-encoded Alzheimer's disease: genetic and clinicopathological description. Ann Neurol 1994;36:362–367 [DOI] [PubMed] [Google Scholar]
- 26.Freire L, Mangin JF. Motion correction algorithms may create spurious brain activations in the absence of subject motion. Neuroimage 2001;14:709–722 [DOI] [PubMed] [Google Scholar]
- 27.Freire L, Mangin JF. Lecture Notes in Computer Science. Berlin: Springer-Verlag; 2002 [Google Scholar]
- 28.Van Dijk KR, Sabuncu MR, Buckner RL. The influence of head motion on intrinsic functional connectivity MRI. Neuroimage 2012;59:431–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erhardt EB, Rachakonda S, Bedrick EJ, Allen EA, Adali T, Calhoun VD. Comparison of multi-subject ICA methods for analysis of fMRI data. Hum Brain Mapp 2010;32:2075–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reiman EM, Langbaum JB, Fleisher AS, et al. Alzheimer's prevention initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis 2011;26(suppl 3):321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andrews-Hanna JR, Snyder AZ, Vincent JL, et al. Disruption of large-scale brain systems in advanced aging. Neuron 2007;56:924–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reiman EM, Quiroz YT, Fleisher AS, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol 2012;11:1048–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clark CM, Davatzikos C, Borthakur A, et al. Biomarkers for early detection of Alzheimer pathology. Neurosignals 2008;16:11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gauthier S, Reisberg B, Zaudig M, et al. Mild cognitive impairment. Lancet 2006;367:1262–1270 [DOI] [PubMed] [Google Scholar]
- 36.Sheline YI, Raichle ME, Snyder AZ, et al. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry 2010;67:584–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braskie MN, Medina LD, Rodriguez-Agudelo Y, et al. Memory performance and fMRI signal in presymptomatic familial Alzheimer's disease. Hum Brain Mapp Epub 2012 Jul 17. [DOI] [PMC free article] [PubMed]
- 38.Thomas JB, Brier MR, Bateman RJ, et al. Functional connectivity in autosomal dominant and sporadic Alzheimer disease. Presented at the Alzheimer's Association International Conference; July 13–18, 2013; Boston
- 39.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron 2009;62:42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer's disease. Lancet Neurol 2011;10:187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.