

Abstract
We report on a patient carrying a 17q21.31 microdeletion and exhibiting many common syndrome features, together with other clinical signs which have rarely or never been described to date. The detected 695-kb 17q21.31 deletion is larger than in most previously reported cases but is still probably the result of recombination between flanking low-copy repeats. Due to the complexity of the patient's clinical condition, together with the presence of 3 previously unreported symptoms, namely chronic anemia, cervical vertebrae arthrosis and vertebrae fusion, this case is an important addition to the existing knowledge about the 17q21.31 microdeletion syndrome.
Key Words: Array-CGH, Cervical arthrosis, Chronic anemia, Intellectual disability, 17q21.31 microdeletion syndrome, Vertebrae fusion
Chromosomal aberrations are a major cause of several syndromic features and congenital malformations. Widespread use of array-CGH technology has enabled the identification of several microdeletion/microduplication syndromes, including the 17q21.31 microdeletion syndrome [Koolen et al., 2006; Shaw-Smith et al., 2006; Varela et al., 2006]. Clinical features of this syndrome include developmental delay, a friendly or amiable behavior, hypotonia, and facial dysmorphisms such as a long face, a tubular or a ‘pear-shaped’ nose with a bulbous nasal tip [Koolen et al., 2008]. Other clinically important symptoms are epilepsy, heart defects and urologic abnormalities [Koolen et al., 2008]. The prevalence of this syndrome was estimated to be 1 in 16,000 individuals [Koolen et al., 2008]. Deletion of the KANSL1 gene has been shown to make a major contribution to the phenotype.
Here, we report on a patient previously diagnosed with Lachiewicz syndrome [Lachiewicz et al., 1985], who, despite showing a 46,XX karyotype by GTG-banding, displayed a complex phenotype that led us suspect a submicroscopic chromosome abnormality.
Case Report
The patient is an 18-year-old Caucasian girl, the eldest child of her mother and the second child of her father. She was born by cesarean section because of a cephalopelvic disproportion, weighed 2,310 g (<3rd percentile) and measured 44 cm (<3rd percentile) in length. Her head circumference was 34 cm (50th percentile). Facial dysmorphisms and clubfoot were also noted at birth. She had failure to thrive and was developmentally delayed: she was hypotonic, sat at 18 months, walked at 2 years and 6 months and spoke her first sentence at 18 months. A clinical examination at age 17 recorded a weight of 38.7 kg (<3rd percentile), a height of 142.5 cm (<3rd percentile) and a head circumference of 55.5 cm (50th percentile). The patient (fig. 1a) has a friendly behavior, nasal speech and moderate intellectual disability. Clinical examination revealed abundant thick straight hair, a triangular face with a high flat forehead, short and abundant eyebrows, ptosis, upslanting palpebral fissures, epicanthic folds, a bulbous nasal tip, anteverted nostrils, a long and deep nasal filter, a preauricular fistula, high palate, discrete microretrognathia, kyphosis, an umbilical hernia, and clubfoot. Her hands are narrow and long with hypoplastic muscles, and she has slender lower limbs. Previous examinations revealed an early cataract and interatrial communication (both surgically corrected), chronic anemia, conductive hearing loss in the left ear, hydronephrosis, ureterocele, and agenesis of the corpus callosum. In addition, a mild cervical vertebrae arthrosis and multiple thoracic and lumbar vertebrae fusions were shown by X-rays (fig. 2). The patient had her first convulsive crisis at the age of 2 years and 8 months which was treated with Tegretol.
Fig. 1.

a Patient at age 17. The pictures show the syndromic aspects of her face. b Array-CGH data showing the deleted region in chromosome 17.
Fig. 2.
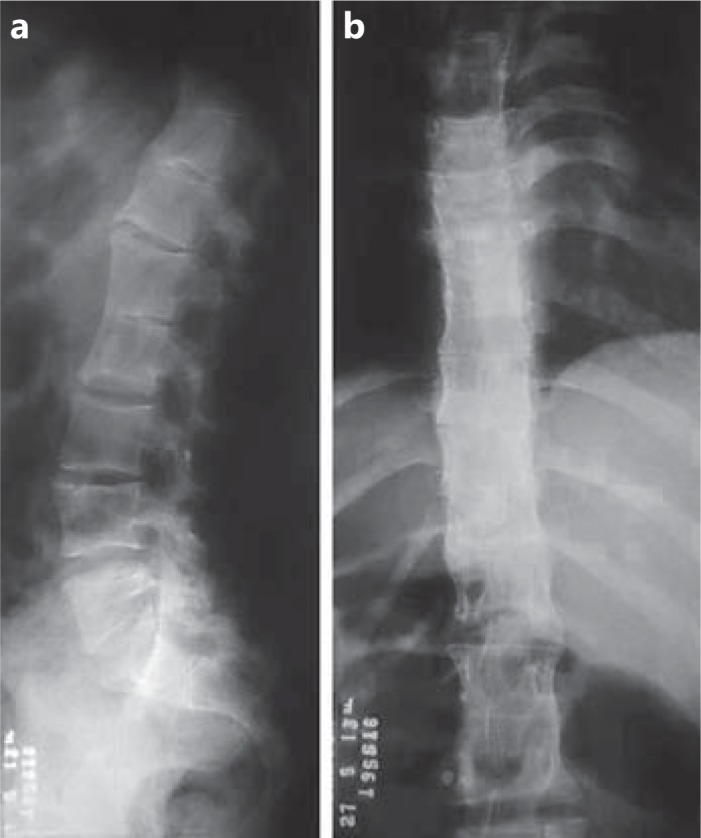
Lumbar (a) and thoracic (b) X-ray showing multiple vertebrae fusions.
The family signed an informed consent form, and the work was approved by the institutional Ethics Committee.
Molecular Analyses
Chromosome analysis of peripheral blood lymphocytes by GTG-banding, at the 550 or greater banding level, revealed a normal 46,XX karyotype. However, array-CGH showed the presence of a submicroscopic chromosomal alteration. The procedure was performed using a whole-genome oligoarray platform consisting of 60,000 oligonucleotides (60K, Agilent Technologies). Labeling, hybridization and post-hybridization washing were performed according to the manufacturer's protocol. A scanned image of the microarray was processed using Feature Extraction software and analyzed with Genomic Workbench software (both from Agilent Technologies), with the statistical algorithm ADM-2, and a sensitivity threshold of 6.7. At least 3 consecutive oligonucleotides had to exhibit aberrant log2 ratios in order to be called by the software as a copy number alteration. Array-CGH analysis identified an interstitial deletion of a segment of ∼695 kb mapped at 17q21.31 in the genomic positions chr17:41012276-41707073 (hg 18) (fig. 1b). The deleted region contains the following genes: MAPT, CRHR1, KANSL1, SPPL2C, and STH, the hypothetical ORF LOC644246 and the non-coding RNA MGC57346.
In addition, the proband's parents are non-consanguineous and there is no family record of hereditary diseases. The mother does not carry the deletion, as shown by array-CGH. Although the father was not available for study, the deletion has previously been reported as pathogenic, and we can assume that it is likely to be a de novo event.
Discussion
The 17q21.31 microdeletion causes a well-described syndrome in the scientific literature, presenting various known clinical features such as developmental delay, neonatal hypotonia, low birth weight and a pear-shaped nose [Koolen et al., 2008] (table 1). Although microdeletion size can vary, most microdeletions are between 500-650 kb and are mediated by non-allelic homologous recombination between flanking low-copy repeats [Koolen et al., 2012]. A minimal chromosomal region has been refined to a segment of 424 kb at chr17:41046729-41470954 (hg 17) and includes the MAPT, CRHR1, KANSL1, SPPL2C, and STH genes [Koolen et al., 2008]. The aforementioned critical region was further narrowed to a 160.8-kb segment (chr17:41310123-41470954, hg 18) that comprises only MAPT, KANSL1 and STH [Dubourg et al., 2010]. Recently, point mutations in KANSL1 have been identified in patients presenting intellectual disability and hypotonia, as well as other symptoms of the 17q21.31 microdeletion syndrome, thus confirming that the deletion of KANSL1 has a major contribution to the phenotype [Koolen et al., 2012; Zollino et al., 2012]. The patient reported in this study shows a deletion within the larger expected size which consists of approximately 695 kb and encompasses the MAPT, CRHR1, KANSL1, STH, and SPPL2C genes, a hypothetical ORF (LOC644246) and a non-coding RNA (MGC57346). We hypothesize that the complexity of the patient's phenotype was due to the larger deletion size.
Table 1.
Clinical features in patients with large 17q.21.31 microdeletions in comparison to the present case and to the main clinical features proposed by Koolen et al. [2008]
| Clinical features | Koolen et al. [2008] |
Wright et al., 2011 |
Dubourg et al. [2010] | Tan et al. [2009] Our patienta | |||
|---|---|---|---|---|---|---|---|
| P1 | P2 | P4 | P7 | P9 | |||
| Size of deletion, kb | 424b | 639 | 519 | 502 | 675 – 810 | ∼800 | 695 |
| Abnormalities of corpus callosum | * | – | – | – | + | + | + |
| Anteverted nostrils | * | – | – | – | – | – | + |
| Blepharophimosis | 36% | – | |||||
| Broad chin | 42% | – | |||||
| Bulbous nasal tip | 95% | + | + | + | + | – | + |
| Cleft palate | 9% | – | |||||
| Cryptorchidism | 78% | – | + | NA | – | + | NA |
| Deafness | * | – | – | sensorineural | – | – | conductive |
| Developmental delay | 100% | + | + | + | + | + | + |
| Dislocation of the hips | 27% | – | |||||
| Ear abnormalities | 59% | + | – | + | + | + | + |
| Epicanthus | 68% | – | – | – | – | – | + |
| Epilepsy | 50% | + | – | – | – | – | + |
| Feeding problems | * | + | + | + | – | ||
| Foot deformities | 27% | – | – | – | – | – | + |
| Friendly/amiable behavior | 89% | – | + | – | + | + | + |
| Hair abnormalities | 55% | + | – | + | – | – | + |
| Heart defects | 27% | – | + | – | + | – | + |
| High/broad forehead | 68% | – | – | – | – | + | + |
| High/narrow palate | 50% | – | – | – | – | – | + |
| Hypermetropia | 36% | – | |||||
| Hypoplastic hand muscles | 29% | + | |||||
| Kidney/urologic defect | 32% | – | + | + | + | – | + |
| Long face | 74% | + | + | + | – | + | + |
| Long fingers | 61% | – | – | – | – | – | + |
| Low birth weight | 27% | – | – | – | – | – | + |
| Microcephaly | 5% | – | |||||
| Narrow hands | 28% | + | |||||
| Nasal speech | 50% | + | |||||
| Neonatal hypotonia | 96% | + | + | + | + | + | |
| Pale irides | 45% | ||||||
| Pectus excavatum | 23% | + | |||||
| Ptosis | 50% | – | |||||
| Scoliosis/kyphosis | 36% | – | – | + | – | + | + |
| Short stature | 18% | – | – | + | – | – | + |
| Skin problems | * | + | + | + | + | – | – |
| Slender lower limbs | 41% | + | |||||
| Strabismus | 45% | + | – | – | – | – | – |
| Tubular/pear-shaped nose | 82% | + | + | + | – | – | + |
| Upward slanted palpebral fissures | 68% | + | + | + | + | ||
| Ventriculomegaly | 38% | – | |||||
NA = Not applicable; + = present; – = not present; * = frequency not established; blank = no information.
The patient also has other features not included in this table, which are: hydronephrosis, discrete microretrognathia, preauricular fistula, umbilical hernia, and chronic anemia.
Minimal critical size proposed by Koolen et al. [2008].
In a study of 22 patients with microdeletions of different sizes, Koolen et al. [2008] described 37 clinical signs and symptoms present in at least 5% of the patients as a basis for the main clinical findings. We compared the frequencies identified by Koolen with the data of previously reported cases with a similarly large (519-810 kb) microdeletion size [Tan et al., 2009; Dubourg et al., 2010; Wright et al., 2011] to that of our patient as shown in table 1.
It is noticeable that with the exception of developmental delay, the clinical presentation in cases with similar microdeletion sizes is different. As can be seen in table 1, the previously described cases with large deletions presented fewer clinical signs than our patient who shows approximately 70% of all known symptoms. Although the deletion size of some of the patients (e.g. P7 and P9) was larger than the deletion observed in our patient, she displayed a significantly expanded clinical condition in relation to the others.
It is also important to keep in mind that the cases in table 1 present deletions relatively larger than the minimal critical region found by Koolen et al. [2008]. This might indicate that there is no effective difference between the characteristics presented/not presented by a patient whose deletion size is smaller or larger than this minimal critical size.
All of the signs and symptoms that our patient presents have previously been seen in reported cases of the 17q21.31 microdeletion syndrome, with the exception of chronic anemia, cervical arthrosis and vertebrae fusion. Our patient presents all features found in more than 50% of patients reported by Koolen et al. [2008], except for ptosis, as well as rarer features like agenesis of the corpus callosum, hydronephrosis (present in only 2 patients to date [Tan et al., 2009; Dubourg et al., 2010]) and conductive deafness (also reported in 2 patients [Tan et al., 2009]).
Here we reported a patient with one of the largest 17q21.31 microdeletions ever described. However, by comparison to patients with similar microdeletions, we were able to conclude that the complexity of the phenotype is not related to the deletion size. The severity of her skeletal malformations suggests an important role of genes mapped to this segment in bone formation. Due to the complexity of her clinical condition and the presence of 3 previously unreported symptoms, namely chronic anemia, cervical arthrosis and vertebrae fusion, this case is an important addition to the existing knowledge about the 17q21.31 microdeletion syndrome.
Acknowledgements
We thank the patient and her family for participating in this study. This work was partially supported by the FAPDF and Ministério da Saúde (PPSUS 2010, Proc 193.000.344/2010) and Decanato de Pesquisa e Pós-graduação, University of Brasilia.
References
- 1.Dubourg C, Sanlaville D, Doco-Fenzy M, Caignec CLE, Missirian C, et al. Clinical and molecular characterization of 17q21.31 microdeletion syndrome in 14 French patients with intellectual disability. Eur J Med Genet. 2010;54:144–151. doi: 10.1016/j.ejmg.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Koolen DA, Vissers LELM, Pfundt R, de Leeuw N, Knight SJL, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 3.Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJL, et al. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45:710–720. doi: 10.1136/jmg.2008.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koolen DA, Kramer JM, Neveling K, Nillesen WM, Moore-Barton HL, et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat Genet. 2012;44:639–641. doi: 10.1038/ng.2262. [DOI] [PubMed] [Google Scholar]
- 5.Lachiewicz AM, Kogan SJ, Levitt SB, Weiner RL. Concurrent agenesis of the corpus callosum and ureteroceles in siblings. Pediatrics. 1985;75:904–907. [PubMed] [Google Scholar]
- 6.Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 7.Tan TY, Aftimos S, Worgan L, Susman R, Wilson M, et al. Phenotypic expansion and further characterization of the 17q21.31 microdeletion syndrome. J Med Genet. 2009;46:480–489. doi: 10.1136/jmg.2008.065391. [DOI] [PubMed] [Google Scholar]
- 8.Varela MC, Krepischi-Santos AC, Paz JA, Knijnenburg J, Szuhai K, et al. A 17q21.31 microdeletion encompassing the MAPT gene in a mentally impaired patient. Cytogenet Genome Res. 2006;114:89–92. doi: 10.1159/000091934. [DOI] [PubMed] [Google Scholar]
- 9.Wright EB, Donnai D, Johnson D, Clayton-Smith J. Cutaneous features in 17q21.31 deletion syndrome: a differential diagnosis for cardio-facio-cutaneous-syndrome. Clin Dysmorphol. 2011;20:15–20. doi: 10.1097/MCD.0b013e32833e8f1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zollino M, Orteschi D, Murdolo M, Lattante S, Battaglia D, et al. Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat Genet. 2012;44:636–638. doi: 10.1038/ng.2257. [DOI] [PubMed] [Google Scholar]