

Abstract
Radical addition processes can be ideally suited for the direct functionalization of heteroaromatic bases, yet these processes are only sparsely used due to the perception of poor or unreliable control of regiochemistry. A systematic investigation of factors affecting the regiochemistry of radical functionalization of heterocycles using alkylsulfinate salts revealed that certain types of substituents exert consistent and additive effects on the regioselectivity of substitution. This allowed us to establish guidelines for predicting regioselectivity on complex π-deficient heteroarenes, including pyridines, pyrimidines, pyridazines and pyrazines. Since the relative contribution from opposing directing factors was dependent on solvent and pH, it was sometimes possible to tune the regiochemistry to a desired result by modifying reaction conditions. This methodology was applied to the direct, regioselective introduction of isopropyl groups into complex, biologically active molecules, such as diflufenican (44) and nevirapine (45).
Introduction
Aromatic heterocycles are essential structures in a large proportion of small molecule drugs. Structural optimization of bioactive leads through systematic chemical alteration is essential for selecting a clinical candidate, and methods to synthesize or elaborate key pharmacophores hold particular significance in the drug discovery process. However, the production of such libraries of compounds can be very challenging, as electron-deficient heteroaromatic bases such as pyrimidines, pyrazines, pyridazines and pyrimidines are often difficult to derivatize directly with standard reactions (Figure 1A).1-4 As a result, the development of mild, direct functionalization methods for the selective late-stage modification of complex heteroarene structures remains an important practical goal.
Figure 1.

Regioselective radical functionalization of heteroarenes.
A universal, but perhaps underappreciated, tendency of a heteroarene (electron-rich or electron-poor) is to react with radical species. Functionalization methods that proceed via radical addition would therefore be expected to be a convenient method for elaborating aromatic heterocycles, yet methodology exploiting this innate reactivity is seldom employed.5 There is a perception that direct radical functionalization processes are frequently low yielding, have unpredictable regioselectivity, and give intractable mixtures of products. Although this reputation is sometimes justified, the problem is compounded because the factors governing the reactivity and selectivity of radical addition to heteroarenes are not widely known or understood.
In our ongoing studies into the functionalization of heteroarenes using alkylsulfinate-derived radicals,6 we have begun to observe certain trends pertaining to regiocontrol that – based upon the similarity to patterns observed in various other radical-mediated functionalization reactions – could be useful in a more general manner. In this full report, an extensive study of the reactivity trends of heteroarene functionalizations employing alkylsulfinate-derived radicals is presented. This work further defines the scope of these reactions, demonstrates that regioselectivity can be tuned according to solvent and pH, and establishes useful, substituent-based reactivity patterns. These studies culminate in the presentation of a hierarchical set of empirically derived, practical guidelines allowing for both prediction and tunability of regioselectivity (Figure 1B). These guidelines provide the framework for further mechanistic studies that promise to increase fundamental understanding of radical-mediated heteroarene functionalization.7.
Background
A major objective at the outset of our studies was to understand the reasons behind seemingly diverse regiochemical results, both in our own work and in studies reported in the literature, with the goal of establishing trends that would allow the prediction of results. One broad conclusion that can be drawn from a variety of different radical-mediated heteroarene functionalization studies8 dating back thirty years or more is that regioselectivity is dominated by the inherent reactivity of the substrate,9 for example the strong preference for pyridines to react at the α and γ positions. Regulating regioselectivity is therefore challenging, but as summarized in Figure 2, there is precedent for innate regiochemical preferences to be influenced by acid (Figure 2A), solvent (Figure 2B), substituents on the heterocycle (Figure 2C), and nature of the radical (Figure 2D).10-18
Figure 2.

Factors influencing regioselectivity in reports of homolytic substitution reactions of heteroarenes (Refs. 10-18).
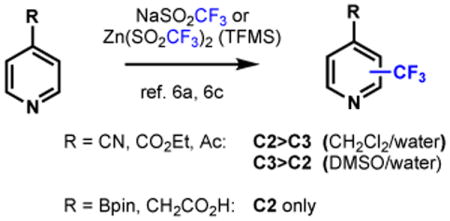
Comparison of these literature results with regiochemical perferences observed in our previous studies using radicals derived from alkylsulfinate salts6 revealed some of the same trends as well as some contrasting observations (Equation 1). Substitution at the α and γ positions of pyridine is generally preferred, although some C3 substitution was observed on pyridines with CN, CO2Et or Ac groups at C4. Intriguingly, however, the ratio of products for these substrates could be altered to favor either C2 or C3 substitution simply by changing the solvent.12b, 15b, 19 Pyridines substituted with CH2COOH or Bpin groups at C4 were fully selective for C2,6c an observation that could not be explained by steric effects, and is in contrast to Minisci's proposal that both electron-donating and electron-withdrawing groups at C4 stabilize the radical adduct arising from attack at C3.14a
Our analysis of the literature underlined the similarity of the trends in regioselectivity observed in a number of different radical functionalization systems, but also revealed examples that were difficult to explain using established knowledge. These considerations led us to undertake further systematic investigations of the factors regulating regiochemical outcomes with the aim to delineate generalizable effects leading to a practical approach to predicting regioselectivity in the radical functionalization of heteroarenes.
 |
(1) |
Results and Discussion
Experimental Design
In-depth study of the regioselectivity of the direct alkylation and fluoroalkylation of heteroarenes using radicals derived from alkylsulfinate salts was initiated, with 1H NMR spectroscopy employed as the primary tool for both product identification and analysis. The 1H NMR spectra of the crude reaction mixtures were exceptionally clean and the aromatic region typically contained only peaks corresponding to starting material and the different product regioisomers. As a result, it was possible to identify products and determine the regioisomeric ratio from these spectra (Figure 3A). Product regiochemistry derived from these spectra was supported by structural assignment after isolation.
Figure 3.

1H NMR analysis of the crude reaction mixture allowed ratios of regioisomeric products and starting material to be quantified (A) and tracked as the reaction progressed (B). The regioisomeric ratio remained constant throughout the reaction (C).
Preliminary studies identified reaction conditions optimal for regiochemical studies. This entailed limiting the occurrence of multiple substitution, which was responsible for the most significant side products observed at higher conversions, and which could alter conclusions about primary reactivity and selectivity. The yields reported below therefore represent intentionally limited values, and not optimized reaction conditions. Temporal profiles of conversion and regioselectivity were monitored in a number of cases, and regioselectivity was found to be constant over the course of the reaction (Figures 3B and 3C). A reasonable mass balance was observed in most cases (Figure 3C). This confirms that selective product degradation that could alter regioselectivity was not significant and that the experimental protocol provides results that accurately reflect the regiochemical preferences in each reaction. (Further experimental details are given in the Supporting Information.)
Reactivity patterns for alkylsulfinate-derived radicals
As a first example, we studied the alkylation of 4-substituted pyridines by radicals derived from alkylsulfinate salts. Figure 4A shows the results of examination of the effect of solvent and pH on the substitution ratio in the isopropylation of 4-cyanopyridine, revealing a pattern reminiscent of that previously observed in Gomberg-Bachmann10 and Minisci8,11-14 chemistry: (i) addition of acid strongly favors reaction at the α position;20 (ii) changing solvent could affect the regioselectivity.
Figure 4.

Factors controlling the regiochemistry of radical addition to pyridines. Reagent and conditions: pyridine (0.125 mmol), zinc sulfinate reagent (1 eq.), TBHP (1.5 eq.), 50 °C, 1.5-2 h. See Supporting Information for more details.
The influence of functional group on regioselectivity was probed by determining the C3:C2 ratio in the isopropylation of C4-substituted pyridines in DMSO (Figure 4B). C3 substituted products were observed only when a C4 π-conjugating, electron-withdrawing group was present, with the C3:C2 ratio roughly corresponding to the electron-withdrawing power of the functionality, as indicated by σ- values21 for these substitu-ents.22 The importance of π-conjugation of the electron-withdrawing group is underlined by the fact that while C3 substitution was observed for the CONH2 derivative, only C2 substitution occurred for the sterically hindered CON(i-Pr)2 group, which cannot act as a conjugating electron-withdrawing group, as it must adopt a conformation orthogonal to the pyridine ring. Non-conjugating groups – such as phenyl, t-butyl, or strongly σ-withdrawing CF3 – did not give C3 products, nor did the conjugating π-donating OMe group.
Figure 4 reveals an ortho directing effect of π-conjugating electron-withdrawing groups at C4, reminiscent of ortho-para effects suggested by other systems,15 and this pattern was supported by further screening. For brevity, in Figure 4 and elsewhere, this ortho-para directed reactivity induced by π-conjugating electron-withdrawing groups is referred to as “conjugate reactivity” because of the similarity of favored regiochemistry to that which would be obtained by a formal “conjugate” or Michael addition at the pyridine ring.
The electronic reasons for the directing effect is under further investigation, but presumably it is due to the ability of the π-conjugating electron-withdrawing group to stabilize the radical intermediate arising from attack at the ortho and para positions. The strong directing effect of π-conjugating electron-withdrawing groups contrasts with the weak or negligible directing effect of π-conjugating electron-donating groups such as OMe, which might also be expected to stabilize the intermediate radical adduct. This may be attributed to the additional captodative stabilization of radicals that can be stabilized by both an electron accepting (such as CN), and an electron donating (the pyridyl nitrogen) group.23
As the nucleophilicity of the radical species decreases24 on moving from i-Pr to CF2H to CF3, the influence of “conjugate reactivity” also decreases, as indicated by the C3:C2 ratio (Figure 4C). This effect also correlates with a decreasing influence of protonation; isopropylation in DMSO showed a complete reversal of selectivity when acid was added, whereas the results with CF3 were virtually unchanged. The more nucleophilic i-Pr radical was also more sensitive to solvent change than either CF3 or CF2H.6a, 6b For i-Pr radical, a striking regioselectivity switch from reaction strongly favoring C2 substitution to strongly favoring C3 substitution could be effected simply by changing solvent (Figure 4A). These results were broadly in line with prior observations.11-14
The relative nucleophilicity of the free radical is implicated in the case of the OMe substrate. While the reactivity trends for the substrates described in Figure 4B for the i-Pr radical were mostly very similar for the CF3 radical, a significant difference was observed for OMe, with a C3:C2 ratio of 1:2 obtained for CF3 compared to C2 only for i-Pr. This result may be rationalized by considering the electrophilic nature of the CF3 radical. It is known that nucleophilic radicals react well with protonated pyridines, but do not react with benzene, and that electrophilic radicals are reactive with arenes, but are unreactive with protonated pyridines.25 Extending this concept to regioselectivity preferences, nucleophilic radicals react at electron-poor sites and electrophilic radicals react at electron-rich sites. Thus, the C3 product arises from the CF3 radical interacting with the substrate as an electrophile at this position.
All the alkyl or fluoroalkyl radicals generated from the alkylsulfinate salts are to a certain extent nucleophilic, as evidenced by their facile reaction with pyridines, but the CF3 radical is the most electrophilic of the examples studied.24 Radical polarity is not always an intrinsic factor, and the behavior can vary between nucleophilic, electrophilic or nonpolar depending on the reaction and substrate involved.8 The nucleophilic i-Pr radical only has useful reactivity when attacking electron-poor sites, but the CF3 radical can give significant conversion when behaving as either a nucleophile or an electrophile. As an example, in Langlois's trifluoromethylation of electron-rich arenes with CF3SO2Na,26 the CF3 radical behaved as an electrophile, yet the same reagent behaves as a nucleophile in the functionalization of pyridines.6a
Working model to explain regioselectivity
Although the mechanisms by which the factors discussed above regulate regiochemistry are not fully understood, the experimentally determined patterns in regiochemistry are observed with sufficient consistency to make it possible to devise a set of empirical guidelines to predict the regioselectivity of substitution. We present here a simple method for explaining regioselectivity on heteroarenes that forms the basis of a surprisingly robust model for predicting reaction outcome.
Our model assumes that regioselectivity of substitution is governed by three major factors (Figure 5): (A) the inherent reactivity of the parent heterocycle (innate reactivity); (B) the reactivity induced by the presence of π-conjugating electron-withdrawing functional groups (“conjugate reactivity”), and (C) the electronic properties of the radical. In addition, we also discuss the role of solvent and acid in determining the relative significance of these factors as well as the importance of a number of “reactivity-modifying factors.”
Figure 5.

Three major factors controlling regiochemistry of homolytic aromatic substitution of pyridines. Green circles highlight positions activated to attack by nucleophilic radicals; blue circles highlight positions activated to attack by electrophilic radicals.
Since the alkylsulfinate-derived radicals discussed here primarily react with pyridines as nucleophiles, to an approximation, the “reactive” sites can be identified by identifying those sites with partial positive (δ+) charge, highlighted with green circles in Figure 5 and all subsequent figures.27 However, as discussed below, in a few cases the trifluoromethyl radical may also be able to react as an electrophile with δ- sites; these are highlighted in blue. Circles of different sizes are used to represent differences in reactivity within the same molecule, with the larger circle representing the preferred site.
Innate reactivity (Figure 5A)
Pyridines are inherently reactive with nucleophiles at the α and γ positions, which carry δ+ charge. The relative intensity of this factor appears to be related to the total π-electron density of the pyridine: electron-rich pyridines such as 4-methoxypyridine are less reactive than the parent pyridine, and electron-poor pyridines such as 4-cyanopyridine have enhanced reactivity. Protonation of the pyridine under acidic conditions will reduce the overall π-electron density of the pyridine ring and therefore increase the relative intensity of the innate reactivity.
Conjugate reactivity (Figure 5B)
The presence of π-conjugating electron-withdrawing groups may be thought of as inducing δ+ charge at the ortho and para sites, which are correspondingly activated to attack by nucleophilic radicals. The relative intensity of this factor appears to be related to solvent.
Radical electrophilicity and nucleophilicity (Figure 5C)
In the majority of cases discussed in this paper the radical, whether trifluoromethyl or isopropyl, behaves as a nucleophile to attack electrophilic (δ+) sites on a heteroarene. Nevertheless, some issues of regiocontrol are best explained by considering the relative nucleophilicity of the radical. Trifluoromethyl radicals are capable of reacting as electrophiles to attack electron-rich (δ-) positions. This usually occurs where there is an electron rich benzenoid ring also present in the molecule, as was previously described for the trifluoromethylation of hydroquinine.6a Occasionally this effect is also seen to influence the regioselectivity on electron deficient heterocycles such as pyridines. The C3 reactivity of a series of C4 substituted pyridines (Figure 4B) illustrates this trend: (i) the π-donating OMe group induces δ- charge at C3, and the pyridine is reactive with CF3 (behaving as an electrophile) at this position; (ii) the π-withdrawing CN group induces δ+ charge at C3, and the pyridine is reactive with i-Pr and CF3 (behaving as a nucleophile) at this position; (iii) the σ-withdrawing CF3 group does not induce significant charge at C3 and the pyridine is effectively unreactive at this position, despite the CF3 group presumably being able to stabilize the radical adduct arising from attack at C3. The consequences of the electrophilicity of trifluoromethyl radical will be revisited later when the regioselectivity of complex structures containing a number of aromatic and heteroaromatic rings are considered, but in general, for the vast majority of small heterocyclic substrates described in this paper, both CF3 and i-Pr radicals show similar regioselectivity trends. The main difference that is observed is that CF3 is more reactive than i-Pr with electron-rich pyridines and that i-Pr radical is more reactive than CF3 radical with the very electron-deficient diazines.
Solvent and acid effects
Attack on a pyridine by a nucleophilic radical may occur at any of the identified δ+ “reactive” sites. If there are no π-conjugating electron-withdrawing groups, only the pyridine α and γ positions will be activated. If there are π-conjugating electron-withdrawing groups present, the relative strengths of innate and conjugate reactivity will determine the ratios of products obtained. In chlorinated solvent/water mixtures, the innate reactivity is typically the dominant factor, and reaction primarily occurs at the α and γ positions, which have higher δ+ charge than the β positions. In DMSO, the balance between conjugate and innate reactivity is altered, such that the “effective” δ+ activation felt by the incoming radical is greatest at the positions ortho and para to the π-conjugating electron-withdrawing group; thus reaction primarily occurs at these sites. In either solvent system, the addition of acid emphasizes the influence of innate reactivity to the extent that it exceeds the influence of conjugate reactivity, even in DMSO (Figure 6A).
Figure 6.

Regioselectivity modifying effect of solvent and radical electrophilicity. Green circles highlight positions activated to attack by nucleophilic radicals; blue circles highlight positions activated to attack by electrophilic radicals. The size of the circle represents relative degree of activation within the same molecule.
Using these concepts, the regioselectivity outcomes from our previous work in alkylsulfinate-derived radical addition to C4 substituted pyridines (Equation 1)6a-6c that were not initially straightforward to explain are now easily rationalized. Pyridines with a C4 π-conjugating electron-withdrawing group, such as Ac, CO2Et or CN, are activated to attack by nucleophilic radicals (δ+ charge) at both the C2 and C3 positions, thereby yielding a regioisomeric mixture. In contrast, the Bpin or CH2CO2H group do not induce either δ+ or δ- charge at C3, so the pyridine is unreactive at this position; as a result exclusive C2 substitution is observed.
The lower susceptibility of CF3 radical to regioselectivity change induced by π-conjugating electron-withdrawing groups or protonation can also be rationalized using this model. The reactivity of the more nucleophilic i-Pr radical can be considered strongly dependent on the δ+ charge of the reactive site, so there is high regioselectivity for the most activated site, whereas the CF3 radical is less dependent on the δ+ charge for reactivity, and therefore retains more reactivity with the less δ+ activated site (Figure 6B). A possible electronic rationale for this preference may be extended from a frontier molecular orbital argument for the increased selectivity of t-butyl radical relative to methyl radical for reaction with electron-poor 4-cyanopyridine compared with electron-rich 4-methoxy-pyridine. The SOMO of the trifluoromethyl radical is lower in energy than the SOMO of the isopropyl radical, so the SOMO-LUMO distance is larger for CF3 than i-Pr. As a result, the i-Pr radical is more strongly affected than the CF3 radical by electronic differences between regioisomeric sites.28
The reason for the change in influence of innate and conjugate reactivity with solvent is unclear. Although it is possible that some component of the solvent dependence is related to the differing pKa of a protonated pyridine in different solvents (pyridine protonation is typically less favorable in DMSO than in water, for example),29 it is likely that a solvent effect unrelated to pKa alters the balance between innate and conjugate reactivity. Minisci observed that the ratio of C2:C4 substitution of protonated pyridines showed a distinct solvent effect,11, 30 and it is known that many other radical reactions exhibit large solvent effects.31 Calculations have also shown that the nucleophilicity and chemical hardness of a radical can vary depending on solvent, either of which could impact the balance between innate and conjugate reactivity.32
Reactivity-modifying factors
While the previous section focused on identifying which sites were capable of reacting, an important factor in predicting regioselectivity is accurately assessing the relative reactivity of multiple “activated” sites. The factors illustrated in Figure 7 and discussed below are termed “reactivity-modifying” because although they can discriminate between activated (δ+) sites, they cannot in general give rise to useful levels of reactivity at a position that is not already activated.
Figure 7.

Regioselectivity-modifying effects of functional groups. Green circles highlight sites activated to nucleophilic attack, red circles represent a site with a functional group induced deactivating effect. The size of the circle represents relative degree of activation/deactivation within the same molecule.
The influence of a variety of functional groups on the reactivity of already activated sites was tested by measuring substitution patterns in otherwise symmetrical (C4-substituted) pyridines. C4 substituted pyridines are reactive at C2 under CHCl3/water conditions that promote innate reactivity. Introducing a C3 group allows the effect of this group on the C2:C6 substitution ratio to be determined. Accordingly, methyl, halogen and methoxy groups were found to function as ortho-activators (Figure 7A). The reason for this is unclear, but possibly relates to a neighboring functional group being able to help stabilize a radical intermediate or transition state. Halogen and methoxy also function as meta-deactivators (highlighted in red), with the effect being particularly strong at the pyridine β and γ positions. Again, the reason for this is unclear but may be due to the π-donor capabilities of these groups.
Substitution at C2 has a particularly strong effect on the reactivity at C6. Electron-withdrawing functionality at C2 typically shut down reaction at C6, a fact that is well illustrated by the exclusive production of mono-substituted products in the trifluoromethylation or difluoromethylation of symmetrical C4-substituted pyridines (Figure 7B).6c In contrast, attempted C2 isopropylation in CHCl3/water/TFA resulted in mono-alkylation being accompanied by concurrent dialkylation; as a result electron-donating functionality at C2 is proposed to be activating at C6.33
Sterics are known to be a significant factor in Minisci reactions34 but appeared to be a secondary consideration in determining regiochemistry for the alkylsulfinate-derived radicals employed in our studies. The preference for para over ortho substitution in “conjugate reactivity” is attributed to steric effects, and the bulky i-Pr radical usually had a stronger preference for para-substitution than CF3 radical. Nevertheless, as shown in Figure 7C, steric effects did not overturn the regioselectivity preference set by functional groups, even when this required substitution in a position with steric crowding.
Predicting the site of radical addition to pyridines
The regioselectivity trends described above operated consistently and reliably across pyridines bearing numerous different functional groups and substitution patterns. Furthermore, the relative importance of each factor followed a consistent hierarchy for each solvent system. Consequently, it is possible to assess whether a pyridine is likely to be reactive at a desired position by considering each of the factors above in turn and combining the activating or deactivating factors. To simplify the thought process for predicting regioselectivity by consideration of the above factors we have designed a flowchart (Figure 8A, with an example35 illustrated in Figure 8B) which guides the reader through the relevant factors in order of importance.
Figure 8.

Flowchart for the prediction of regioselectivity of alkylsulfinate-derived radical functionalization of pyridines. Color code: green circles, sites activated to nucleophilic attack; red circles, sites subject to a functional group induced deactivating influence; yellow circles, otherwise activated sites that have reduced or negligible activity due to substituent-induced deactivation. The size of the circle represents relative degree of activation within the same molecule. Further examples of regioselectivity prediction are in the Supporting Information.
In Step 1, the innate sites of the heterocycle are identified. Step 2 considers those sites that are reactive because of their orientation relative to a π-conjugating electron withdrawing group. The sites identified in these two steps may be considered “activated” and are highlighted in green. A further level of analysis (Step 3) considers other factors that may modify the reactivity of these “activated sites” but will not usually alter the regioselectivity preferences of an unactivated site. These modifying factors may reinforce an existing preference, or exert a deactivating effect (highlighted in red) on an otherwise activated site – in this case the “activated” site will have reduced or negligible reactivity (highlighted in yellow).
The final level of prediction (Step 4) considers the solvent system being used for the reaction, and how this affects the relative importance of innate reactivity (Step 1) and conjugate reactivity (Step 2). In general, in CHCl3/water or CH2Cl2/water solvent systems (with or without acid), or in DMSO/acid the significantly reactive sites are the innate sites identified in Step 1, and in DMSO (neutral) the significantly reactive sites are the “conjugate” sites identified in Step 2.
An example of how this analysis might be used to predict the regioselectivity is illustrated for the case of methyl 3-methylpicolinate as shown in Figure 8B (further details of the regioisomeric ratio of this reaction are given in Figure 10).35 Proceeding through the steps of our model, we find that the analysis correctly predicts the trend of the C4 product being preferred in CHCl3/water, and increased preference for C5 reactivity in DMSO.
Figure 10.

Tuning regioselectivity with solvent choice. (For key to highlighting circles see Figures 6 and 7.) Reagents and conditions: standard conditions as previously described. Forcing conditions for compound 30.6c See Supporting Information for details.
These guidelines are based upon regioselectivity ratios obtained in reactions with the set of commercially available pyridines tested, but the trends observed have proven to be applicable to unfamiliar substrates and functional groups (vide infra, for example, 43-46 in Figure 12). In general, the predictions of regioselectivity obtained by this method closely matched the experimental results, although there are some cases where the predicted values fit less closely, reflecting the fact that guidelines seeking to be general will necessarily simplify information concerning the relative contributions of different factors. For example, the result shown in Figure 8B shows that the increased preference for conjugate reactivity in DMSO for isopropyl radical is not sufficient to fully outweigh the innate reactivity in the case of this substrate. It is interesting to compare this result with that for the nitrile analog 16 (see Figure 9B). The CN group is a more strongly electron-withdrawing π-conjugating group than is CO2Me, which enhances the influence of conjugate reactivity (see Figure 4B). Isopropylation of 3-methylpicolinonitrile in DMSO gave an enhanced preference for the C5 product compared to methyl 3-methylpicolinate (C5:C4 = 2.6:1 for 16 compared to 1:1 for 3).
Figure 12.

Regioselectivity remains predictable on complex substrates. (For key to highlighting circles see Figures 6-8.) Reagents and conditions: standard conditions as previously described. All examples gave >9:1 selectivity for the regioisomer shown. aD-MSO solvent. bCHCl3/H2O solvent mixture.
Figure 9.

Substrates tested to assess combined directing effects on complex pyridines. (For key to highlighting circles see Figures 6 and 7.) Reagents and conditions: pyridine (0.125 mmol), zinc sulfinate reagent (TFMS or IPS) (2 eq.), TBHP (3 eq.), 50 °C, 12–16 h. See Supporting Information for more details.
Testing the model
A selection of substrates used to develop and test these guidelines is shown in Figure 9. Regioselectivity was examined with both IPS (i-Pr radical) and TFMS (CF3 radical) to represent both the most nucleophilic and the most electrophilic from our alkylsulfinate salts. Where a result is listed for only one radical species or solvent, this usually reflects a lack of reactivity, or product instability.36 This study focused on two different reaction conditions: (i) CHCl3/water (with the addition of TFA to enhance reactivity for the i-Pr examples), and (ii) DMSO without addition of acid.37 Typically CHCl3/water38 solvent gave higher yields for electron-rich pyridines, and DMSO gave higher yields for electron-poor pyridines.
Particular points of note include: (i) an interesting example of the meta-deactivating effect of chlorides may be seen by comparing the unreactive 26 (Fig 9C) with the successful functionalization of the methyl analogue (15 and 16, Fig 9B); (ii) methoxy proved to be a stronger ortho-activator than bromide on 10 (Fig 9A), in keeping with the high electronegativity of oxygen relative to bromine;17 (iii) i-Pr gave very low yields (albeit with the same general regioselectivity pattern) on the most electron-rich pyridines (7–9 and 13, Fig 9A) as expected for a nucleophilic radical; (iv) neither position in 12 (Fig 9B) is very activated under the reaction conditions, resulting in low yield and selectivity – the C5 position has little reactivity when not in DMSO solvent, and the C4 position has limited reactivity due to the meta-deactivating methoxy group, even under acidic conditions. Importantly, these results show that it is possible to predict which substrates are likely to fail or be reluctant to react.
As described previously, the relative contribution of innate and conjugate reactivity is dependent on solvent and pH. As a consequence, in examples where the innate and conjugate reactivities activate different sites, it is sometimes possible to tune the reactivity to favor a specific site, simply by changing solvent, as shown in Figure 10. Note the slightly lower selectivity for C5 over C4 substitution in 29 relative to 18 and 20, reflecting the absence of a group that meta-deactivates the C4 position.
Extension of regioselectivity trends to diazines
The regioselectivity of radical isopropylation of various functionalized and unfunctionalized diazines was examined to assess whether the functional group-dependent regioselectivity observed in pyridines could be extended to other heteroarene systems (Figure 11).39 DMSO was used as solvent to achieve synthetically useful conversions of products on these substrates. The pattern observed with the diazines was similar to that for pyridines: (i) the parent heterocycle had an innate regioselectivity preference for a particular position; (ii) π-conjugating, electron-withdrawing groups induced a preference for reaction at the ortho and para-positions, and could allow functionalization at otherwise unreactive sites; (iii) halides and methyl groups did not activate the para position, but did exert an ortho-directing effect. The influence of conjugate reactivity on the innate preference is weaker compared with the pyridines, and the preference of the functional group typically influenced, but did not override, the innate preference.
Figure 11.

Extension of regioselectivity study to diazines: the same general principles apply, although the innate preference of the parent heterocycle is harder to overcome. (For key to highlighting circles, see Figures 6 and 7.) Reagents and conditions: heterocycle (0.125 mmol), IPS (2 eq.), TBHP (3 eq.), DMSO, 50 °C, 12–16 h. See Supporting Information for more details.
2-Acylpyrazine derivatives gave the para-substituted products as a single regioisomer both for isopropyl (33), and the trifluoromethyl6a and difluoroethyl6d examples reported previously. Similar regioselectivity effects have also been seen in Minisci alkylations and acylations of 2-acyl pyrazines.40 Pyridazines had an innate reactivity at the C4/C5 (β) positions. One of these two activated positions could be favored over the other with an ortho-directing functional group in the α position. The innate reactivity preference was by far the dominant factor, with even a strongly electron withdrawing nitrile group (37) having a limited capability to activate the para C6 position. Pyrimidines had an innate reactivity for C4, which could be partially overridden to give significant quantities of the C2 or C5 products with a π-conjugating electron-withdrawing group in the para-position (cf 39 with 40; 41 with 42).
The innate sites of reactivity to radical functionalization for pyrimidines and pyridazines identified here are not unique to the alkylsulfinate-derived radical system; the same preferences have been observed in Minisci-type alkylations and acylations of diazine substrates.40a, 41
Testing on complex biologically active substrates
An important goal of our predictive model is to demonstrate generality for more complex heteroarenes such as those that might be encountered in pharmaceutical or agrochemical research. These compounds are often difficult to elaborate due to problems of solubility, the presence of other reactive functional groups, or steric crowding. Several small heterocyclic drugs, herbicides, pesticides, and natural products were identified as substrates likely to give good reactivity and regiocontrol based upon the previously described guidelines. It was intended that these would provide a simulation of the size and complexity of the kind of molecules that the alkylsulfinate reagents might be used to functionalize.
Figure 12 shows that isopropylation of these compounds in DMSO solvent gave products that very closely followed the expected results. In all cases the predicted product was the major regioisomer obtained, with a ratio of at least 9:1; in most cases the reaction was fully selective for the predicted product.
The pesticide picolinafen (43) isopropylates exclusively at C5, closely following the precedent set by simple pyridines 18 and 20. The elucidation of the regioselectivity guidelines focused on a limited selection of functional groups such as esters, nitriles and methoxy for which a wide variety of disubstituted pyridines are commercially available, but the trends observed are generalizable to other functional groups in the same class. Thus the π-conjugating electron-withdrawing amide group functions as an ortho-para director, and the aryl ether behaves as an ortho-activator. The exclusive regioselectivity of radical isopropylation of the herbicide diflufenican (44) is also explained in this manner, bearing in mind that like the methoxy group, an aryl ether can function as a meta-deactivator.
Nevirapine (45), an anti-retroviral drug, shows 9:1 selectivity for substitution at C9 on the nicotinamide ring, rather than at C2 on the electron-rich pyridine. This illustrates two important points: (i) the strong preference of the nucleophilic i-Pr radical to react with the polar, electron-deficient pyridine ring, instead of the electron-rich pyridine; (ii) the ability to predict regioselectivity for a molecule bearing the previously unexamined amine group, based on the reasoning that this σ-withdrawing, π-donating functionality should function as an ortho-activator and meta-deactivator, as do methoxy and halide functionalities.
The exclusive regioselectivity of substitution of ethyl β-carboline-3-carboxylate (46) shows the very strong deactivation of a pyridine C2 position by a strongly electron-withdrawing group at C6. The reason for the amine failing to act as a meta-deactivator, as predicted in this case, is under further investigation. It is likely that the meta deactivation effect is dependent on the π-donor capability of the functional group, and that this is reduced by participation of the nitrogen lone-pair in the indole system.
Voriconazole (47), an anti-fungal medicine, is isopropylated exclusively at the pyrimidine C4 position. One position is selected over three other open heterocyclic sites α to nitrogen. The sensitivity of regiocontrol that is possible in radical functionalization processes is underlined by the fact that the C2 position of a pyrimidine is also reactive to radicals. Indeed, C2 substitution occurred exclusively for fenarimol (47), where the C4 position is too sterically crowded to react.
Influence of the electrophilic character of CF3 radical
Our guidelines for predicting regioselectivity identify the sites on heteroarenes that are most reactive towards attack by radicals acting as nucleophiles. This works well for simple heteroarenes because despite their different electronic properties, both trifluoromethyl and isopropyl radicals primarily behave as nucleophiles when attacking electron-deficient substrates such as pyridines and diazines. A limitation of this prediction method may arise when considering more complex structures with several potentially reactive arenes and heteroarenes. In these cases, the site of reaction may be dependent on the properties of both the radical and (hetero)arene. In the examples shown in Figure 12, the nucleophilic i-Pr radical invariably reacted with the most electron-deficient ring, whereas regioselectivity for the more electrophilic trifluoromethyl radical was more subtly controlled.
In diflufenican (49) there is a reactive pyridine that competes with the relatively unreactive arenes bearing electron-withdrawing groups. Thus, trifluoromethylation occurred exclusively at the pyridine C6 position, analogously to the i-Pr radical. On the other hand, trimethoprim (50), which has an unreactive, electron-deficient heterocycle (neither amine can donate electron density to the unsubstituted position) competing with a very reactive trimethoxybenzene, is trifluromethylated almost exclusively on the arene. The previously reported results on differing sites of reactivity for difluoromethylation and trifluoromethylation of varenicline and dihydroquinine can be explained by using a similar argument.6b
Further investigations aim towards predicting the point at which trifluoromethyl radical will switch from nucleophilic to electrophilic behavior, but our investigations so far suggest that the analysis described in Figure 8 generally provides a good model for predicting the regioselectivity of alkylsulfinate-mediated radical addition to electron-deficient heteroarenes. Nevertheless, an important caveat is that when considering trifluoromethylation of molecules containing electron-rich arenes, or electron-deficient heterocycles that are fused to benzenoid or five-membered heteroaromatic rings, the possibility of the trifluoromethyl radical behaving as an electrophile to attack nucleophilic sites should also be considered.
Generalization to the borono-Minisci radical arylation
Examination of substitution patterns in literature examples of homolytic substitution of heteroarenes suggested that the principles of predicting and controlling regiochemistry for alkylsulfinate-derived radicals might be generalizable to other radical-mediated C–H functionalizations. A preliminary study was undertaken to assess the extent to which the findings in this report could be generalized to direct arylation of heteroarenes.
Arylboronic acids or alkyltrifluoroborates can be used for the direct functionalization of electron-deficient heteroarenes via a radical substitution process.42 The observed regiochemistry is broadly in line with that for typical for Minisci-type reactions (i.e., an overwhelming preference for the α and γ positions). However, in the “borono-Minisci” arylation42a of heterocycles there were unexplained regiochemical results. The direct arylation of C4-substituted pyridines gave mixtures of C2 and C3 products for the 4-cyano and 4-chloro derivatives, yet the 4-trifluoromethyl and 4-(1,3-dioxolan-2-yl) derivatives were substituted only at the C2 position (Figure 13A). These results are understandable based upon the model described earlier (Figures 5 and 6), noting that aryl radicals are slightly more nucleophilic than trifluoromethyl radicals, and may also react as weakly electrophilic or nonpolar species.24
Figure 13.

Extension of regioselectivity prediction to “borono-Minisci” direct arylation. Reagents and conditions: heterocycle (0.125 mmol), p-tolylboronic acid (1.5 eq.), AgNO3 (20 mol%), K2S2O8 (3 eq.), 50 °C, 3–16 h.
The original reaction conditions42a used a 1:1 CH2Cl2:H2O solvent in the presence of TFA. By increasing the reaction temperature from room temperature to 50 °C, we were able to get appreciable conversions without the presence of acid. NMR studies of the regioselectivity of direct arylation of 4-cyanopyridine showed a familiar trend: (i) using DMSO rather than a chlorinated solvent38 increased the proportion of “conjugate reactivity” to give the C3 product; (ii) as seen with CF3, addition of acid showed only a slight increase in production of the “innate” C2 product (Figure 13B). The regioselectivity of arylation of a selection of more complex pyridines were assessed and similar regioselectivity patterns to those found for alkylsulfinate-derived radicals were observed (Figure 13C).
These preliminary results provide a precedent that the regioselectivity guidelines that we developed may be generally useful for other radical-mediated direct functionalization processes, and they show that it may be possible to apply information from the solvent dependence of regioselectivity in the alkylsulfinate system to tune regioselectivity in the functionalization of heteroarenes by other types of radicals.
Conclusions
Factors affecting the regioselectivity of radical functionalization of pyridines were investigated systematically with a view to understanding, predicting and tuning the regiochemical outcome. A hierarchical model to rationalize the observed regioselectivity is proposed based upon experimental results and literature reports of related reactions. The principles of this model for regiocontrol have been distilled into a set of guidelines that summarize the stepwise process for predicting the regioselectivity of direct C–H functionalization of electron-deficient heteroarenes using radicals derived from alkylsulfinate salts. An advantage of alkylsulfinate salts as a means for radical generation compared with more traditional techniques is the facile ability to modify solvent and pH to influence the regioselectivity of substitution. This empirical model provides a method of explaining and predicting regiochemistry that closely fits with experimental observations.
The regioselectivity trends observed for pyridines were replicated for other heteroarenes, including pyrimidines and pyridazines. The results obtained with alkylsulfinate salts correspond with published data describing other radical direct functionalizations of heterocycles, such as Minisci chemistry. The potential general applicability of these observations was illustrated by similar regioselectivity effects with the “borono-Minisci” radical arylation. The guidelines established for simple pyridines proved to be equally valid for more complex heterocyclic drugs and agrochemicals.
Further studies to investigate the regioselectivity principles described in this report are ongoing and will be reported in due course. Application of the guidelines described here should aid in the increased application of radical-mediated direct functionalization processes in modern pharmaceutical and agricultural chemistry.
Supplementary Material
Acknowledgments
We thank L. Pasternack and D.-H. Huang for NMR spectroscopic assistance, B. Maryanoff for advice on a draft of this manuscript, and M. R. Collins (Pfizer), A. G. O'Brien, R. D. Baxter, Y. Ishihara, W. R. Gutekunst and H. Lundberg for advice and useful discussion. We are grateful to M. R. Collins (Pfizer) and the Yu, Shenvi and Boger labs for loan of chemicals, and M. Eastgate (BMS) for samples of nevirapine and diflufenican. Financial support for this work was provided by US NIH/NIGMS (GM-073949) and the US-UK Fulbright Commission (postdoctoral fellowship for F.O.).
Abbreviations
- Bpin
4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl
- DFMS
bis(((difluoromethyl)sulfinyl)oxy)zinc
- DMSO
dimethylsulfox-ide
- EDG
electron-donating group
- EWG
electron-withdrawing group
- IPS
bis(((isopropyl)sulfinyl)oxy)zinc
- TBHP
tert-butyl hydroperoxide
- TFA
trifluoroacetic acid
- TFMS
bis(((trifluoromethyl)sulfinyl)oxy)zinc
Footnotes
Supporting Information. Experimental procedures and analytical data for all new compounds, including 1H, 13C, and 19F NMR files. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For an overview of standard functionalization methods, see Joule JA, Mills K. Heterocyclic Chemistry. 5th. Wiley: Chichester; 2010.
- 2.For some recent examples of SNAr chemistry, see: Chen Q, du Jourdin XM, Knochel P. J Am Chem Soc. 2013;135:4958–4961. doi: 10.1021/ja401146v.Bull JA, Mousseau JJ, Pelletier G, Charette AB. Chem Rev. 2012;112:2642–2713. doi: 10.1021/cr200251d.
- 3.For examples of functionalization via heteroarene metallation, see: Haag B, Mosrin M, Ila H, Malakhov V, Knochel P. Angew Chem, Int Ed. 2011;50:9794–9824. doi: 10.1002/anie.201101960.Schlosser M, Mongin F. Chem Soc Rev. 2007;36:1161–1172. doi: 10.1039/b706241a.Gros PC, Fort Y. Eur J Org Chem. 2009;2009:4199–4209.Queguiner G, Marsais F, Snieckus V, Epsztajn J. Adv Heterocycl Chem. 1991;52:187–304.Hartung CG, Snieckus V. Modern Arene Chemistry. Wiley; 2004. pp. 342–344.Miller RE, Rantanen T, Ogilvie KA, Groth U, Snieckus V. Org Lett. 2010;12:2198–2201. doi: 10.1021/ol100493v.
- 4.For recent examples of metal-catalyzed methodology for the C-H functionalization of pyridines see: Murphy JM, Liao X, Hartwig JF. J Am Chem Soc. 2007;129:15434–15435. doi: 10.1021/ja076498n.Takagi J, Sato K, Hartwig JF, Ishiyama T, Miyaura N. Tetrahedron Lett. 2002;43:5649–5651.Liu T, Shao X, Wu Y, Shen Q. Angew Chem, Int Ed. 2012;51:540–543. doi: 10.1002/anie.201106673.Nakao Y, Yamada Y, Kashihara N, Hiyama T. J Am Chem Soc. 2010;132:13666–13668. doi: 10.1021/ja106514b.Wasa M, Worrell BT, Yu JQ. Angew Chem, Int Ed. 2010;49:1275–1277. doi: 10.1002/anie.200906104.Ye M, Gao GL, Edmunds AJF, Worthington PA, Morris JA, Yu JQ. J Am Chem Soc. 2011;133:19090–19093. doi: 10.1021/ja209510q.Ye M, Gao GL, Yu JQ. J Am Chem Soc. 2011;133:6964–6967. doi: 10.1021/ja2021075.Berman AM, Lewis JC, Bergman RG, Ellman JA. J Am Chem Soc. 2008;130:14926–14927. doi: 10.1021/ja8059396.Campeau LC, Rousseaux S, Fagnou K. J Am Chem Soc. 2005;127:18020–18021. doi: 10.1021/ja056800x.Cho SH, Hwang SJ, Chang S. J Am Chem Soc. 2008;130:9254–9256. doi: 10.1021/ja8026295.Lewis JC, Bergman RG, Ellman JA. J Am Chem Soc. 2007;129:5332–5333. doi: 10.1021/ja070388z.Nakao Y, Kanyiva KS, Hiyama T. J Am Chem Soc. 2008;130:2448–2449. doi: 10.1021/ja710766j.
- 5.For a review on applications of Minisci chemistry in medicinal chemistry see: Duncton MAJ. MedChemComm. 2011;2:1135–1161.; Intramolecular examples of homolytic aromatic substitutions on heterocycles are more commonly used in synthesis, see for example: Bowman WR, Storey JMD. Chem Soc Rev. 2007;36:1803–1822. doi: 10.1039/b605183a.Harrowven DC, Sutton BJ, Coulton S. Org Biomol Chem. 2003;1:4047–4057. doi: 10.1039/b309331j.
- 6.(a) Ji Y, Brückl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc Natl Acad Sci U S A. 2011;108:14411–14415. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fujiwara Y, Dixon JA, Rodriguez RA, Baxter RD, Dixon DD, Collins MR, Blackmond DG, Baran PS. J Am Chem Soc. 2012;134:1494–1497. doi: 10.1021/ja211422g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fujiwara Y, Dixon JA, O'Hara F, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herle B, Sach N, Collins MR, Ishihara Y, Baran PS. Nature. 2012;492:95–99. doi: 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhou Q, Ruffoni A, Gianatassio R, Fujiwara Y, Sella E, Shabat D, Baran PS. Angew Chem, Int Ed. 2013;52:3949–3952. doi: 10.1002/anie.201300763. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) O'Hara F, Baxter RD, O’Brien AG, Collins MR, Dixon JA, Fujiwara Y, Ishihara Y, Baran PS. Nat Protoc. 2013;8:1042–1047. doi: 10.1038/nprot.2013.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The zinc sulfinate reagents used in this report, and other related alkylsulfinate salts, are commercially available from Aldrich, with the following catalog numbers: TFMS (trifluoromethylation reagent) 771406; DFMS (difluoromethylation reagent) 767840; IPS (isopropylation reagent) L511161.
- 8.For a comprehensive review of the Minisci reaction, which is the most widely studied system, see: Punta C, Minisci F. Trends Heterocycl Chem. 2008;13:1–68.
- 9.Brückl T, Baxter RD, Ishihara Y, Baran PS. Acc Chem Res. 2012;45:826–839. doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For an example of use of an acid additive to induce a regiochemical preference in Gomberg-Bachmann phenylation of pyridine, and calculations that showed that this selectivity correlated with the localization energy, see: Lynch BM, Chang HS. Tetrahedron Lett. 1964;5:2965–2968.Dou HJM, Lynch BM. Tetrahedron Lett. 1965;6:897–901.Brown RDJ. Chem Soc. 1956:272–275.
- 11.For a study on the solvent effect in the α to γ regioselectivity of substitution of protonated pyridines for a variety of radicals, see: Minisci F, Vismara E, Fontana F, Morini G, Serravalle M, Giordano C. J Org Chem. 1987;52:730–736.
- 12.The more pronounced solvent effect observed for more nucleophilic radicals was attributed to the enhanced contribution from polar forms of the intermediate radical adduct, see: Minisci F, Bernardi R, Bertini F, Galli R, Perchinummo M. Tetrahedron. 1971;27:3575–3579.Palla G. Tetrahedron. 1981;37:2917–2919.
- 13.For a study of the substituent effect on the rate of C2 exclusive substitution of C4-substituted pyridines, see: Minisci F, Mondelli R, Gardini GP, Porta O. Tetrahedron. 1972;28:2403–2413.; The rate of C2 substitution of C4-substituted pyridines closely followed the electron density, with the exception of the OMe and Cl substrates. This was attributed to the “enhanced electron-releasing effects” associated with the C4 substituent lone pair interacting with the pyridine nitrogen, see: Belli ML, Illuminati G, Marino G. Tetrahedron. 1963;19:345–355.
- 14.For a study of substituent effect on the rate of radical phenylation of C4 substituted pyridines, see: Minisci F, Vismara E, Fontana F, Morini G, Serravalle M, Giordano C. J Org Chem. 1986;51:4411–4416.; The rate at C3 was generally higher than that at C2 under neutral conditions, but when protonated, the rate at C2 far exceeded that at C3. See also a similar result showing that the increase in reactivity of pyridine on protonation is due to enhanced rate at the C2 and C4 positions, while the rate at C3 shows little change: Bonnier JM, Court J. Compt Rend. 1967;265:C133–136.
- 15.For an observation of an apparent ortho-para directing effect in the cyclohexylation or dioxanylation of acetyl, ester or cyano substituted pyridines under neutral conditions, which was attributed to a preference for reactivity at positions where the unpaired electron can be delocalized into the substituent for additional stability, see: Tiecco M. In: NATO Advanced Research Workshop on Substituent Effects in Radical Chemistry. Viehe HG, Janousek Z, Merényi R, editors. Vol. 189. D. Reidel Publishing Company; Louvain-la-Neuve, Belgium: 1986. pp. 435–442.Chianelli D, Testaferri L, Tiecco M, Tingoli M. Tetrahedron. 1982;38:657–663.; For a related result by Palla, see reference 12b. Very low yields of selective C3 substitution were reported for the reaction of 4-cyanoquinoline with dioxanyl and a-amidoalkyl radicals generated from solvent. This was attributed to the CN group activating homolytic substitution more than the nuclear nitrogen.
- 16.For an example of radical methylation of 3-picoline showing a strong preference for C2 substitution, see: Abramovitch RA, Kenaschuk K. Can J Chem. 1967;45:509–513.
- 17.For an example of substituent effects in the Minisci alkylation of unsymmetrical 3,6-disubstituted pyridines, see: Cowden CJ. Org Lett. 2003;5:4497–4499. doi: 10.1021/ol035814+.; The regioselectivity was attributed to a preference for reaction ortho to the more electronegative substitutent.
- 18.Studies into the regioselectivity of radical substitution of arenes has also shown substituent effects, see: Shelton JR, Uzelmeier CW. J Am Chem Soc. 1966;88:5222–5228.
- 19.The previous reports describing the production of C3 products involve altering the means of radical generation to in situ hydrogen abstraction from a vast excess of solvent. Alkylsulfinate salts may be used to append a variety of alkyl and fluoroalkyl groups to heterocycles, including several which would not be plausible to generate from solvent excess, such as the pharmaceutically valuable CF3 and CF2H groups.
- 20.All the reactions appeared to become acidic during the reaction, and addition of base (calcium carbonate) hindered reaction progress. Nevertheless, addition of stoichiometric quantities of strong acid had a significant effect on rate and regioselectivity.
- 21.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. [Google Scholar]
- 22.Sterics will also play a role in determining the observed C3:C2 ratio, but this appears to be a relatively minor affect
- 23.(a) Viehe HG, Janousek Z, Merényi R, Stella L. Acc Chem Res. 1985;18:148–154. [Google Scholar]; (b) Viehe HG, Merényi R, Stella L, Janousek Z. Angew Chem Int Ed Engl. 1979;18:917–932. [Google Scholar]; (c) we thank a referee for this suggestion. [Google Scholar]
- 24.De Vleeschouwer F, Van Speybroeck V, Waroquier M, Geerlings P, De Proft F. Org Lett. 2007;9:2721–2724. doi: 10.1021/ol071038k. [DOI] [PubMed] [Google Scholar]
- 25.Minisci F. In: NATO Advanced Research Workshop on Substituent Effects in Radical Chemistry. Viehe HG, Janousek Z, Merényi R, editors. Vol. 189. D. Reidel Publishing Company; Louvain-la-Neuve, Belgium: 1986. pp. 391–434. [Google Scholar]
- 26.Langlois BR, Laurent E, Roidot N. Tetrahedron Lett. 1991;32:7525–7528. [Google Scholar]
- 27.It is important to note that the δ+ sites identified in this manner indicate only that the position may be capable of reacting with a nucleophilic radical, not that it must do so. Furthermore, the magnitude of the S charge on a position does not generally relate to relative reactivity in any straightforward manner. Simple calculations of Hückel charges failed to adequately predict regioselectivity, as did examination of the proton and carbon NMR spectra of substrates. Although more sophisticated calulations may be able to better model the reaction, our aim was to provide a method of predicting regioselectivity and reaction outcome in a quick and simple manner
- 28.(a) Fleming I. Frontier Orbitals and Organic Chemical Reactions. 1st. Wiley; London: 1976. pp. 192–193. [Google Scholar]; (b) Fleming I. Molecular Orbitals and Organic Chemical Reactions: Reference Edition. 1st. Wiley; London: 2010. pp. 284–285. [Google Scholar]; The ionization potential of trifluoromethyl radical is reported as 9.05 eV in Asher RL, Ruscic B. J Chem Phys. 1997;106:210–221. corresponding to a SOMO energy of -9.05 eV. The SOMO energy of a secondary alkyl radical (s-Bu) is given as -7.4 eV in (a)
- 29.Chmurzyhski L. J Heterocycl Chem. 2000;37:71–74. [Google Scholar]
- 30.Minisci F, Vismara E, Morini G, Fontana F, Levi S, Serravalle M, Giordano C. J Org Chem. 1986;51:476–479. [Google Scholar]
- 31.Litwinienko G, Beckwith ALJ, Ingold KU. Chem Soc Rev. 2011;40:2157–2163. doi: 10.1039/c1cs15007c. [DOI] [PubMed] [Google Scholar]
- 32.De Vleeschouwer F, Geerlings P, Proft F. Theor Chem Acc. 2012;131:1–13. [Google Scholar]
- 33.This does not address the overwhelming preference for production of 2,5-dialkylated products in DMSO, or the similar pattern observed under non-protonative conditions by Tiecco and Testaferri
- 34.For a clear example of the impact of steric effects on the regioselectivity of the Minisci reaction, see: Pfleger K, Fuchs W, Pailer M. Monatsh Chem. 1978;109:597–602.
- 35.Further examples of the prediction and explanation of regiochemistry for 1, 11, 12, 15, 16, 17, 18, 28 and 29 may be found in the Supporting Information
- 36.NMR analysis of the reaction mixture for the 2-chloro derivatives 1 and 22 both showed only starting material and products for i-Pr substitution, but when purification was attempted after workup, numerous products were obtained. It is possible that the 2-chloro functionality is unstable on silica with electron-rich alkyl pyridine derivatives
- 37.Although DMSO/H2SO4 had given a promising C2:C3 ratio in early testing (Figure 3), there was a reduction in yield associated with removal of DMSO by aqueous washing from small, water soluble pyridine products. As a result the use of DMSO was limited to cases where “conjugate reactivity” was specifically needed. The use of DMSO/acid solvent mixtures may, however, be useful if the molecule under consideration has poor solubility in chlorinated solvent/water mixtures
- 38.Chloroform was substituted for dichloromethane to accommodate the increase in reaction temperature and facilitate NMR studies. This substitution did not make a significant difference in the results obtained
- 39.Trifluoromethyl radical did not show synthetically useful reactivity with many of these electron-poor diazines; therefore, although the same regioselectivity trends as described for i-Pr radical were observed, these results were not reported in this study. Electrophilic radicals were generally less regioselective for the most polar (C4) site on pyridazines and pyrimidines, although the same preference was still followed. Examination of the literature also showed that, for the limited range of examples showing substitution of a diazine with a somewhat electrophilic radical, a similar pattern to that observed with pyridines was seen
- 40.(a) Rao CJ, Agosta WC. J Org Chem. 1994;59:2125–2131. [Google Scholar]; (b) Sato N, Kadota H. J Heterocycl Chem. 1992;29:1685–1688. [Google Scholar]; (c) Opletalová V, Patel A, Boulton M, Dundrová A, Lacinová E, Převorová M, Appeltauerová M, Coufalová M. Collect Czech Chem Commun. 1996;61:1093–1101. [Google Scholar]; (d) Tada M, Totoki S. J Heterocycl Chem. 1988;25:1295–1299. [Google Scholar]
- 41.(a) Heinisch G, Lötsch G. Tetrahedron. 1985;41:1199–1205. [Google Scholar]; (b) Altenbach RJ, Adair RM, Bettencourt BM, Black LA, Fix-Stenzel SR, Gopalakrishnan SM, Hsieh GC, Liu H, Marsh KC, McPherson MJ, Milicic I, Miller TR, Vortherms TA, Warrior U, Wetter JM, Wishart N, Witte DG, Honore P, Esbenshade TA, Hancock AA, Brioni JD, Cowart MD. J Med Chem. 2008;51:6571–6580. doi: 10.1021/jm8005959. [DOI] [PubMed] [Google Scholar]; (c) Sakamoto T, Ono T, Sakasai T, Yamanaka H. Chem Pharm Bull. 1980;28:202–207. [Google Scholar]; (d) Sakamoto T, Sakasai T, Yamanaka H. Chem Pharm Bull. 1980;28:571–577. [Google Scholar]; (e) Brumfield MA, Agosta WC. J Am Chem Soc. 1988;110:6790–6794. [Google Scholar]; (f) Kos NJ, van der Plas HC, van Veldhuizen A. Recl Trav Chim Pays-Bas. 1980;99:267–270. [Google Scholar]; (g) Togo H, Aoki M, Kuramochi T, Yokoyama M. J Chem Soc, Perkin Trans 1. 1993:2417–2427. [Google Scholar]; (h) Togo H, Ishigami S, Fujii M, Ikuma T, Yokoyama M. J Chem Soc, Perkin Trans 1. 1994:2931–2942. [Google Scholar]; (i) Heinisch G, Lötsch G. Heterocycles. 1984;22:1395–1402. [Google Scholar]; (j) Danter WR, Lau CK. Compounds and method for the treatment of cancer. International Patent PCT/CA2008/00229 [Google Scholar]; (k) Heinisch G, Matuszczak B, Mereiter K, Soder J. Heterocycles. 1995;41:1461–1470. [Google Scholar]; (l) Haider N, Mavrokordatou E, Steinwender A. Synth Commun. 1999;29:1577–1584. [Google Scholar]; (m) Ruggiero A, Fuchter MJ, Kokas OJ, Negru M, White AJP, Haycock PR, Hoffman BM, Barrett AGM. Tetrahedron. 2009;65:9690–9693. [Google Scholar]
- 42.(a) Seiple IB, Su S, Rodriguez RA, Gianatassio R, Fujiwara Y, Sobel AL, Baran PS. J Am Chem Soc. 2010;132:13194–13196. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Molander GA, Colombel V, Braz VA. Org Lett. 2011;13:1852–1855. doi: 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Singh PP, Aithagani SK, Yadav M, Singh VP, Vishwakarma RA. J Org Chem. 2013;78:2639–2648. doi: 10.1021/jo302797r. [DOI] [PubMed] [Google Scholar]; (d) Wang J, Wang S, Wang G, Zhang J, Yu XQ. Chem Commun. 2012;48:11769–11771. doi: 10.1039/c2cc35468c. [DOI] [PubMed] [Google Scholar]; (e) Presset M, Fleury-Brégot N, Oehlrich D, Rombouts F, Molander GA. J Org Chem. 2013;78:4615–4619. doi: 10.1021/jo4005519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.