

Abstract
Multipotent human adipose tissue mesenchymal stromal cells (hAMSCs) are promising therapy vehicles with tumor-homing capacity that can be easily modified to deliver cytotoxicity activating systems in the proximity of tumors. In a previous work, we observed that hAMSCs are very effective delivering cytotoxicity to glioma tumors. However, these results were difficult to reconcile with the relatively few hAMSCs surviving implantation. We use a bioluminescence imaging (BLI) platform to analyze the behavior of bioluminescent hAMSCs expressing HSV-tTK in a U87 glioma model and gain insight into the therapeutic mechanisms. Tumor-implanted hAMSCs express the endothelial marker PECAM1(CD31), integrate in tumor vessels and associate with CD133-expressing glioma stem cells (GSC). Inhibition of endothelial lineage differentiation in hAMSCs by Notch1 shRNA had no effect on their tumor homing and growth-promoting capacity but abolished the association of hAMSCs with tumor vessels and CD133+ tumor cells and significantly reduced their tumor-killing capacity. The current strategy allowed the study of tumor/stroma interactions, showed that tumor promotion and tumor-killing capacities of hAMSCs are based on different mechanisms. Our data strongly suggest that the therapeutic effectiveness of hAMSCs results from their association with special tumor vascular structures that also contain GSCs.
Introduction
Glioblastoma multiforme is a highly malignant brain tumor, with patient median survival of less than a year.1,2 This poor prognosis is related to the capacity of gliomas to induce neovascularization and infiltrate normal brain tissue following vascular tracks, leading to tumor microsatellites that grow distant from the main tumor mass.3,4
Despite major improvements in surgical technology, the high brain and spinal cord infiltration capacity of glioma cells prevents their complete resection. Increasing malignancy of gliomas also correlates with an increase in cellularity and a poorly organized tumor vasculature. Insufficient blood supply, hypoxic areas and ultimately the formation of necrosis limit drug diffusion and result in higher resistance to chemo and radiotherapy.5
Recent studies have shown that a subpopulation of tumor cells with stem cell characteristics (GSCs), including self-renewal capacity, is responsible for tumor initiation, progression, and infiltration of adjacent normal parenchyma and is also responsible for tumor recurrences after surgery, radio and chemoresistance.6 It has been described that in brain tumors, GSCs occupy special structures next to capillaries, a niche similar to that of neural stem cells that is critical for their maintenance.7,8
Some therapeutic approaches avoiding systemic exposure to cytotoxic agents have been based on the so-called “sucide gene therapy”. In these procedures, a gene coding for an enzyme that converts a nontoxic prodrug into a lethal compound is delivered to the tumor where it destroys not only the delivery vehicle but also neighboring cells (bystander effect).9 Thus far, one of the most successful enzyme systems tested in vitro and in animal models has been the HSV-thymidine kinase (HSV-tk) (highest number of entrances in Pubmed). This enzyme has a high affinity for the deoxyguanosine analog ganciclovir (GCV)10,11 prodrug and converts it to the phosphorilated form deoxyguanosine triphosphate, a polymerase-I inhibitor and DNA chain terminator. Incorporation of the nucleotide analog in the growing DNA chains, results in cell death. Gap junction-mediated uptake of the activated prodrug and other cell products extends and amplifies the toxic effect, killing multiple neighboring tumor cells.12,13 A key factor determining the efficiency of this therapy is the system used to deliver the activating enzyme to the tumor proximity.
Mesenchymal stromal cells (MSCs) have recently emerged as potential delivery vehicles for HSV-tk due to their glioma-homing capacity, following intra-arterial or intracranial injections.14 Human adipose tissue mesenchymal stromal cells (HAMSCs) can be genetically manipulated with relative ease;15 have the capacity to modulate immune and inflammatory reactions16 and do not generate tumors when implanted during long periods of time.17 In addition, their abundance in human adipose tissue and their ease of preparation make hAMSCs particularly attractive drug delivery vehicles. Therapeutic strategies based on the use of hAMSCs genetically modified to express CD-UPRT and HSV-tk, have shown to exert inhibition of glioma growth.18,19,20
It has been suggested that bone marrow-derived MSCs associate with vascular structures in rat glioma tumors where they appear to integrate and express pericyte markers.21,22 In our own work, luciferase-eGFP expressing hAMSCs implanted in a human U87 glioma model in mice, also associated with tumor vascular structures, but expressed the CD31 endothelial marker.19 In our studies, implantation in tumors of hAMSCs expressing the HSV-tTK cytotoxic gene resulted in a noticeable increase in tumor growth in the absence of GCV. Nevertheless, induction of toxicity by GCV treatment resulted in dramatic killing of tumor cells. These results lead us to suggest that integration in the tumor vascular system of endothelial lineage differentiated hAMSCs placed them in a strategic position to promote tumor growth, but also to deprive the tumor of nutrients when their cytotoxic capacity was activated by GCV administration.
Notch1 signaling is involved in a number of processes including vascular/endothelial differentiation. Previous studies showed that inhibition of Notch signaling by a γ-secretase inhibitor abolishes differentiation of bone marrow-derived MSC.23 In the current work, we use hAMSCs stably transduced to express αNotch1 shRNA to explore the interaction of hAMSCs with the tumor, in an effort to better understand the basis of their therapeutic effectiveness.
Results
Bioluminescence and fluorescence labeling of hAMSCs
To analyze the behavior of tumor cells and implanted hAMSCs, we adopted a dual-luciferase labeling strategy that allowed independent imaging of cell number, distribution and differentiation.
Human glioblastoma U87 tumor cells were genetically modified, when required, by transduction with a lentiviral vector for stable expression of a chymeric reporter with Photinus pyralis luciferase (PLuc) and enhanced green fluorescent protein (EGFP) activities, generating PL-G-U87 tumor cells.
To genetically modify hAMSCs, the cells were transduced with a lentiviral vectors for stable expression of a trifunctional chimerical reporter including Renilla reniformis luciferase (RLuc), red fluorescent protein (RFP), and herpes virus-truncated thymidine kinase (tTK) activities regulated by the CMV promoter (RL-R-tTK-AMSCs).
For cell differentiation studies, positively transduced hAMSCs expressing RFP were selected by FACS and labeled with a second lentiviral chimerical construct comprising the chimerical coding sequence for Photinus luciferase and Green Fluorescent Protein (PLuc-GFP), under transcriptional regulation by the PECAM-promoter (PECAM:PL-G/RL-R-tTK-hAMSCs). Due to the use of noncrossreacting substrates, coelenterazin and luciferin, both luciferases can be imaged independently. Expression of RFP and GFP allows detection of implanted and differentiated cells by fluorescence confocal microscopy in histological preparations. Moreover, changes in PECAM-regulated luciferase expression could be quantified in relation to RLuc activity regulated by the constitutive CMV promoter, a measure for cell number, avoiding in this manner potential artifacts due to cell death or proliferation.
In experiments where the contribution of hAMSC differentiation to tumor therapy was analyzed, we inhibited the Notch signaling pathway by transducing hAMSCs with a lentivirus for expression of αNotch1 shRNA; what generated αNotch1-RL-R-tTK-hAMSCs and αNotch1-PECAM:PL-G/RL-R-tTK-hAMSCs. Two additional sets of control cells, Ф-RL-R-tTK-hAMSC and Ф-PECAM:PL-G/RL-R-tTK-hAMSC, transduced with the empty pLKO.1-puro vector were also generated.
In vitro validation of vascular differentiation-inhibition by αNotch1 shRNA
A Matrigel assay was used to assess the effect of αNotch shRNA on tube formation capacity of hAMSCs. While positive control PECAM:PL-G/RL-R-tTK-hAMSCs and mock transduced Ф-PECAM:PL-G/RL-R-tTK-hAMSCs are capable of forming tubes in Matrigel, αNotch1-PECAM:PL-G/RL-R-tTK-hAMSC completely lack tube formation capacity (three different αNotch1 clones were tested with similar results, data not shown) Figure 1a. The clone with the highest level of tube formation inhibition was selected for further experiments. Real-time PCR quantification of mRNA from the same cells following the Matrigel tube formation assay, showed that the level of mRNA expression for Notch1 and other endothelial specific markers, including the endothelial marker PECAM, was significantly reduced in αNotch1-PECAM:PL-G/RL-R-tTK-hAMSC relative to positive control PECAM:PL-G/RL-R-tTK-hAMSCs and to mock transduced Ф-PECAM:PL-G/RL-R-tTK-hAMSC (Figure 1b). However, no significant reduction in the expression level of the Notch1-independent BGLAP/Osteocalcin gen was observed in either of the three cell types. These results validated the Matrigel assay results showing that αNotch1 shRNA effectively inhibits expression vascular lineage genes in these cells.
Figure 1.

In vitro tube formation by hAMSC requires Notch1 expression. Phase contrast photomicrographs (a) of untransduced (left); αNotch1 shRNA transduced (middle) and mock ΦshRNA transduced (right) PECAM:PL-G/RL-R-tTK-hAMSCs incubated in Matrigel for 4 hours. (b) RNA extracted from cells in matrigel was assayed by RT-PCR to determine transcription the levels of the indicated endothelial (ILK, SDF1, EGR3,PECAM-1, VEGFA-1, Notch-1) and bone BGLAP/Octeocalcin differentiation markers. The histogram shows % fold change relative to the control un-transduced PECAM:PL-G/RL-R-tTK-hAMSCs. *P < 0.05; n = 3.
In vivo imaging of vascular lineage differentiation inhibition by Notch1 silencing
Unlabeled U87 glioma tumor cells were implanted in the brain of SCID mice together with either double-labeled αNotch1-PECAM:PL-G/RL-R-tTK-hAMSCs or double-labeled Ф-PECAM:PL-G/RL-R-tTK-hAMSCs and monitored by bioluminescence imaging (BLI) (Figure 2). After 7 days postimplantation, imaging of cell distribution and number shows that a large proportion of both types of implanted cells have disappeared or dispersed from the implantation sites (Figure 2 a2, b2). However, a subpopulation of the implanted cells appears to remain within the tumor. For these tumor-associated cells, imaging of the endothelial differentiation reporter PECAM-PLuc shows little activity from αNotch1-PECAM:PL-G/RL-R-tTK-hAMSCs but a considerable increase from Ф-PECAM:PL-G/RL-R-tTK-hAMSC cells transduced with empty control vector (Figure 2 a4, b4). For each of the two cell types, the level of differentiation can be quantified by calculating the ratio of light photons produced by the differentiation reporter (PECAM-PLuc) divided by those generated by the constitutively expressed reporter (CMV-RLuc) representing cell number. As shown in the histogram (Figure 2c), αNotch-inhibited cells increased their endothelial differentiation level by a 4x factor in the 7-day implantation period, while mock inhibited cells increased by a 57-fold factor in the same time period. These results indicate that while differentiation to the endothelial lineage was active in control mock inhibited hAMSCs, it was effectively reduced (more than 10x) in cells transduced with αNotch shRNA.
Figure 2.

Endothelial differentiation of hAMSCs in U87 glioma tumors requires Notch1 expression. hAMSCs expressing PECAM-promoter regulated PLuc-GFP and CMV-promoter regulated RL-R-tTK were transduced with either αNotch1 shRNA or a mock ΦshRNA to produce αNotch1-PECAM:PL-G/RL-R-tTK-hAMSCs and control Φ-PECAM:PL-G/RL-R-tTK-hAMSCs, respectively. Independent mixtures comprising unlabeled U87 glioma cells and one of the above cell types in a ratio of 1:4 (U87:hAMSC) were implanted at specific brain coordinates and imaged to determine cell number and distribution (a1,3; b1,3), or differentiation to the endothelial lineage (a2,4 and b2,4) by BLI of Renilla and Photinus luciferases, respectively. Pseudocolor images are superimposed on black and white dorsal images of recipient mice. Color bars illustrate relative light intensities of PLuc (right) and RLuc (left); low: blue and black; high: red and blue, respectively. The ratio between photons acquired from Photinus luciferase and Renilla luciferase generated images (PLuc/RLuc) was used to evaluate differentiation of implanted cells to the endothelial lineage (c). *P < 0.05; n = 4.
Following BLI analysis, mice were perfused using a high (2,000,000 MW) fluorescein-conjugated dextran, sacrificed and fixed in formaldehyde. Analysis by fluorescence laser confocal microscopy of tumor slices revealed RFP expressing Ф-PECAM:PL-G/RL-R-tTK-hAMSCs with endothelial morphology and in close association with tumor vascular structures labeled with fluorescein (Figure 3a top). However, RFP expressing αNotch1-PECAM:PL-G/RL-R-tTK-hAMSCs were found less frequently associated with vascular structures (Figure 3a bottom) as shown by the histogram in Figure 3b. These results indicate that inhibition of endothelial lineage differentiation by αNotch shRNA also interferes with the participation of hAMSCs in the formation of tumor vascular structures. The endothelial, but not pericytic lineage of hAMSCs in the tumor vasculature, was further supported by immunohistochemical analysis showing (see Supplementary Figure S1 and Supplementary Materials and Methods online) that Ф-PECAM:PL-G/RL-R-tTK-hAMSCs implanted in combination with unlabeled U87 cells, expressed the PECAM1 protein that could be detected with an endothelial specific antibody, but did not express SM22 antigen, a generally accepted pericyte specific marker.
Figure 3.
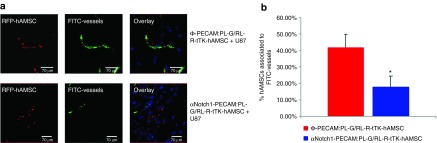
Association of hAMSCs with tumor microvessels is Notch1 dependent. Representative laser confocal microscope images of slices from brain tumors implanted with αNotch1-PECAM:PL-G/RL-R-tTK-hAMSCs or control Φ-PECAM:PL-G/RL-R-tTK-hAMSCs. (a) Microphotographs show the location of RFP expressing control Φ-PECAM:PL-G/RL-R-tTK-hAMSCs (top) and inhibited αNotch1-PECAM:PL-G/RL-R-tTK-hAMSCs (bottom) relative to FITC-labeled microvessel (gree). (b) Histogram representing the percentage of red hAMSCs associated to FITC-labeled microvessel for Φ-PECAM:PL-G/RL-R-tTK-hAMSCs and αNotch1-PECAM:PL-G/RL-R-tTK-hAMSCs. Bars represent mean ± SEM of percent values. *P < 0.05; n = 25 (tumor slices analyzed).
Human AMSCs expressing HSV tTK gene are effective vehicles to deliver bystander cytotoxic effect to U87 glioma cells in vitro independently of Notch1 expression
Human glioma U87 cells were transduced with a lentiviral construct for the expression of Photinus Luciferase and GFP as a chimerical protein regulated by the CMV promoter. Following selection by FACS, positively transduced glioma cells (PL-G-U87) were mixed in a 1:5 proportion with either Ф-RL-R-tTK-hAMSCs or αNotch1-RL-R-tTK-hAMSCs, cultivated as described, and treated with the prodrug GCV 4 mg/ml or PBS. To monitor the effect of GCV on cell number, cocultures were subjected to BLI following luciferin administration. Mixtures of Ф-RL-R-tTK-hAMSCs or αNotch1-RL-R-tTK-hAMSCs plus PL-G-U87 cells grow well in the absence of GCV (Figure 4). However, GCV treatment had a clear cytotoxic effect and resulted in a nearly complete abolition of PL-G-U87 cell growth (*P < 0.01), regardless of the accompanying hAMSC type. This result indicates that both Ф-RL-R-tTK-hAMSCs and αNotch1-RL-R-tTK-hAMSCs are equivalently effective at delivering a cytotoxic effect to U87 cells, and that such effect, when assayed in vitro is independent of their vascular differentiation capacity.
Figure 4.
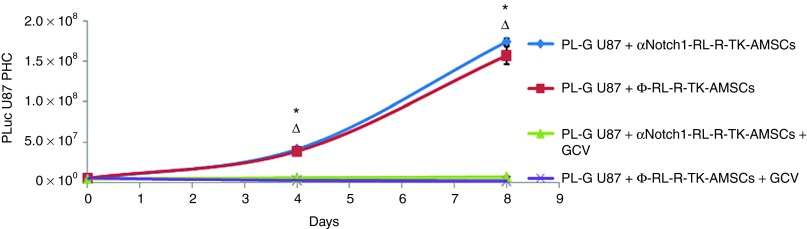
Expression of TK cytotoxicity is independent of Notch1 expression. PL-G-U87cells were grown during the indicated times in combination with either Φ-RL-R-tTK-hAMSCs or αNotch1-RL-R-tTK-hAMSCs in a 5:1 proportion, and treated with GCV (4 μg/ml), as indicated. Cell viability was evaluated by quantification of luciferase activity by BLI at the indicated times, and expressed as PL-G-U87 PHCs. The graph shows mean ± SEM, *P < 0.01 Φ-RL-R-tTK-hAMSCs+GCV-treated group versus Φ-RL-tTK-hAMSC control group, ΔP < 0.01 αNotch1-RL-R-tTK-hAMSCs+GCV-treated group versus αNotch1-RL-R-tTK-hAMSC control group, n = 3 for each group.
The capacity of tTK expressing hAMSCs to deliver bystander cytotoxic killing to U87 tumors is dependent on their capacity to differentiate to the endothelial lineage
Next, we explored the relation between vascular differentiation of hAMSCs and their capacity to deliver cytotoxic therapy to U87 tumors. For this, we compared the therapeutic effectiveness of Ф-RL-R-tTK-hAMSC and αNotch1-RL-R-tTK-hAMSC in a model of glioblastoma described previously.19 A group of 12 6-week-old SCID mice was stereotactically inoculated in the brain with 4 × 104 PL-G-U87 alone or with a mixture of 4 × 104 PL-G-U87 and either 1.6 × 105 Ф-RL-R-tTK-hAMSCs or the same number of αNotch1-RL-R-tTK-hAMSCs. Light emission by PL-G-U87 cells was monitored by BLI beginning on the implantation day, and every week thereafter. Seven days after implantation, the treatment with the GCV prodrug was initiated as described in methods (Animal procedures and experimental design). Representative BLI results (Figure 5b) and their quantitative evaluation, based on counting image-captured photons, showed (Figure 5a) that growth of U87 tumors with either Ф-RL-R-tTK-hAMSC or αNotch1-RL-R-tTK-hAMSC cells was similar up to the initiation of GCV treatment on day 6 and considerably faster (75-fold) than that of tumors without hAMSCs. However, by day 14, only 7 days after initiation of GCV treatment, the number of cells in tumors with Ф-RL-R-tTK-hAMSCs had dropped considerably and by day 21, the number of tumor cells had been reduced by 94% relative to that at day 6. Tumors with αNotch1-RL-R-tTK-hAMSCs responded with only a 36% drop after GCV treatment in 21 days, and showed a slight growth trend within the same time period. These results show that the tumor growth promoting capacity of hAMSCs and their capacity to deliver bystander therapy are based on independent mechanisms and that vascular differentiation of hAMSCs is required for delivery of bystander therapy.
Figure 5.

Delivery of cytotoxicity by HAMSC to U87 gliomas in vivo is Notch1 dependent. Glioma PL-G-U87 cells (4 × 104) mixed with either αNotch1-RL-R-tTK-hAMSCs or Φ-RL-R-tTK-hAMSCs (1.6 × 106), or no-hAMSCs, were implanted (methods) in the brain of mice, and at day 7 p.i. a daily GCV treatment (100 mg/kg) was initiated. Tumor growth was monitored at implantation day and every 7 days thereafter. (a) The graph shows changes in tumor generated luciferase activity calculated from the recorded images, represented as log10 mean ± SEM. *P < 0.05; n = 8. (b) Composite pseudo-color BLI images from representative mice.
Glioma hAMSC interactions
In an attempt to understand the mechanism by which the Ф-RL-R-tTK-hAMSCs + GCV treatment achieves the observed high degree of therapeutic effect, we studied the association of therapeutic cells and GSC in tumors. 6-week-old SCID mice were stereotactically inoculated with a mixture of 4 × 104 PL-G-U87 cells and either 1.6 × 105 Ф-RL-R-tTK-hAMSCs or the same number of αNotch1-RL-R-tTK-hAMSCs. One week later, mice were sacrificed and brains harvested, fixed, prepared for immunohistochemestry and stained with a monoclonal antihuman CD133 antibody, a glioma stem cell marker. Fluorescence confocal microscope analysis was used to count associations between red fluorescent hAMSCs, green fluorescent tumor cells and green fluorescent tumor cells also expressing the CD133 antigen. Our results showed frequent GFP expressing tumor cells that positively stained with the GSC marker, and were also in close association with RFP expressing Ф-RL-R-tTK-hAMSCs (42.6 ± 5.5%) (Figure 6a, top). Although associations were also detected in the tumors inoculated with αNotch1-RL-R-tTK-hAMSCs (Figure 6a, bottom), in the latter case, they were significantly less frequent (18.68 ± 5.53%) P < 0.05 (Figure 6b). There was no difference in the total number of GSC and hAMSCs in either case. These results suggest that the interaction between hAMSCs and tumor stem cells is related to the vascular differentiation capacity of the former.
Figure 6.

Co-localization of hAMSCs and U87 cancer stem cells in gliomas. (a) Tumors generated by implanting 4 × 104 PL-G-U87 cells and either 1.6 × 105 Φ-RL-R-tTK-hAMSCs (top row) or the same number of αNotch1-RL-R-tTK-hAMSCs (bottom row) were excised after one week of growth, sectioned and incubated with an anti-human-CD133 ab. Representative laser scanning confocal microscope images show hAMSCs (red fluorescent), U87 glioma cells (green fluorescent) and CD133+ GSCs (grey). Hoechst nuclear stain in blue. Insets are higher magnification images corresponding to the boxed regions in merged images emphasizing interactions between CD133+ GSCs (white arrows) and hAMSCs; Scale bar= 20 μm. (b) Histogram representing the percentage of PL-G-U87 GSCs associated to hAMSC in the case of Φ-RL-R-tTK-hAMSCs and αNotch1-RL-R-tTK-hAMSCs. Bars represent mean ± SEM of percent values. *P < 0.05; n = 15.
Additionally, RFP-expressing Ф-RL-R-tTK-hAMSCs were also found in association with GFP-expressing tumor cells that positively stained with an antibody against Sox2, another GSC marker (see Supplementary Figure S2 and Supplementary Materials and Methods online).
Discussion
MSC from adipose tissue and bone marrow have been used as therapy-delivering vehicles in various types of experimental cancers.24,25,26,27
In previous work, we showed that the inclusion of hAMSCs expressing HSV thymidine kinase and luciferase reporters in prostate10 and glioma tumors19 results in a significant promotion of tumor growth compared with tumors without hAMSCs. However, induction of cytotoxicity by administration of the prodrug GCV results in very effective tumor killing, capable of reducing tumor cell burden by a 10-4 factor relative to untreated controls. Surprisingly, while at implantation time, the proportion of hAMSCs to U87 cells was 4:1, due to the rapid growth of tumor cells and the replicative quiescence of hAMSCs, by the start of GCV therapy, the proportion of hAMSCs to tumor cells was only 1:13. Further analysis showed that hAMSCs differentiated within tumors to the endothelial lineage, expressing the CD31 marker and integrating in the tumor vascular system where they adopt an endothelial phenotype.19 Previous studies have reported that bone marrow-derived MSCs implanted in tumors express pericyte but not endothelial markers, and integrate to the tumor vasculature.21 However, MSCs from bone marrow and adipose tissue have also been reported to differentiate to the endothelial lineage and express endothelial markers.28,29
In the current work, we explore the interaction between hAMSCs and U87 glioma in a SCID mouse model, to understand the basis of hAMSC effectiveness to deliver cytotoxic tumor therapy.
Deletion of DII4 receptor or Notch1 during embryogenesis results in vascular anomalies and lethality.30,31 Notch1 signaling is required for endothelial cell differentiation, and inhibition of this pathway in bone marrow-derived MSC has been shown to block their differentiation into functional endothelial cells, and the expression of endothelial-specific markers.23,32
We first showed that, by using αNotch1 shRNA to interfere with Notch1 signaling, we were able to abolish the network formation capacity of hAMSCs in an in vitro matrigel assay, a good indicator of vasculogenic potential.33,34 Loss of network formation capacity was accompanied by significant reduction in the expression of endothelial markers ILK, SDF1, EGR3, PECAM, and Notch itself, in the same cells, but not of osteocalcin, a Notch1-independent bone differentiation marker.
We then modified hAMSCs to express two different bioluminescence reporter systems, one (CD31-PLuc-eGFP) regulated by the CD31 promoter and the other (CMV-RLuc-RFP) regulated by CMV promoter, to measure endothelial differentiation and cell number, respectively. This double-labeling strategy allowed us to evaluate changes in the expression of the differentiation reporter relative to that for cell number, minimizing potential artifacts due to cell death or proliferation.
Luciferase tagged cells were also transduced with lentiviral vectors for the expression of either αNotch shRNA, or an empty ФshRNA, what allowed us to generate hAMSCs that were impaired to differentiate to the endothelial lineage and the corresponding unimpaired control cells, respectively.
By monitoring the activity of luciferase reporters in control Ф-hAMSCs or αNotch-hAMSCs independently implanted with U87 cells in the brain of SCID mice, we were able to observe that a large proportion of the hAMSCs implanted with tumors die or disappear. However, while Ф-hAMSCs that remained associated with the tumor suffered a pronounced increase in the activity of the CD31-regulated luciferase, this was not the case when Notch1 signaling was inhibited in αNotch-hAMSCs. This result was further supported by fluorescence confocal microscope analysis of tissue slices from the same tumors which showed that control Ф-hAMSCs associated with tumor vascular structures significantly more frequently than did αNotch inhibited hAMSCS. These data are supported by a recent study that showed that inhibition of Notch1 deregulated endothelial sprouting and proliferation in a VEGFR3 dependent way, without VEGFR2 signalling.35
While these experiments showed that hAMSC differentiation to the endothelial lineage and association to the vascular system in U87 tumors is Notch1 dependent, they did not exclude the possibility that some hAMSCs in the tumor vascular system may also differentiate to pericytes. However, by staining the slices of U87 tumor generated with RFP expressing hAMSCs with either antihuman PECAM1 or SM22 antibodies, we found that red fluorescent protein expressing hAMSCs exclusively expressed the PECAM1 but not the SM22 antigen, excluding their pericytic lineage.
Notch1 signaling is not required for HSV thymidine kinase expression and we were able to show that in the presence of GCV both, control and αNotch shRNA inhibited hAMSCs are equally cytotoxic, effectively killing U87 tumor cells in vitro. This result allowed us to explore the relation between vascular differentiation and tumor killing capacity of hAMSCs in vivo. Photinus luciferase expressing U87 tumors (PLuc-U87) were implanted together with mock inhibited hAMSC, αNotch inhibited hAMSCs, or no hAMSCs, and allowed to grow during 7 days. Following this tumor establishment period, GCV treatment was initiated and continued during 2 additional weeks. BLI of tumors in live mice clearly showed that both mock-inhibited and αNotch-inhibited hAMSCS had a very effective tumor growth promoting capacity during the first week without GCV, in comparison with tumors without hAMSCs. Although GCV treatment rapidly halted growth of tumors with both hAMSC types, this effect of GCV was significantly more pronounced (96%) in tumors containing mock-inhibited hAMSCs, than in those containing Notch-inhibited hAMSCs (36%) P < 0.05, clearly showing that Notch1 signaling is required for an effective delivery of cytotoxicity by hAMSCs. The residual tumor-killing effect of αNotch-inhibited hAMSCs is likely the result of a lower than 100% efficiency on Notch-signaling inhibition by αNotch shRNA. Moreover, in experiments allowing the visualization of the tumor vascular system, it was possible to show that inhibition of Notch1 in hAMSCs also significantly reduced their association with vascular structures.
Surprisingly, it was also clear from these results that the powerful tumor-promoting effect on orthotopically implanted U87 tumors exerted by hAMSCs is independent of the Notch-signaling pathway.
The high efficacy of tTK-expressing hAMSCs at killing tumor cells suggested that the vascular structures with which these cells interact may also comprise important functions responsible for tumor maintenance other than the vascular system. We used an antibody against the CD133 surface antigen, a stemness marker also expressed by neural stem cells, and by at least a fraction of the glioma stem cell population, to evaluate their association with hAMSCs.
Our results showed a large proportion (42%) of CD133+ U87 cells (putative GSCs) associate with hAMSCs. Moreover, a significantly larger proportion of CD133+ U87 cells associate with RFP expressing hAMSCs than with Notch1-inhibited hAMSCS (42 vs. 18.6%, respectively; P < 0.05). Again, incomplete silencing of hAMSCs by shNotch RNA may be responsible for the observed association of αNotch-hAMSCs with CD133+ GSCs. Thus, a likely explanation for the effectiveness of hAMSC-based therapy could be that by homing to vascular structures that also contain the tumor stem cells, tTK expressing hAMSCs occupy a privileged location from which they can deliver effective cytotoxic damage to the tumor.
Thus, it would appear that: hAMSCs genetically modified to express HSV thymidine kinase are very effective vehicles to deliver bystander suicide therapy to U87 tumors in the presence of the GCV prodrug. However, in the absence of the GCV prodrug, hAMSCs are potent stimulators of tumor growth.
Notch1 expression, required for differentiation of hAMSCs to the endothelial lineage, is also required for their association with tumor vascular structures, with CD133+ U87 (putative glioma stem cells) and for effective bystander killing of tumor cells.
The effectiveness of cytotoxic hAMSCs to deliver tumor therapy is the result of their capacity to associate with privileged vascular structures also populated by tumor stem cells, possibly a cancer stem cell niche, and a privileged location for the delivery of therapy. Nevertheless, other mechanisms related with inflammation and induction of proapoptotic genes as recently proposed may also play an important role in this process.36
Materials and Methods
Vector constructs. Five lentiviral constructs were used to label cells: hrl-mrfp-tk vector containing a chimeric construct of the RLuc reporter gene, the monomeric red fluorescent protein (mRFP1) and a truncated version of the herpes simplex virus thymidine kinase gene sr39tk in a PHR vector, was constructed as described;37 pRRL-PLuc-IRES-eGFP vector containing Photynus pyralis luciferase reported gene (PLuc) and a enhanced green fluorescent protein, was kindly provided by L. Alvarez-Vallina; pLox:Pecam-Luc:eGFP vector for expression of chymeric Photinus pyralis luciferase and enhanced green fluorescent protein under control of the endothelial specific promoter PECAM (CD31) was constructed as described;38 Notch homolog 1 MISSION shRNA Lentiviral Transduction Particle (NM_017617 Sigma, Steinheim, Germany) for expression of anti-Notch1 shRNA in a PLKO.1-puro backbone vector; MISSION pLKO.1-puro Empty Vector Control Plasmid (SHC001, Sigma).
Lentiviral particle production. Production of viral particles was performed using human embryonic kidney cells 293T (ATCC, CRL-11268) grown in Dulbecco's Modified Eagle's Medium-high glucose (DMEM-hg) (Sigma), 10% heat-inactivated fetal bovine serum (FBS) (Sigma), 2 mmol/l L-glutamine (Sigma), 50 units/ml penicillin/streptomycin (Sigma), and 2 mmol/l HEPES. The day previous to transfection, 3 × 106 cells were seeded on 10 cm2 poly-D-lysine (Sigma) treated plates. 6 μg of each lentiviral transfer vector, were mixed with 2 μg of viral envelope plasmid (pMD-G-VSV-G) and 4 μg of packaging construct (pCMV DR8.2) in 250 μl of 150 mmol/l NaCl and then mixed with 48 μl of 1 mg/ml polyethylene amine (Polyscience, Warrington, PA) in 250 μl of 150 mmol/l NaCl, and incubated at room temperature (RT) for 20 minutes. This DNA solution was then added drop wise to the plate containing the 293T cells plus medium, swirled gently and incubated for 16 hours at 37 °C with 5% CO2. The following day, the transfection solution was removed, the cells were rinsed with PBS 1X, and medium without FBS was added to the cells. Following a 48-hour incubation, the supernatant was collected, centrifuged at 2000 rpm to remove cell debris, and filtered through a 0.45 μm low protein-binding filter (Corning, Bath, UK). Cell supernatants were directly used for transduction of cell cultures.
Tumor and primary cell culture. hAMSCs were isolated from adipose tissue derived from cosmetic subdermal liposuctions, with patient consent, as described previously.10 Liposuction samples were obtained after written informed consent by anonymous donors from Hospital de la Santa Creu i Sant Pau, Barcelona, Spain. Work with human samples was approved by the Ethical Committee of Clinical Investigation of Hospital Santa Creu i Sant Pau, Barcelona, Spain; and Bioethical Subcommittee of Superior Council of Scientific Research. Briefly, lipoaspirate was suspended in 1X collagenase type I (Invitrogen, Carlsbad, CA) solution and incubated at 37 °C and inactivated by addition of DMEM + 10% FBS. hAMSC were isolated by the plastic adherence technique. hAMSCs were grown in DMEM-hg with 20% FBS (Hyclone, Logan, UT), 2 mmol/l L-glutamine (Sigma) and 50 units/ml penicillin/strepotomycin (Sigma), expanded for until 70% of confluence.
Selected adherent cells were expanded in the same medium and functionally characterized by demonstrating their capacity for in vitro differentiation to the chondrogenic, adipogenic and osteogenic lineages. The endothelial differentiation capacity of hAMSC in vitro and in vivo were described in previous articles.38,39,40 In addition, putative MSCs were also characterized by immunodetection and FACS analysis of characteristic surface markers (see Supplementary Figure S3 online).
Human glioma cells U87 (ATCC, HTB-14), were grown in nutrient mixture DMEM/Hams F12 HAM containing 10% heat-inactivated FBS (Sigma), 2 mmol/l L-glutamine (Sigma) and 50 units/ml penicillin/strepotomycin (Sigma).
Cell transduction and labeling with lentiviral vectors. For viral transductions, log phase cell cultures were infected by mixing with viral stocks, using 10 μg/ml polybrene (Sigma) followed by incubated for 24–48 hours (MOI = 20). Cells labeled with luciferase-fluorescent protein vectors were selected by FACS, and cells with the highest 5% fluorescence collected. Cells were transduced with viral stocks of either the RLuc-RFP-tTK, or with both, first with the RLuc-RFP-tTK and then with the pLox:Pecam-Luc:eGFP.
Three shRNA clones of Notch homolog 1 MISSION shRNA Lentiviral Transduction Particles were used for hAMSC transduction and assayed by Matrigel TM (BD Bioscience Inc, Franklin Lakes, New Jersey, EUA). for inhibition of hAMSCs differentiation to endothelial lineage (TRCN0000003360; TRCN0000003359; TRCN0000003361). TRCN0000003360 clone showed the finest inhibition and was used for following experiments. Cells were transduced using the previously described protocol and positive clone were selected by culture in puromycin containing medium (3.5 μg/ml).
Glioma U87 cells were labeled with luciferase-fluorescent reporters by infection with pRRL-PLuc-IRES-eGFP viral stock (MOI = 20), as described above. Positively transduced cells were selected by FACS as described above.
RNA extraction and real-time PCR. Total RNA was extracted from cultured hAMSC, αNotch1-RL-R-tTK-hAMSC, Ф-RL-R-tTK-hAMSC using RNAeasy mini kit (Qiagen, Dusseldorf, Germany). 1 μg of total RNA was reverse transcribed using the Revertaid First Strand cDNA Synthesis Kit (Fermentas, Germany) and product cDNA was then RT-PCR amplified, using the ABI PRISM 7000 (Applied Biosystems, Foster City, CA). The Glyceraldehyde-3-phosphate dehydrogenase gene was used as internal control (Hs99999905_ml). A pre-amplification stage was performed with TaqMan PreAmp Master Mix (Applied Biosystems). FAM-labeled primer/probes were purchased from Applied Biosystems: EGR-3 (Hs00231780_m1); CD31 (Hs00169777_m1); ILK (Hs00177914_m1); SD1-α (Hs00930455_m1); VEGFa1 (Hs00173626_m1); Notch1 (Hs01062014_m1); negative control Osteocalcin BGLAP (Hs01587813_g1). The threshold cycle (Ct) method was used to quantify relative expression for each gene using GAPDH as endogenous reference.
In vitro BLI determination of luciferase activity. For BLI of tissue culture plates, medium was removed from the wells, the wells were rinsed twice with PBS 1x, and imaged immediately following addition of 100 µl/well of PLuc or RLuc substrate stock reagent (Caliper, Hopkinton, MA and Prolume, Pinetop, AZ). Imaging of PLuc and RLuc activities was performed in consecutive days.
For imaging, plates are placed in the detection chamber of a high-efficiency ORCA-2BT Imaging System (Hamamtsu Photonics, Hamamatsu City, Japan) provided with a C4742-98-LWG-MOD camera fitted with a 512 × 512 pixel charge couple device cooled at -80 °C. Images were acquired during a predetermined time using 1 × 1 array (binning 1 × 1). In order to register the position of the light signal, an additional image was obtained using a white light from a lamp in the detection chamber. Recorded light events were calculated using the Wasabi image analysis software and expressed as PHC after discounting the background signal produced by wells without cells. The net number of PHCs in the area of interest was calculated using the formula: PHCs = (total number of PHCs in the area of interest)-((number of pixels in the area of interest) × (average background PHCs per pixel)).
Pseudocolor images were generated using arbitrary color bars representing standard light intensity levels for PLuc (blue: lowest; red: highest) and for RLuc (black: lowest; blue: highest).
Animal procedure and experimental design. Adult 6–8-weeks-old SCID mice were purchased from (Charles Rivers, Wilmington, MA) and kept under pathogen-free conditions in laminar flow boxes. Animal maintenance and experiments were performed in accordance with established guidelines from the Catalan Government and protocol num. 4565 approved by Direcció General del Medi Natural, Generalitat de Catalunya. Animals were anesthetized by i.p. injections of xylazine (Henry Schein, Melville, NY) 3.3 mg/kg and ketamine (Merial, Duluth, GA) 100 mg/kg. Subsequently, mice were mounted in a stereotactic frame (Stoelting, Wood Dale, IL) and their heads were secured using a nose clamp and two ear bars. A skin flap was lifted to expose the skull surface and further anesthetized, by injection of fentanil (Kernpharma, Barcelona, Spain). For stereotactic cell implantation, a burr hole was drilled and the cell suspensions was injected at a 250 nl/min rate using a Hamilton syringe series 700 at the previously indicated coordinates. Following surgery, the animals were placed in individual recovery cages and supplied with buprenorfine (Buprex, Schering Plough SA, Madrid, Spain) in the drinking water. GCV sodium (Cymevene, Roche, Basel, Switzerland) was injected i.p. daily at a dose of 100 mg/kg.41,42 Control animals were inoculated with the same volume of PBS.
In vivo bioluminescent imaging. In vivo BLI of engrafted SCID mice was performed as described previously.10 Briefly, mice were anesthetized i.p. and then injected i.p. with 150 μl of luciferin (Caliper) (16.7 mg/ml in saline) or through tail vein with 25 μl of benzyl coelenterazine (1 mg/ml in 50/50 propilenglycol/ethanol) (Prolume) diluted in 125 μl of water. When BLI was performed after brain surgery, no anesthesia was required. Animals were placed in the detection chamber of the high efficiency ORCA-2BT Imaging System and images acquired from the dorsal direction during 5 minutes period or less when required. A second image of the animal was obtained using a white-light source inside the detection chamber, to register the position of the luminescence signal. To increase detection sensitivity, the readout noise of the recorded signal was reduced by adding together the light events registered by arrays of 8 × 8 adjacent pixels that are read simultaneously (binning 8 × 8) of the camera charge couple device. Mice were imaged weekly during experiments. Quantification and analysis of photons recorded in images was done using the Wasabi image analysis software (Hamamatsu Photonics) as described above.
Histology. Brains from sacrificed mice were harvested, washed with physiological serum and fixed with 5% paraformaldehyde (Sigma) during 48 hours. Brains were then washed with PBS 1x, embedded in OCT (Tissue-Tek, Barcelon, Spain), sliced in 10 μm sections and mounted on glass slides. Hoechst staining was performed for detection of cell nuclei. Immunohistochemical detection of endothelial or cancer stem cells was performed on 10 μm thick section using a primary mouse anti-human CD133 (1:50 dilution) (R&D Systems, Madrid, Spain) or primary rabbit anti-human Sox2 (1:50 dilution) (Cell signaling, Barcelona, Spain). A secondary antimouse or antirabbit antibody conjugated with Cy5 (3 μg/ml) (Jackson ImmunoResearch, Suffolk, UK) was used for detection of bound primary antibodies, and sections counterstained with Bisbenzimide (Hoechst, Sigma).
Fluorescence angiography and laser confocal microscope observation of brain implanted hAMSCs. For tumor microvessel imaging, mice were anesthetized and injected trough the lateral tail vein with 200 μl (10 mg/ml) of a high molecular weight (2,000,000 MW) FITC-conjugated dextran Sigma (St Louis, MO). Ten minutes after, the injection mice were sacrificed and brains were retrieved and fixed in 5% paraformaldehyde during 24 hours. The fixed brains were sliced and analyzed for microvessel formation by fluorescence confocal microscopy.
Fluorescence confocal microscopy. EGFP and RFP-expressing cells were detected by excitation at 488 and 553 nm, respectively, using a confocal laser scanning microscopy (Leica TS1 SP2). Secondary antibodies conjugated with Cy5 were detected by excitation at 649 nm.
Statistical analysis. Student's unpaired two-tailed t-test was used for comparison between groups. Descriptive statistics were performed with SPSS Statistics (15.0.1 version, SPSS Inc., Chicago, IL). Data are presented as mean ± SEM and considered significant at P < 0.05 or P < 0.01.
SUPPLEMENTARY MATERIAL Figure S1. Confocal microscopy analysis was performed on slices from brain tumors implanted with Φ-PECAM:PL-G/RL-R-tTK-hAMSC to determine coexpression of grafted cells with endothelial PECAM1 and pericytic SM22 markers. hAMSCs expressing CMV-promoter regulated eGFP (green) also labeled by (a) an antihuman PECAM antibody (gray) but (b) did not express SM22 antibody. (c, d) Control immunohistochemistry was performed on human miocardium tissue confirming both antibodies specificity. Figure S2. Slices from brain tumors implanted with Φ-RL-R-tTK-hAMSCs incubated with an antihuman-Sox2 ab. Representative laser scanning confocal microscope images show hAMSCs (red fluorescent), U87 glioma cells (green fluorescent) and Sox2+GSCs (grey). Hoechst nuclear stain in blue; Scale bar=20 µm. Figure S3. Characterization of the human adipose tissue-derived MSCmultilineage differentiation capacity into adipogenic, chondrogenic and osteogenic lineages. Images show differentiated cell cultures following staining with, from left to right, (a) Oil red O, Alizarin red S, and Alcian blue. (b) Flow cytometry analysis of marker expression pattern, positive for CD73 and CD105, as well as negative for CD34 and CD31. Materials and Methods.
Acknowledgments
We thank Dr SS Gambhir and Dr Carmelo Bernabeu for the kind donation of the CMV-Rluc-RFP and PECAM-1 promoter, respectively. This work was supported by SAF2009-07102 (Ministerio de Ciencia e Innovacio), SAF2009-07315E Acción Explora (Ministerio de Ciencia e Innovación), Red Tematica de Investigacion Cooperativa TerCel (Instituto de Salud Carlos III).
Supplementary Material
References
- Argyriou AA, Kalofonos HP. Molecularly targeted therapies for malignant gliomas. Mol Med. 2009;15:115–122. doi: 10.2119/molmed.2008.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner JC, Brown PD, O'Neill BP, Meyer FB, Wetmore CJ, Uhm JH. Central nervous system tumors. Mayo Clin Proc. 2007;82:1271–1286. doi: 10.4065/82.10.1271. [DOI] [PubMed] [Google Scholar]
- Castro MG, Cowen R, Williamson IK, David A, Jimenez-Dalmaroni MJ, Yuan X, et al. Current and future strategies for the treatment of malignant brain tumors. Pharmacol Ther. 2003;98:71–108. doi: 10.1016/s0163-7258(03)00014-7. [DOI] [PubMed] [Google Scholar]
- Winkler F, Kienast Y, Fuhrmann M, Von Baumgarten L, Burgold S, Mitteregger G, et al. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia. 2009;57:1306–1315. doi: 10.1002/glia.20850. [DOI] [PubMed] [Google Scholar]
- Amberger-Murphy V. Hypoxia helps glioma to fight therapy. Curr Cancer Drug Targets. 2009;9:381–390. doi: 10.2174/156800909788166637. [DOI] [PubMed] [Google Scholar]
- Dong J, Huang Q. Targeting glioma stem cells: enough to terminate gliomagenesis. Chin Med J. 2011;124:2756–2763. [PubMed] [Google Scholar]
- Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Christensen K, Schrøder HD, Kristensen BW. CD133 identifies perivascular niches in grade II-IV astrocytomas. J Neurooncol. 2008;90:157–170. doi: 10.1007/s11060-008-9648-8. [DOI] [PubMed] [Google Scholar]
- Altaner C. Prodrug cancer gene therapy. Cancer Lett. 2008;270:191–201. doi: 10.1016/j.canlet.2008.04.023. [DOI] [PubMed] [Google Scholar]
- Vilalta M, Dégano IR, Bagó J, Aguilar E, Gambhir SS, Rubio N, et al. Human adipose tissue-derived mesenchymal stromal cells as vehicles for tumor bystander effect: a model based on bioluminescence imaging. Gene Ther. 2009;16:547–557. doi: 10.1038/gt.2008.176. [DOI] [PubMed] [Google Scholar]
- Miletic H, Fischer YH, Giroglou T, Rueger MA, Winkeler A, Li H, et al. Normal brain cells contribute to the bystander effect in suicide gene therapy of malignant glioma. Clin Cancer Res. 2007;13 22 Pt 1:6761–6768. doi: 10.1158/1078-0432.CCR-07-1240. [DOI] [PubMed] [Google Scholar]
- Mesnil M, Piccoli C, Tiraby G, Willecke K, Yamasaki H. Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins. Proc Natl Acad Sci USA. 1996;93:1831–1835. doi: 10.1073/pnas.93.5.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matuskova M, Hlubinova K, Pastorakova A, Hunakova L, Altanerova V, Altaner C, et al. HSV-tk expressing mesenchymal stem cells exert bystander effect on human glioblastoma cells. Cancer Lett. 2010;290:58–67. doi: 10.1016/j.canlet.2009.08.028. [DOI] [PubMed] [Google Scholar]
- Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–3318. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- Reiser J, Zhang XY, Hemenway CS, Mondal D, Pradhan L, La Russa VF. Potential of mesenchymal stem cells in gene therapy approaches for inherited and acquired diseases. Expert Opin Biol Ther. 2005;5:1571–1584. doi: 10.1517/14712598.5.12.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J, et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102:3837–3844. doi: 10.1182/blood-2003-04-1193. [DOI] [PubMed] [Google Scholar]
- Vilalta M, Dégano IR, Bagó J, Gould D, Santos M, García-Arranz M, et al. Biodistribution, long-term survival, and safety of human adipose tissue-derived mesenchymal stem cells transplanted in nude mice by high sensitivity non-invasive bioluminescence imaging. Stem Cells Dev. 2008;17:993–1003. doi: 10.1089/scd.2007.0201. [DOI] [PubMed] [Google Scholar]
- Huang Q, Pu P, Xia Z, You Y. Exogenous wt-p53 enhances the antitumor effect of HSV-TK/GCV on C6 glioma cells. J Neurooncol. 2007;82:239–248. doi: 10.1007/s11060-006-9279-x. [DOI] [PubMed] [Google Scholar]
- Alieva M, Bagó JR, Aguilar E, Soler-Botija C, Vila OF, Molet J, et al. Glioblastoma therapy with cytotoxic mesenchymal stromal cells optimized by bioluminescence imaging of tumor and therapeutic cell response. PLoS ONE. 2012;7:e35148. doi: 10.1371/journal.pone.0035148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altanerova V, Cihova M, Babic M, Rychly B, Ondicova K, Mravec B, et al. Human adipose tissue-derived mesenchymal stem cells expressing yeast cytosinedeaminase::uracil phosphoribosyltransferase inhibit intracerebral rat glioblastoma. Int J Cancer. 2012;130:2455–2463. doi: 10.1002/ijc.26278. [DOI] [PubMed] [Google Scholar]
- Bexell D, Gunnarsson S, Tormin A, Darabi A, Gisselsson D, Roybon L, et al. Bone marrow multipotent mesenchymal stroma cells act as pericyte-like migratory vehicles in experimental gliomas. Mol Ther. 2009;17:183–190. doi: 10.1038/mt.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bexell D, Scheding S, Bengzon J. Toward brain tumor gene therapy using multipotent mesenchymal stromal cell vectors. Mol Ther. 2010;18:1067–1075. doi: 10.1038/mt.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Liu X, Chen J, Zacharek A, Cui X, Savant-Bhonsale S, et al. Simvastatin enhances bone marrow stromal cell differentiation into endothelial cells via notch signaling pathway. Am J Physiol, Cell Physiol. 2009;296:C535–C543. doi: 10.1152/ajpcell.00310.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SA, Hwang SK, Wang KC, Cho BK, Phi JH, Lee JY, et al. Therapeutic efficacy and safety of TRAIL-producing human adipose tissue-derived mesenchymal stem cells against experimental brainstem glioma. Neuro-oncology. 2011;13:61–69. doi: 10.1093/neuonc/noq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetzkendorf J, Mueller LP, Mueller T, Caysa H, Nerger K, Schmoll HJ. Growth inhibition of colorectal carcinoma by lentiviral TRAIL-transgenic human mesenchymal stem cells requires their substantial intratumoral presence. J Cell Mol Med. 2010;14:2292–2304. doi: 10.1111/j.1582-4934.2009.00794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C, Kumar S, Chanda D, Kallman L, Chen J, Mountz JD, et al. Cancer gene therapy using mesenchymal stem cells expressing interferon-beta in a mouse prostate cancer lung metastasis model. Gene Ther. 2008;15:1446–1453. doi: 10.1038/gt.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ding Q, Wu Z, Jiang H, Fang Z. Therapeutic potential of human mesenchymal stem cells producing IL-12 in a mouse xenograft model of renal cell carcinoma. Cancer Lett. 2010;290:157–166. doi: 10.1016/j.canlet.2009.08.031. [DOI] [PubMed] [Google Scholar]
- Li M, Yu J, Li Y, Li D, Yan D, Ruan Q. CXCR4+ progenitors derived from bone mesenchymal stem cells differentiate into endothelial cells capable of vascular repair after arterial injury. Cell Reprogram. 2010;12:405–415. doi: 10.1089/cell.2009.0088. [DOI] [PubMed] [Google Scholar]
- Konno M, Hamazaki TS, Fukuda S, Tokuhara M, Uchiyama H, Okazawa H, et al. Efficiently differentiating vascular endothelial cells from adipose tissue-derived mesenchymal stem cells in serum-free culture. Biochem Biophys Res Commun. 2010;400:461–465. doi: 10.1016/j.bbrc.2010.08.029. [DOI] [PubMed] [Google Scholar]
- Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK. Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111:1826–1832. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurugi-Kobayashi T, Itoh H, Schroeder T, Nakano A, Narazaki G, Kita F, et al. Adrenomedullin/cyclic AMP pathway induces Notch activation and differentiation of arterial endothelial cells from vascular progenitors. Arterioscler Thromb Vasc Biol. 2006;26:1977–1984. doi: 10.1161/01.ATV.0000234978.10658.41. [DOI] [PubMed] [Google Scholar]
- Ribeiro MJ, Phillips DJ, Benson JM, Evatt BL, Ades EW, Hooper WC. Hemostatic properties of the SV-40 transfected human microvascular endothelial cell line (HMEC-1). A representative in vitro model for microvascular endothelium. Thromb Res. 1995;79:153–161. doi: 10.1016/0049-3848(95)00101-v. [DOI] [PubMed] [Google Scholar]
- Shao R, Bao S, Bai X, Blanchette C, Anderson RM, Dang T, et al. Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol Cell Biol. 2004;24:3992–4003. doi: 10.1128/MCB.24.9.3992-4003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, et al. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012;484:110–114. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- Cihova M, Altanerova V, Altaner C. Stem cell based cancer gene therapy. Mol Pharm. 2011;8:1480–1487. doi: 10.1021/mp200151a. [DOI] [PubMed] [Google Scholar]
- Ray P, De A, Min JJ, Tsien RY, Gambhir SS. Imaging tri-fusion multimodality reporter gene expression in living subjects. Cancer Res. 2004;64:1323–1330. doi: 10.1158/0008-5472.can-03-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagó JR, Aguilar E, Alieva M, Soler-Botija C, Vila OF, Claros S, et al. In vivo bioluminescence imaging of cell differentiation in biomaterials: a platform for scaffold development. Tissue Eng Part A. 2013;19:593–603. doi: 10.1089/ten.tea.2012.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila OF, Bagó JR, Navarro M, Alieva M, Aguilar E, Engel E, et al. Calcium phosphate glass improves angiogenesis capacity of poly(lactic acid) scaffolds and stimulates differentiation of adipose tissue-derived mesenchymal stromal cells to the endothelial lineage. J Biomed Mater Res A. 2013;101:932–941. doi: 10.1002/jbm.a.34391. [DOI] [PubMed] [Google Scholar]
- Roura S, Bagó JR, Soler-Botija C, Pujal JM, Gálvez-Montón C, Prat-Vidal C, et al. Human umbilical cord blood-derived mesenchymal stem cells promote vascular growth in vivo. PLoS ONE. 2012;7:e49447. doi: 10.1371/journal.pone.0049447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll NM, Chase M, Chiocca EA, Tanabe KK. The effect of ganciclovir on herpes simplex virus-mediated oncolysis. J Surg Res. 1997;69:413–417. doi: 10.1006/jsre.1997.5089. [DOI] [PubMed] [Google Scholar]
- Marconi P, Tamura M, Moriuchi S, Krisky DM, Niranjan A, Goins WF, et al. Connexin 43-enhanced suicide gene therapy using herpesviral vectors. Mol Ther. 2000;1:71–81. doi: 10.1006/mthe.1999.0008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.