

Abstract
Human islet transplantation can be a permanent treatment of type 1 diabetes if the immune rejection and primary nonfunction (PNF) of transplanted islet grafts were properly addressed. In this study, we determined whether cotransplantation of human bone marrow-derived mesenchymal stem cells (hBMSCs) could prevent immune rejection and improve human islet transplantation in a humanized NOD scid gamma (NSG) mouse model. Human immunity was rebuilt and maintained in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice up to 13 weeks after intraperitoneal injection of mature human peripheral blood mononuclear cells (PBMCs). The blood glucose control and the levels of serum insulin and c-peptide clearly indicated a better outcome of islet transplantation when islets were cotransplanted with hBMSCs. hBMSCs actively interacted with interleukin-10 (IL-10)-producing CD14+ monocytes to suppress the proliferation and activation of T cells in the PBMC/hBMSC coculture and prevent the T cell recruitment into the transplantation site. hBMSCs also increased the percentage of immunosuppressive regulatory T cells (Tregs) and prevented the cytokine-induced loss-of-function of human islets. Taken together, our studies demonstrated that transplantation of islets with hBMSCs is a promising strategy to improve the outcome of human islet transplantation.
Introduction
Since its first introduction in the late 1990s, Edmonton Protocol for human islet transplantation has helped more than 500 type 1 diabetic patients worldwide. However, its wide application is still hindered by two major barriers: the immune rejection from the organ recipients and the primary nonfunction (PNF) of islet grafts. Immune rejection describes a process where transplanted islets are attached, recognized, and attacked by the host immune system, whereas the PNF is characterized as the loss of islet viability and function caused by nonimmune reactions, such as the disruption of islet microvasculature during islet isolation and purification process, hypoxia in the core of islet grafts, and production of inflammatory cytokines at the transplantation sites.
Despite the administration of immunosuppressive drugs such as tacrolimus, sirolimus, and mycophenolic acid and the recent improvement in islet isolation, preparation, and transplantation, insulin independence is rarely sustained for long term after islet transplantation mostly due to inadequate immunosuppression. Several strategies such as gene therapy and cell therapy have been proposed to address this issue. Gene therapy, which relies on “vectors” to deliver therapeutic genes into human islets, have faced serious problems such as the low transfection efficiency of nonviral vectors and the increasing safety concerns of viral vectors.1,2 Cell therapy, especially stem cell therapy, on the other hand, has recently met great success as a novel regenerative medicine to support solid organ transplantation including human islet transplantation.3,4 Among all types of stem cells, mesenchymal stem cells (MSCs) are given special interest for their self-renewal potential, multilineage capacities, paracrine effects (trophic mediator), and immune modulatory effects,5,6 making it a great candidate for improving human islet transplantation. MSCs, mostly found in bone marrow, adipose, and umbilical cord blood, are one of the most extensively studied adult stem cells used in treating degenerative diseases as well as solid organ transplantation.7 Unlike embryonic stem cells or induced pluripotent stem cells, adult stem cells show restricted proliferation and lineage differentiation, and consequently have little risk of inducing tumor.5
MSC-based therapy has been used to improve human islet transplantation from several aspects. Ding et al. reported the immunosuppressive effects of MSCs by secreting soluble factors.8 Korbutt et al. reported that coculture with MSCs improved islet viability and function against proinflammatory cytokines.9 Ito et al. and other researchers reported that MSCs were capable of promoting the revascularization of islets in vivo.3,10 MSCs can be isolated from the diabetic patient, expanded in vitro, and infused back into the patient to exert immunosuppressive effect. Therefore, MSCs are more biocompatible to the patient and friendlier to transplanted islets than the immunosuppressive drugs which are usually toxic to islet function. However, the translation to a real clinical breakthrough is slow and the data from limited clinical trials seems vague and controversial, probably due to the lack of the human immunity in most of the previous studies. Hereby, we evaluated the effectiveness of human bone marrow derived mesenchymal stem cells (hBMSCs) from a third-party donor to improve the outcome of human islet transplantation in a diabetic humanized NOD scid gamma (NSG) mouse model. We plan to answer two questions by this study: (i) the immunomodulatory effect of hBMSCs on adoptively transferred human immunity to protect islets in vivo and (ii) the tropic effect of hBMSCs to support islet function.
Results
hBMSCs suppressed the activation and proliferation of peripheral blood mononuclear cells
Primary hBMSCs exhibit a spindle-shaped fibroblastic morphology after ex vivo expansion (Supplementary Figure S1a). The hBMSCs maintained in our lab were positive for human leukocyte antigen (HLA) class I and negative for HLA-DR, Fas ligand (FasL), CD14, CD80, and CD86 (Supplementary Figure S1b), which is consistent with the literatures.11
Peripheral blood mononuclear cells (PBMCs) were isolated from human buffy coats. We first tested the immunomodulatory effect of hBMSCs on PBMCs in a mixed lymphocyte reaction. Carboxyfluorescein diacetate succinimidyl ester (CFSE) was used to determine the proliferation of PBMCs in the presence or absence of hBMSCs. Briefly, CFSE passively diffuses into cells, retained within cells, and covalently couples to intracellular molecules. Once the CFSE-labeled cell undergoes mitosis, the daughter cells were determined by the progressive halving of CFSE fluorescence following each division. The parent generation was identified as the rightmost peak in the histogram of flow cytometry, while successive generations were the peaks to the left of the parent peak. The percentage of the successive generations was used as a marker for the proliferation of PBMCs. More cell proliferation resulted in higher percentage of successive generation. In this experiment, the PBMC/hBMSC coculture was stimulated with phytohemagglutinin (PHA) (5 µg/ml) and allowed to proliferate for 2 days. The coincubation with hBMSCs strongly inhibited the proliferation of PBMCs as indicated by the area of successive generations at 24 hours after coincubation (Figure 1a). The total number of PBMCs in the coculture was also significantly less than the PBMCs cultured alone (Figure 1b). Moreover, less interferon-γ (IFN-γ), interleukin-2 (IL-2), and IL-2sRα (a soluble subunit of IL-2 receptor) were detected in the coculture, suggesting less T cell activation (Figure 1c). We then determined whether the immunosuppression required direct contact between PBMCs and hBMSCs. Surprisingly, hBMSCs were equally potent to inhibit the proliferation of PBMCs and the expression of IFN-γ, IL-2, and IL-2sRα in a transwell system (Figure 1d–f), suggesting a predominant paracrine model. In other words, hBMSCs inhibited PBMCs by secreting soluble factors.
Figure 1.
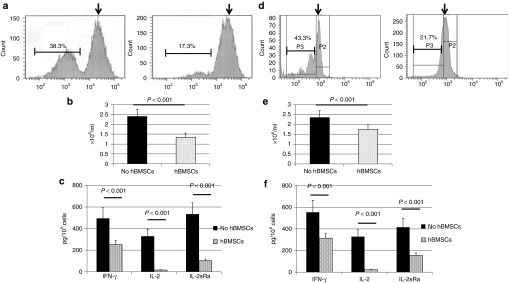
hBMSCs prevented the proliferation and activation of peripheral blood mononuclear cells (PBMCs). (a) The percentage of successive generations of PBMCs when stimulated with phytohemagglutinin (1 µg/ml) for 1 day and allowed to proliferate for 2 more days. PBMCs were labeled with carboxyfluorescein diacetate succinimidyl ester and subjected to analysis using flow cytometry. Black arrows indicate the parent generation. Left, PBMCs alone; right, PBMCs with hBMSCs. (b) The total number of PBMCs as determined by an automatic cell counter. (c) The levels of IL-2, IL-2sRα, and IFN-γ in the medium as determined by enzyme-linked immunosorbent assay. The levels of cytokines were normalized with total cell number. (d–f) Repeat of the previous experiments (a–c) using a transwell system. n = 3. Data are presented as the mean ± SD, n = 6. hBMSCs, human bone marrow-derived mesenchymal stem cells; IFN-γ, interferon-γ IL-2, interleukin-2.
IL-10 is a well-documented immunosuppressive factor by suppressing T cell activation, preventing antigen presenting process, and inhibiting proinflammatory cytokines. We previously reported marginal IL-10 expression (~20 pg/ml) in the medium of primary hBMSCs.12 However, IL-10 expression was significantly boosted (300–400 pg/ml) in the PBMC/hBMSC coculture, despite the existence of PHA-stimulation (Figure 2a). The inhibition of IL-10 pathway partially restored the production of IFN-γ, IL-2, and IL-2sRα by the stimulated T cells in the presence of hBMSCs (Figure 2b), indicating the crucial role of IL-10 in the immunomodulatory effects of hBMSCs. We next determined which subpopulation of PBMCs produced IL-10. Neither T cells nor B cells have been reported to produce potent IL-10. We found that CD14+ monocytes were required for IL-10 expression in the PBMC/hBMSC coculture since CD14+ monocytes-depleted PBMCs produced significantly less IL-10 (Figure 2c). Real-time reverse transcription-PCR also demonstrated that IL-10 transcription was activated in CD14+ monocytes instead of T cells or B cells, suggesting that the signal transduction between hBMSCs and monocytes might be the first step toward the production of IL-10 and subsequent immune suppression.
Figure 2.
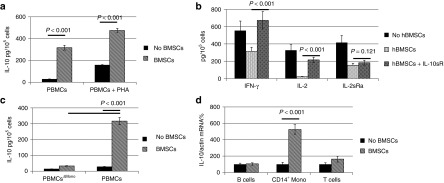
CD14+ monocytes and IL-10 were required for the immunomodulatory effect of hBMSCs. PBMCs were stimulated with PHA (1 µg/ml) for 1 day and cocultured with hBMSCs for 2 more days. (a) IL-10 was highly expressed by the PBMC/hBMSC coculture but not by PBMCs alone. (b) The inactivation of PBMCs mediated by hBMSCs can be reversed by adding IL-10sR. (c) Monocyte depleted PBMCs (PBMCsΔMono) failed to produce high level of IL-10 in the PBMC/hBMSC coculture. (d) IL-10 mRNA was highly expressed in CD14+ monocytes but not B cells and T cells in the PBMC/hBMSC coculture. n = 3. BMSCs, bone marrow stromal cells; hBMSCs, human bone marrow-derived mesenchymal stem cells; IFN-γ, interferon-γ IL, interleukin; PBMCs, peripheral blood mononuclear cells; PHA, phytohemagglutinin.
Some literatures showed positive expression of FasL on the surface of hBMSCs and hypothesized that hBMSCs promoted the apoptosis of T cells and subsequent immune tolerance.13,14 However, we did not detect FasL expression on the surface of hBMSCs maintained in our lab (Supplementary Figure S1b). Annexin V staining demonstrated that hBMSCs had no effect on the apoptosis of T cells in the coculture (Supplementary Figure S3). Our results was supported by the results from Ryan et al.6
hBMSCs improved islet function against inflammatory cytokines
Besides immune rejection, islets also suffer from the PNF which is the loss of islet function by nonimmune reactions. Although still in debate, inflammatory cytokines seem to play important roles in the PNF of human islets because raised inflammatory cytokines was accompanied with most post-transplantation challenges including hypoxia in the core of islet grafts, inflammation in the transplantation site, and the disruption of vasculature.1 In this study, we used a cocktail of inflammatory cytokines (50 ng/ml tumor necrosis factor-α, 5 ng/ml IL-1β, and 50 ng/ml IFN-γ) to mimic the PNF-related challenges to human islets. Without inflammatory cytokines, the presence of hBMSCs in the coculture did not affect the insulin release from human islets when exposed to stimulatory glucose. Inflammatory cytokines significantly impaired the first phase (the first peak appears immediately after stimulatory glucose) and the total insulin release of human islets after 4 days (Figure 3a). However, the presence of hBMSCs in the coculture significantly ameliorated the impairment caused by inflammatory cytokines (Figure 3b), suggesting a protective effect of hBMSCs.
Figure 3.

hBMSCs protected the function of human islets against the inflammatory cytokines. (a) The dynamic insulin release from human islets. Briefly, 50 islets from each group were perifused with basal glucose (1.67 mmol/l) for 60 minutes, stimulatory glucose (16.7 mmol/l) for 30 minutes, and finally basal glucose until the insulin release reversed to the basal level. The flow rate was maintained at 1 ml/minute and the temperature was maintained at 37 °C. Samples were collected once per 2 minutes and analyzed for insulin concentration. For the cytokine treatment groups, islets, alone or with hBMSCs, were stimulated with a cytokine cocktail (50 ng/ml tumor necrosis factor-α, 5 ng/ml IL-1β, and 50 ng/ml interferon-γ) for 4 days prior to perifusion. (b) Cumulative insulin release calculated from dynamic insulin release profile on the left. n = 3. hBMSCs, human bone marrow-derived mesenchymal stem cells.
hBMSCs improved islet allograft survival in humanized NSG mice
The PBMCs isolated from concentrated human buffy coat were composed of T lymphocytes (~55%) and B lymphocytes (~40%) (Supplementary Figure S2a). Potent human IgG in the mouse blood stream and evident human CD3+ T cells in the spleen sections were detected at around 2 weeks after the intraperitoneal injection of PBMCs (5 × 106/mouse), suggesting the successful establishment of human immunity (Supplementary Figure S2b,c). In contrast, human IgG and CD3+ T cells were not detected in NOD.CB17-Prkdcscid/J (NOD-SCID) mice because NOD-SCID mice expelled the xenogeneic immunocytes constantly.15
The coadministration of streptozotocin (STZ) and PBMCs usually led to strong T cell alloreactivity and subsequent graft versus host diseases (GVHDs) and death. In this study, we first used a long-term injection of low doses of STZ to induce diabetes. Mice were mostly diabetic after 3 weeks. Then, 500 handpicked human islets were transplanted under the kidney capsule of each diabetic NSG mouse with hBMSCs at serial ratios (islet: hBMSC = 1:0, 1:50, 1:100, and 1:200). At 4 weeks after successful islet transplantation, PBMCs (5 × 106/mouse) were injected intraperitoneally to establish the human immunity. This strategy helped us to identify the settlement of human immunity by comparing blood glucose before and after PBMC injection and to avoid STZ-stimulated GVHD. Glucose measurement showed that all mice restored normoglycemia during the first 4 weeks after islet transplantation, indicating successful islet engraftment and function (Figure 3a). However, the blood glucose of some mice reversed to hyperglycemia after PBMC infusion, probably due to the destruction of islet grafts by the established human immunity. Briefly, 11/11 mice receiving islets only were back to diabetes in the first 2 weeks after PBMC infusion. In contrast, 10/11, 6/12, and 2/10 mice were back to diabetes when the islets were transplanted with hBMSCs (islets: hBMSCs = 1:50, 1:100, and 1:200), respectively (Figure 3b). Dose dependent improvement of the outcome of human islet transplantation was observed by increasing the ratio of hBMSCs, suggesting the protective role of hBMSCs against the adoptively transferred human immunity.
Intraperitoneal glucose tolerance test was performed 2 weeks after the PBMC injection to determine the function of islets in real time. Despite the difference in the nonfasted blood glucose level between different groups, all mice displayed similar baseline blood glucose levels following overnight fasting. Blood glucose levels increased immediately following glucose (2 g/kg) injection, peaked at 15 minutes in all groups, and decreased over time (Figure 4c). The mice receiving islet/hBMSC cotransplantation showed faster and better response to the stimulatory glucose compared with the mice receiving islets alone (Figure 4c), suggesting a better glucose control. At the end of the study, the mice were anesthetized to collect blood to measure serum human insulin and human c-peptide levels by enzyme-linked immunosorbent assay (ELISA). The levels of human insulin and human c-peptide of the mice receiving islet/hBMSC cotransplantation were significantly higher than those of the mice transplanted with islets alone (Figure 4d).
Figure 4.

The outcome of human islet transplantation with hBMSCs in diabetic humanized NOD scid gamma (NSG) mice. (a) The blood glucose levels of all mice after successful islet transplantation. For each mouse, 500 handpicked islets (150–300 μm in diameter) were cotransplanted with primary hBMSCs at serial ratios (islet: hBMSC = 1:0, 1:50, 1:100, and 1:200) under the kidney capsule of diabetic NSG mice. The blood glucose level of each mouse was monitored using a glucometer weekly. An intraperitoneal injection of peripheral blood mononuclear cells (PBMCs, 5 × 106/mouse) was given 4 weeks after islet transplantation. At 17 weeks after islet transplantation, the graft-bearing kidneys were then removed from some mice to validate the function of islet grafts by the return of blood glucose levels to ≥400 mg/dl. (b) The Kaplan–Meier plot for the percentage of insulin-independent mice of each group after PBMC injection. The mouse with blood glucose level <200 mg/dl for two consecutive measurements was identified as insulin independent. P value was calculated using Logrank test as compared with the islet-alone group using R version 2.15.2. (c) Intraperitoneal glucose tolerance test was done with the mice receiving islet alone and islet/hBMSC cotransplantation (islet: hBMSC = 1:200) to determine the islet function at 2 weeks after the injection of PBMCs. (d) The serum insulin and c-peptide of the mice receiving islet alone and islet/hBMSC cotransplantation (islet: hBMSC = 1:200) at the end of study. Data are presented as the mean ± SD, n = 9. hBMSCs, human bone marrow-derived mesenchymal stem cells; PBMC, peripheral blood mononuclear cell.
hBMSCs prevented T cell infiltration into the transplantation site and increased regulatory T cells
The kidney bearing islets was isolated at the end of study and subjected to immunofluorescence staining. The location and function of islet grafts were identified by staining for insulin. The insulin content under the kidney capsules of the mice receiving islet/hBMSC cotransplantation was significantly higher than those receiving islets only (Figure 5a). Moreover, the number of CD3+ T cells that infiltrated into the transplantation site was significantly reduced in the presence of hBMSCs (Figure 5a). Taken together, these data suggested that hBMSCs prevented T cell infiltration into the transplantation site and prevented the destruction of islets in vivo.
Figure 5.

Human bone marrow-derived mesenchymal stem cells (hBMSCs) prevented T cell infiltration into the transplantation site. (a) The immune fluorescence staining of the kidney section bearing human islets alone (left) or with hBMSCs (right). Briefly, three mice in each group were killed at 2 weeks after the injection of peripheral blood mononuclear cells (PBMCs, 5 × 106/mice), or 6 weeks after islet transplantation. Insulin was stained in red to identify the transplanted human islets, while CD3+ T cells were stained in green. The white outlines indicated the boundary of functional islets. The slides were counter-stained by 4′,6-diamidino-2-phenylindole (blue) which passed through membrane and stained the nuclei specifically. Scale bar represents 200 µm. All experiments were done in triplicates and a representative picture was shown. (b) The percentage of CD4+CD25+FoxP3+ regulatory T cells (Tregs) recovered from the blood of NOD scid gamma (NSG) mice at 2 weeks after the injection of PBMCs. Briefly, CD4+ T cells were isolated from the total blood of NSG mice using Dynabeads at the end of study. The percentage of CD25+FoxP3+ Tregs among the total CD4+ T cells was determined by flow cytometry. Q2 represents the Tregs. (c) The percentage of Tregs of the total CD4+ T cells recovered from PBMC/hBMSC coculture. Briefly, PBMCs (1 × 106) were stimulated with phytohemagglutinin (1 µg/ml) for 24 hours and cocultured with hBMSCs (5 × 104) in a transwell for 5 more days. Total CD4+ T cells were isolated using Dynabeads at the end of the study. The CD4+CD25+FoxP3+ Tregs were stained using Human Regulatory T cells 3-color kit and the percentage of Tregs in the total CD4+ T cells was calculated using flow cytometry.
The expansion of regulatory T cells (Tregs) might play an important role in the immune tolerance of transplanted organs in vivo.16 Moreover, hBMSCs were recently reported to increase the proportion of Tregs.17,18 To further explore whether such mechanism can explain the reduced infiltration and destruction by alloreactive T cells into the transplantation site, we isolated all CD3+ T cells and detected the percentage of Tregs from the blood stream of NSG mice. Despite the low cell recovery, significantly higher percentage of CD3+ CD25+ Foxp3+ Tregs was detected in the blood stream (Figure 5b), which was consistent with the hBMSC/T cells coculture study in vitro (Figure 5c). Taken together, these results suggested that hBMSC-mediated increase in the proportion of Tregs might contribute to the immune tolerance of islet transplantation.
Discussion
Edmonton protocol for human islet transplantation, which was developed more than 20 years ago, failed to maintain insulin independence in the long term in most patients. Despite the development of new immunosuppressive regimens and new combination therapy, graft rejection remains a major challenge for human islet transplantation. Other challenges include the hypoxic environment, secretion of inflammatory cytokines and apoptosis of islet cells during isolation, transportation, and storage process, all of which are summarized as the PNF of islet grafts.
MSC-based cell therapy has attracted wide attention in regenerative medicine and tissue engineering. There are two major strategies of using MSCs for islet transplantation: (i) directing the differentiation of MSCs to replenish the lost islets and (ii) use of MSCs to nourish islet grafts and mitigate immune response. For the first strategy, several groups have reported the generation of islet-producing cells and islet-like clusters by the in vitro transdifferentiation of MSCs.19,20 However, this strategy usually required long-term in vitro induction and failed to generate enough islets for clinical study and the risk of tumorigenicity was somehow overlooked. For the second strategy, MSCs constitutively secreted multiple factors, which are capable of regulating insulin production, revascularization, immune rejection, and apoptosis of human islets. This strategy does not require in vitro transdifferentiation and huge amount of MSCs. Most importantly, MSCs can be isolated from and infused back into the same patient to exert the therapeutic potential as indicated by several clinical studies.21 Therefore, in this study, we selected the second strategy for improving human islet transplantation.
The pattern of the surface markers has great impact on the function of hBMSCs and provided useful information to the following studies. Our hBMSCs were positive for HLA class I and negative for CD14 (Supplementary Figure S1b), which concurred with previous report and suggested hBMSCs can be recognized and destroyed by the cytotoxic T cells.6 hBMSCs were also negative for costimulatory factors such as CD80 and CD86 (Supplementary Figure S1b), which might explain why hBMSCs were unable to stimulate the multiplication of helper T (Th) cells and the subsequent antibody production. Generally, the lack of costimulatory factors (CD80 and CD86) led to an immature antigen presenting to the Th cells and subsequent immune tolerance.22 Despite that some groups showed that hBMSCs expressed FasL and promoted the apoptosis of T cells through direct contact,13,23 our hBMSCs were negative for FasL (Supplementary Figure S1b), which was supported by the results from other groups6,24 and confirmed by the mixed lymphocyte reaction that hBMSCs did not promote the apoptosis of T cells (Supplementary Figure S3). The expression of HLA class II on the surface of hBMSCs is still unclear. While some groups demonstrated that hBMSCs were positive for HLA class II,22 the studies from our and other groups suggested that hBMSCs were negative for HLA class II which explained the hypoimmunogenicity of MSCs (Supplementary Figure S1b).6 This discrepancy is probably due to the difference in culture conditions and the sources of hBMSCs and can only be solved if a standard operating protocol is developed and maintained among all research groups.
T cells are the most important cells mediating the recognition and destruction of human islets. T cells can be activated by PHA and the activation of T cells is characterized by the production of IL-2, IL-2R, and IFN-γ. In the mixed lymphocyte reaction, hBMSCs prevented PHA-induced proliferation and activation of PBMCs by secreting the soluble factors, among which IL-10 may play an important role (Figure 1d–f and 2b). However, despite potent IL-10 expression in the PBMC/hBMSC coculture, significantly less IL-10 was expressed by hBMSCs and PBMCs when they were cultured alone (Figure 2a),12 suggesting that the communications between hBMSCs and a subpopulation of PBMCs were required for the production of immunosuppressive IL-10.25,26 We identified CD14+ monocytes as the prerequisite of the hBMSC-mediated immune suppression (Figure 2c,d), which is consistent with previous reports that monocytes or monocyte-derived dendritic cells were deeply involved in the hBMSC-mediated T cell suppression.27,28 First, hBMSCs secreted soluble factor(s) and activated CD14+ monocytes to express IL-10. Then IL-10 suppressed the subsequent proliferation and activation of T cells. Such two-step immunosuppression could be an important feature for the cotransplantation of human islets and hBMSCs as the hBMSCs only exert a local effect to protect human islets without impairing the host immunity systemically. However, these results should be interpreted with caution because (i) the role of IL-10 in the hBMSC-mediated immunosuppression is controversial as some groups observed that the inhibition of IL-10 did not reverse and even further suppressed T cell proliferation25,27 and (ii) the soluble factors secreted by hBMSCs in this process remained unknown. Besides IL-10, other immunosuppressive factors such as hepatocyte growth factor, transforming growth factor-β, and prostaglandin E2 may also play important roles in the hBMSC-mediated immunosuppression.6,12,29 It is highly possible that the immunomodulatory effect of hBMSCs is mediated through a network of molecular pathways instead of a single pathway.
Next, we evaluated the interactions of hBMSCs with human islets and human immunity in vivo. In fact, several groups have previously shown the improvement of islet transplantation using MSCs, either through cotransplantation or intravenous infusion. For example, Ito et al. reported that MSCs improved the outcome of islet transplantation by promoting the revascularization of islet grafts.10 Ding et al. reported MSCs prevented the rejection of allogenic islets by matrix metalloproteinases (MMP2 and MMP9).8 Yeung et al. reported that MSCs protected human islets from inflammatory cytokines.9 However, the lack of human immunity in most of these studies caused limited understanding of the immune mediated graft loss. Yet the mechanism of hBMSC-mediated protection to human islets in the presence of human immunity has not been thoroughly understood. Although xenogeneic human islet transplantation in immunocompetent rodents has been reported,30 the distinct differences in the immune system between rodent and human hindered the translation into a real clinical advance. The humanized mouse developed by Dr Leonard Schultz is a “state of the art” animal model for research related to human immunity.15 First, it is a perfect model to study the development of type 1 diabetes, which is an autoimmune disease. Second, the interaction between transplanted islets and the adoptively transferred human immunity provide useful information about immune rejection after human islet transplantation. The human immunity was established in NSG mice by injection of human PBMCs and validated by the presence of human IgG in the blood steam and CD3+ T cells in the mouse spleen (Supplementary Figure S2). The adoptively transferred human immunity posed a challenge, which is similar to the clinical immune rejection, to the transplanted human islets. Compared with previous studies using immunodeficient animals,10,12 the duration of insulin independence was significantly reduced in humanized NSG mice, as evidenced by the rapid loss of islet and T cell infiltration (Figures 4a and 5a). Cotransplantation with hBMSCs significantly prolonged the duration of insulin independence in the presence of human immunity (Figure 4a). The intraperitoneal glucose tolerance test and the serum levels of human insulin and c-peptide further validate the improved islet function in the islet/hBMSC cotransplantation group (Figure 4c,d). The interactions between hBMSCs and T cells may be of critical importance to the protection to human islets. We demonstrated that hBMSCs “switched on” the production of immunosuppressive IL-10 by CD14+ monocytes to prevent the proliferation and activation of T cells in coculture (Figure 1), which is consistent with the report from François et al.31 However, François et al. reported that the hBMSCs needed to be “activated” by tumor necrosis factor-α and IFN-γ to exert immunosuppressive effect, which is different from our results that the media derived from hBMSCs were equally potent to suppress the activation of PBMCs and to activate the IL-10 transcription in CD14+ monocytes (Supplementary Figure S4). Such discrepancy could be caused by the various sources of hBMSCs. In vivo results showed that hBMSCs prevented the infiltration of CD3+ T cells into the transplantation site, probably due to the reversal to T cell activation (Figure 5a). Besides, we also found increased immunosuppressive Tregs in the blood stream of humanized NSG mice receiving islet/hBMSC cotransplantation, suggesting locally injected hBMSCs still had the potential to exert a considerable systemic effect (Figure 5b). Similar results were found in the mixed lymphocyte reaction and supported by the results from Weng et al. (Figure 5c).18 However, further explorations are still required to unveil the underlying pathways to the proliferation of Treg.
Besides the immune modulatory effect, hBMSCs improved insulin production of human islets against inflammatory cytokines in an islet perifusion assay (Figure 3), suggesting the role of hBMSC as a trophic mediator to prevent the PNF caused by inflammatory cytokines. These results might be due to the cytoprotective factors produced by hBMSC, such as hepatocyte growth factor, MMP2, and MMP9. hBMSCs also produce angiogenic factors such as hepatocyte growth factor and vascular endothelia growth factor to promote the revascularization, which has been explored in depth in immune deficient animal model by us and other groups.10,12
Based on our results, we hereby propose three possible mechanisms for hBMSC-mediated islet tolerance (Figure 6): (i) hBMSCs inhibit alloreactive T cells by the immature antigen presenting and/or activating CD14+ monocytes to produce IL-10; (ii) hBMSCs promote the proliferation of Tregs through soluble factors; and (iii) hBMSCs prevent the PNF of islets. The protection of human islets by hBMSCs in the presence of human immunity may be a result of the synergistic combination of all three mechanisms.
Figure 6.

Schematic illustration why hBMSCs prevent immune rejection in human islet transplantation. BMSC, bone marrow stromal cell; hBMSCs, human bone marrow-derived mesenchymal stem cells; IL-10, interleukin-10; PBMCs, peripheral blood mononuclear cells; Tregs, regulatory T cells.
Despite the evidences that hBMSCs significantly improved islet transplantation in humanized NSG mice, the data should be interpreted with caution as several issues needed to be resolved. First, we did not characterize the interactions of hBMSCs with B cells and natural killer cells. Such characterization may uncover the new aspects of hBMSCs-mediated immune tolerance and is necessary in the future. The second issue is the GVHD caused by the infused mature immunocytes. Most humanized NSG mice or even NRG-Akita mice, which simultaneously develop diabetes without STZ injection, will develop GVHD in 5–7 weeks after the infusion of PBMCs,15 providing us a limited window to evaluate the efficacy of hBMSC cotransplantation. The amount of infused PBMCs has great effect on the onset of GVHD. The dose of 5 × 106 PBMCs was reported to be relatively safe to induce significant GVHD and adopted for this study.15 We did not detect significant GVHD-like symptoms including weight loss, hunched posture, ruffled fur, reduced mobility, and tachypnea in the mice receiving islet/hBMSC cotransplantation, which might be due to the presence of hBMSCs. However, we did observe GVHDs in the mice receiving islets alone from 5 weeks after the infusion of PBMCs. Humanized mice generated from hematopoietic stem cells may resolve the issue of GVHDs.32 The third issue is the route of administration of hBMSCs. Many reports have shown that the systemic administration of hBMSCs prevented the onset of type 1 diabetes. However, following systemic injection, hBMSCs may be sequestered in the lung and other sites. In this study, we chose islet/hBMSC cotransplantation to exert a local effect, which will have more impact on the future clinical application because the immunity of the patients receiving islet transplantation will not be compromised. Previous studies from us and other groups also ruled out the possibility of hBMSC migration and demonstrated that hBMSC cotransplantation stayed in the proximity of islet grafts and helped the engraftment and revascularization of islet grafts.3,10 Last but not the least, despite the fact that hBMSCs from the third party is equally potent in protecting human islets from adoptively transferred human immunity (Figure 5a), the mismatch in HLA typing remains a concern for future clinical application. To better fit the scope of personalized medicine, hBMSCs and PBMCs isolated from the same subject would be preferred. This requires further collaboration of institutions with hospitals.
In summary, our studies demonstrated that cotransplantation with hBMSCs can be a promising strategy to improve the current status of human islet transplantation. The hBMSC served as a major role of immune modulator to protect islets from immune destruction while also as a trophic mediator to prevent the PNF of islet grafts after transplantation.
Materials and Methods
Materials. NSG and NOD-SCID mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in pathogen-free microisolator cages. All animal use was in accordance with the guidelines from National Institutes of Health and the Institutional Animal Care and Use Committees of the University of Tennessee Health Science Center. Human PBMCs were isolated from fresh buffy coats (Lifeblood South, Memphis, TN) by gradient centrifugation using Ficoll-Paque (GE Healthcare, Little Chalfont, UK). Before use, the preparations of PBMCs were characterized by FlowCellect Human Lymphocyte ZAP-70 Characterization Kit (Millipore, Billerica, MA). To establish human immunity, PBMCs were injected intraperitoneally into NSG mice (5 × 106/mouse) as described before.33 The settlement of T cells in the spleen was assessed 2 weeks after injection by immunofluorescence staining. The function of B cells was assessed by measuring human IgG concentration in the mouse blood. Human islets were received from Integrated Islet Distribution Program (Duarte, CA). Primary hBMSCs were purchased from Thermo Fisher Scientific (Waltham, MA), passaged, and frozen in our lab. Before use, hBMSCs were characterized by the surface markers including HLAs-DR, HLA class I, CD14, CD80, CD86, and FasL (or CD95L) (Abcam, Cambridge, MA).
Mixed lymphocyte reaction. The proliferation of PBMCs was determined using CFSE proliferation assay (Molecular Probes, Invitrogen). CFSE passively diffuses into cells, retained by the cells throughout division, and can be used to differentiate the successive generations from the parental generations. Briefly, CFSE-labeled PBMCs (1 × 106) were stimulated with PHA (1 µg/ml) for 24 hours and cocultured with hBMSCs (5 × 104) in the same well or in a transwell system for indicated time. In the transwell system, hBMSCs were adherent at a 0.4-µm transwell cell culture inserts (Corning, Tewksbury, MA), while the PBMCs were cultured outside the insert in a cell culture plate. The media were carefully monitored to rule out the penetration of PBMCs into the inserts. The division history of PBMCs was analyzed by flow cytometry. The percentage of proliferated cells was calculated by dividing the number of cells undergoing divisions with the number of total cells in the cultures. The total number of PBMCs was measured using T4 Automatic Cell Counter (Nexcelom, Lawrence, MA). The extent of T cell activation was determined by measuring the level of IL-2, IL-2sRα, and IFN-γ in the medium using ELISA. IL-10 level in the medium was also determined by ELISA. Total T cells, CD4+ T cells, CD19+ B cells, and CD14+ monocytes were isolated or depleted from PBMCs using specific Dynabeads for each population, respectively (Invitrogen, Carlsbad, CA). The mRNA levels of IL-10 in each subpopulation were determined by real-time reverse transcription-PCR and normalized by the levels of β-actin. The percentage of CD4+ CD25+ FoxP3+ Tregs was determined by Human Regulatory T cells 3-color kit (RnD System, Minneapolis, MN) using flow cytometry. To study whether a direct contact between hBMSCs and PBMCs was required for the immunosuppression of hBMSCs, a transwell system was set up as described before.
Islet viability study. Primary hBMSCs (5 × 105) were cocultured with 500 islets in a 10-cm dish of CMRL-1066 medium with 10% fetal bovine serum for 2 days. Then, the islet/hBMSC coculture was stimulated with a cytokine cocktail (50 ng/ml tumor necrosis factor-α, 5 ng/ml IL-1β, and 50 ng/ml IFN-γ) for 4 days and the insulin secretion from human islets was quantified using a dynamic islet perifusion assay. Briefly, 50 islets from each group were handpicked and loaded onto a Swinnex 13 chamber (Millipore, Burlington, MA) and perifused with Krebs-Ringer bicarbonate HEPES buffer of the following composition (in mmol/l): 129 NaCl, 5 NaHCO3, 4.8 KCl, 1.2 KH2PO4, 1.2 MgSO4, 2.5 CaCl2, and 10 HEPES at pH 7.4. The flow rate was maintained at 1 ml/minute with a peristaltic pump (Themo Fisher, Waltham, MA) and the temperature was maintained at 37 °C with a solution heater (Warner Instruments, Hamden, CT). Islets were first perifused with basal glucose (1.67 mmol/l) for 60 minutes and stimulated with glucose (16.7 mmol/l) for 30 minutes, and finally perifused with basal glucose (1.67 mmol/l) until insulin release reversed to the basal level. Samples were collected once per 2 minutes through an automatic fraction collector (Waters, Milford, MA) and analyzed for insulin content by ELISA.
Islet transplantation. A long-term low-dose STZ injection was used to induce diabetes in NSG mice. Briefly, STZ (70 mg/kg) was injected intraperitoneally to NSG mice in the first day of the first week and the third week. Animals were mostly diabetic after 3 weeks as indicated by two consecutive measurements of blood glucose ≥400 mg/dl using a glucometer. Then 500 handpicked human islets (150–300 µm in diameter) were cotransplanted with primary hBMSCs at serial ratios (islet: hBMSC = 1:0, 1:50, 1:100, and 1:200) under the kidney capsule of diabetic NSG mice. The nonfasted glucose levels and body weights of all the mice were measured from the pricked tail of each animal up to 15 weeks after transplantation. PBMCs (5 × 106/mouse) were infused intraperitoneally into these mice at 4 weeks after islet transplantation. At the end of study (17 weeks after islet transplantation), the graft-bearing kidneys were removed from some mice to confirm the function of islet grafts by the return of blood glucose levels to ≥400 mg/dl for 2 consecutive days and all mice were subjected to euthanasia. Serum was collected to measure the level of human insulin and c-peptide by ELISA. Whole blood was collected and human CD4+ T cells were isolated using Dynabeads. The percentage of CD4+ CD25+ FoxP3+ Tregs was determined by flow cytometry.
Intraperitoneal glucose tolerance test. Two weeks after islet transplantation, glucose tolerance was analyzed in overnight-fasted mice as described by Garcia-Ocana et al.34 Briefly, the mice were subjected to intraperitoneal injection of glucose at 2 g/kg of body weight. Blood samples were obtained from the snipped tail at 15, 30, 60, 90, 120, and 180 minutes after injection and analyzed for glucose levels using a glucometer.
Immunofluorescence staining. Mice receiving islets alone and islets cotransplanted with primary hBMSCs were killed 4 weeks after transplantation. The kidneys bearing islets were isolated, washed with phosphate-buffered saline, fixed in 4% paraformaldehyde overnight, and embedded in optimal cutting temperature compound. Frozen sections of 5 µm thickness were cut. To detect insulin-positive human islets, the slides were stained with guinea pig anti-insulin primary antibody (1:200) at 4 °C overnight and Alexa Fluor 568 conjugated goat anti-guinea pig secondary antibody (1:500; Invitrogen) at room temperature for 1 hour. To detect CD3+ T cells, the slides were stained with rabbit anti-CD3 primary antibody (1:500) at 4 °C overnight and Dylight 488-conjugated goat anti-rabbit secondary antibody (1:500; Abcam) at room temperature for 1 hour. Slides were counter-stained with 4′,6-diamidino-2-phenylindole. Three independent images were analyzed using ImageJ to quantify the fluorescence intensity.
Statistical analysis. Statistical significance of the difference between the two groups was determined by unpaired t test and Logrank test in the analysis of blood glucose levels after islet transplantation.
SUPPLEMENTARY MATERIAL Figure S1. Characterization of human bone marrow-derived mesenchymal stem cells (hBMSCs). Figure S2. Human immunity can be rebuilt in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice but not NOD.CB17-Prkdcscid/J (NOD-SCID) mice using human peripheral blood mononuclear cells (PBMCs). Figure S3. Human bone marrow-derived mesenchymal stem cells (hBMSCs) did not induce significant apoptosis to the cocultured peripheral blood mononuclear cells (PBMCs) as determined by Annexin V-FITC staining. Figure S4. Human bone marrow-derived mesenchymal stem cells (hBMSCs) did not need to be conditioned by peripheral blood mononuclear cells (PBMCs) to exert immunosuppressive effect. Materials and methods.
Acknowledgments
We thank the National Institutes of Health (NIH) for financial support (RO1 DK69968). We thank Meifeng Lu and Ernestine Hayes, University of Tennessee Health Science Center, for their support in histology and animal studies, respectively. http://www.uthsc.edu/pharmacy/rmahato. The authors declared no conflict of interest.
Supplementary Material
References
- Narang AS, Mahato RI. Biological and biomaterial approaches for improved islet transplantation. Pharmacol Rev. 2006;58:194–243. doi: 10.1124/pr.58.2.6. [DOI] [PubMed] [Google Scholar]
- Mahato RI, Henry J, Narang AS, Sabek O, Fraga D, Kotb M, et al. Cationic lipid and polymer-based gene delivery to human pancreatic islets. Mol Ther. 2003;7:89–100. doi: 10.1016/s1525-0016(02)00031-x. [DOI] [PubMed] [Google Scholar]
- Wu H, Ye Z, Mahato RI. Genetically modified mesenchymal stem cells for improved islet transplantation. Mol Pharm. 2011;8:1458–1470. doi: 10.1021/mp200135e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Popp FC, Koehl GE, Piso P, Schlitt HJ, Geissler EK, et al. Immunomodulatory effects of mesenchymal stem cells in a rat organ transplant model. Transplantation. 2006;81:1589–1595. doi: 10.1097/01.tp.0000209919.90630.7b. [DOI] [PubMed] [Google Scholar]
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- Ryan JM, Barry FP, Murphy JM, Mahon BP. Mesenchymal stem cells avoid allogeneic rejection. J Inflamm (Lond) 2005;2:8. doi: 10.1186/1476-9255-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hematti P. Role of mesenchymal stromal cells in solid organ transplantation. Transplant Rev (Orlando) 2008;22:262–273. doi: 10.1016/j.trre.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Xu D, Feng G, Bushell A, Muschel RJ, Wood KJ. Mesenchymal stem cells prevent the rejection of fully allogenic islet grafts by the immunosuppressive activity of matrix metalloproteinase-2 and -9. Diabetes. 2009;58:1797–1806. doi: 10.2337/db09-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung TY, Seeberger KL, Kin T, Adesida A, Jomha N, Shapiro AM, et al. Human mesenchymal stem cells protect human islets from pro-inflammatory cytokines. PLoS ONE. 2012;7:e38189. doi: 10.1371/journal.pone.0038189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Itakura S, Todorov I, Rawson J, Asari S, Shintaku J, et al. Mesenchymal stem cell and islet co-transplantation promotes graft revascularization and function. Transplantation. 2010;89:1438–1445. doi: 10.1097/tp.0b013e3181db09c4. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lu W, Mahato RI. Mesenchymal stem cells as a gene delivery vehicle for successful islet transplantation. Pharm Res. 2011;28:2098–2109. doi: 10.1007/s11095-011-0434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazar J, Thomas M, Bezrukov L, Chanturia A, Pekkurnaz G, Yin S, et al. Cytotoxicity mediated by the Fas ligand (FasL)-activated apoptotic pathway in stem cells. J Biol Chem. 2009;284:22022–22028. doi: 10.1074/jbc.M109.032235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–3843. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- Brehm MA, Shultz LD, Greiner DL. Humanized mouse models to study human diseases. Curr Opin Endocrinol Diabetes Obes. 2010;17:120–125. doi: 10.1097/MED.0b013e328337282f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa F, Hester J, Goto R, Nadig SN, Goodacre TE, Wood K. Ex vivo-expanded human regulatory T cells prevent the rejection of skin allografts in a humanized mouse model. Transplantation. 2010;90:1321–1327. doi: 10.1097/TP.0b013e3181ff8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Liu R, Shi D, Liu X, Chen Y, Dou X, et al. Mesenchymal stem cells induce mature dendritic cells into a novel Jagged-2-dependent regulatory dendritic cell population. Blood. 2009;113:46–57. doi: 10.1182/blood-2008-04-154138. [DOI] [PubMed] [Google Scholar]
- Weng J, He C, Lai P, Luo C, Guo R, Wu S, et al. Mesenchymal stromal cells treatment attenuates dry eye in patients with chronic graft-versus-host disease. Mol Ther. 2012;20:2347–2354. doi: 10.1038/mt.2012.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LB, Jiang XB, Yang L. Differentiation of rat marrow mesenchymal stem cells into pancreatic islet β-cells. World J Gastroenterol. 2004;10:3016–3020. doi: 10.3748/wjg.v10.i20.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnieli O, Izhar-Prato Y, Bulvik S, Efrat S. Generation of insulin-producing cells from human bone marrow mesenchymal stem cells by genetic manipulation. Stem Cells. 2007;25:2837–2844. doi: 10.1634/stemcells.2007-0164. [DOI] [PubMed] [Google Scholar]
- Koç ON, Gerson SL, Cooper BW, Dyhouse SM, Haynesworth SE, Caplan AI, et al. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol. 2000;18:307–316. doi: 10.1200/JCO.2000.18.2.307. [DOI] [PubMed] [Google Scholar]
- Chan JL, Tang KC, Patel AP, Bonilla LM, Pierobon N, Ponzio NM, et al. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-γ. Blood. 2006;107:4817–4824. doi: 10.1182/blood-2006-01-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodie TA, Blickarz CE, Devarakonda TJ, He C, Dash AB, Clarke J, et al. Systematic analysis of reportedly distinct populations of multipotent bone marrow-derived stem cells reveals a lack of distinction. Tissue Eng. 2002;8:739–751. doi: 10.1089/10763270260424105. [DOI] [PubMed] [Google Scholar]
- Sato H, Kuwashima N, Sakaida T, Hatano M, Dusak JE, Fellows-Mayle WK, et al. Epidermal growth factor receptor-transfected bone marrow stromal cells exhibit enhanced migratory response and therapeutic potential against murine brain tumors. Cancer Gene Ther. 2005;12:757–768. doi: 10.1038/sj.cgt.7700827. [DOI] [PubMed] [Google Scholar]
- Rasmusson I, Ringdén O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit lymphocyte proliferation by mitogens and alloantigens by different mechanisms. Exp Cell Res. 2005;305:33–41. doi: 10.1016/j.yexcr.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105:2214–2219. doi: 10.1182/blood-2004-07-2921. [DOI] [PubMed] [Google Scholar]
- Groh ME, Maitra B, Szekely E, Koç ON. Human mesenchymal stem cells require monocyte-mediated activation to suppress alloreactive T cells. Exp Hematol. 2005;33:928–934. doi: 10.1016/j.exphem.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, Yu XD, et al. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. 2005;105:4120–4126. doi: 10.1182/blood-2004-02-0586. [DOI] [PubMed] [Google Scholar]
- Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood. 2008;111:1327–1333. doi: 10.1182/blood-2007-02-074997. [DOI] [PubMed] [Google Scholar]
- Boumaza I, Srinivasan S, Witt WT, Feghali-Bostwick C, Dai Y, Garcia-Ocana A, et al. Autologous bone marrow-derived rat mesenchymal stem cells promote PDX-1 and insulin expression in the islets, alter T cell cytokine pattern and preserve regulatory T cells in the periphery and induce sustained normoglycemia. J Autoimmun. 2009;32:33–42. doi: 10.1016/j.jaut.2008.10.004. [DOI] [PubMed] [Google Scholar]
- François M, Romieu-Mourez R, Li M, Galipeau J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther. 2012;20:187–195. doi: 10.1038/mt.2011.189. [DOI] [PubMed] [Google Scholar]
- Brehm MA, Bortell R, Diiorio P, Leif J, Laning J, Cuthbert A, et al. Human immune system development and rejection of human islet allografts in spontaneously diabetic NOD-Rag1null IL2rγnull Ins2Akita mice. Diabetes. 2010;59:2265–2270. doi: 10.2337/db10-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harui A, Kiertscher SM, Roth MD. Reconstitution of huPBL-NSG mice with donor-matched dendritic cells enables antigen-specific T-cell activation. J Neuroimmune Pharmacol. 2011;6:148–157. doi: 10.1007/s11481-010-9223-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ocana A, Takane KK, Reddy VT, Lopez-Talavera JC, Vasavada RC, Stewart AF. Adenovirus-mediated hepatocyte growth factor expression in mouse islets improves pancreatic islet transplant performance and reduces β cell death. J Biol Chem. 2003;278:343–351. doi: 10.1074/jbc.M207848200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.