

Abstract
Chemical-induced read through of premature stop codons might be exploited as a potential treatment strategy for genetic disorders caused by nonsense mutations. Despite the promise of this approach, only a few read-through compounds (RTCs) have been discovered to date. These include aminoglycosides (e.g., gentamicin and G418) and nonaminoglycosides (e.g., PTC124 and RTC13). The therapeutic benefits of these RTCs remain to be determined. In an effort to find new RTCs, we screened an additional ~36,000 small molecular weight compounds using a high-throughput screening (HTS) assay that we had previously developed and identified two novel RTCs, GJ071, and GJ072. The activity of these two compounds was confirmed in cells derived from ataxia telangiectasia (A-T) patients with three different types of nonsense mutation in the ATM gene. Both compounds showed activity comparable to stop codons (TGA, TAG, and TAA) PTC124 and RTC13. Early structure-activity relationship studies generated eight active analogs of GJ072. Most of those analogs were effective on all three stop codons. GJ071 and GJ072, and some of the GJ072 analogs, appeared to be well tolerated by A-T cells. We also identified another two active RTCs in the primary screen, RTC204 and RTC219, which share a key structural feature with GJ072 and its analogs.
Introduction
Approximately 30% of human disease-causing alleles are nonsense mutations.1 To date, there is no efficient treatment for most genetic disorders. Certain compounds can induce read through of premature termination codons (PTCs), which allows translation of full-length protein. The read through–induced protein is often functional, even when it contains a missense amino acid.2,3,4,5 Thus, chemical-induced read through of stop codons caused by nonsense mutations has great potential as a treatment strategy. This read-through strategy could be especially useful for diseases such as ataxia telangiectasia (A-T) and cystic fibrosis. The severity of these diseases may be significantly reduced by restoring even a small amount of affected protein.3,6,7,8,9,10,11,12,13
Despite the promise of this approach, only a few read-through compounds (RTCs) have been described to date. These include aminoglycosides (e.g., gentamicin and G418) and nonaminoglycosides (e.g., PTC124, RTC13, RTC14, and tylosin). Aminoglycosides demonstrate read-through efficiency in many genetic disease models, such as cystic fibrosis,3,10,14,15 Duchenne muscular dystrophy,2,16 Hurler syndrome,17 spinal muscular atrophy,18 and A-T.19 However, the therapeutic benefits of aminoglycosides in clinical trials remain unclear.20,21,22 The systemic toxicity of most commercial aminoglycosides in mammals further diminishes their clinical potential.23,24 The macrolide tylosin has RT activity in prokaryotes and is being further evaluated in patients with somatic APC mutations in colon cancer.25 A nonaminoglycoside RTC, PTC124, was developed by high-throughput screening (HTS) using a luciferase-based reporter assay.13,26 A phase I clinical study in cystic fibrosis showed that PTC124 was well tolerated and more effective than aminoglycosides.27 However, PTC124's phase 2b clinical study in Duchenne muscular dystrophy patients failed to show significant improvement in the 6-minute walk distance or dystrophin expression.28 Moreover, PTC124 is not equally effective with all three stop codons, working best on the TGA stop codon.26 This selective activity would limit the number of patients who might benefit from PTC124. Moreover, PTC124 does not effectively cross the blood–brain barrier, a critical factor for treating neurological disorders such as A-T, Alzheimer diseases, and the CNS effects encountered in Hurler patients. Therefore, successfully developing new RTCs with optimized efficacy and low toxicity holds great promise for numerous genetic diseases that are caused by nonsense mutations. Furthermore, the potential therapeutic benefit of a small molecular read-through (SMRT) compound may also extend to cancer-prone individuals carrying highly penetrant nonsense mutations in genes, such as BRCA1, BRCA2, and CHEK2, if such medications were proven safe for long-term prophylactic therapy, and cancer-preventing benefits could be convincingly documented.
In an effort to develop novel RTCs, we previously described two SMRT compounds, RTC13 and RTC14, that can read through nonsense mutations in both the ATM and dystrophin genes.6 We further demonstrated that RTC13 has in vivo read-through activity in mdx mice (a Duchenne muscular dystrophy model carrying a TAA nonsense mutation in exon23 of the dystrophin gene) that induces dystrophin protein.12 Recently, we were able to generate a series of active RTC13 analogs that are currently being characterized in different animal models with nonsense mutations.29 Herein, we report a second series of SMRT chemicals, exemplified by GJ071 and GJ072, that are comparable to or better than RTC13 or PTC124. We further describe two additional compounds identified in our primary screen, RTC204 and RTC219, that share a key structural element and read-through activity with GJ072 and its analogs.
Results
HTS identified two novel small molecular weight RTCs
We previously developed a luciferase-independent HTS assay, protein transcription-translation (PTT)–enzyme-linked immunosorbent assay (ELISA), that allowed us to screen small molecular weight libraries.6 The PTT-ELISA was based on an in vitro transcription and translation system driven by a plasmid containing “region 5” of the ATM gene (coding sequence for 403-1986 aa) with a TGA C mutation (c.5623C>T; Arg1875X). This assay has proven very reliable in that most “hits” previously selected by the assay have had RT activity. In this study, we screened ~36,000 additional compounds and were able to identify two novel active parent compounds, GJ071 and GJ072. Both compounds have structures different from any previously reported RTCs (structures shown in Figure 1a). Their in vitro read-through activity on a TGA mutation was subsequently confirmed by PTT-ELISA in dose-response experiments (Figure 1b). This activity was again confirmed in tertiary assays with A-T cells carrying each of the three types of nonsense mutations (see below).
Figure 1.

HTS of chemical libraries identified two novel SMRT compounds, GJ071 and GJ072. (a) Chemical structures of GJ071 and GJ072. (b) In vitro read-through activity was measured by PTT-ELISA using a plasmid containing a disease-causing ATM mutation, 5623C>T (TGA C). The compounds were tested independently. G418 was used as positive control for each experiment (not shown). The experiments were repeated multiple times; a representative experiment is shown here and raw RLU numbers are used. *P ≤ 0.05 as compared with untreated control. A-T, ataxia telangiectasia; SMRT, small molecular read through.
RTCs induced read through of different PTCs in A-T cells
To characterize the cellular read-through activity of GJ071 and GJ072, we selected three A-T lymphoblastoid cell lines derived from patients containing different types of homozygous ATM nonsense mutations: AT153LA (c.8977C>T, TGA A, Arg2993X), AT229LA (c.3102T>G, TAG A, Tyr1034X), and AT187LA (c.5908C>T, TAA G, Gln1970X). The A-T cells were treated with each RTC for 4 days and then harvested to measure ATM kinase activity, which is often completely deficient in A-T cells. Both GJ071 (Figure 2a) and GJ072 (Figure 2b) induced ATM kinase on both TGA and TAG stop codons and restored ATMpSer1981 autophosphorylation and SMC1pSer966 transphosphorylation as measured by FACS. Notably, GJ071 and GJ072 were also active in A-T cells with a homozygous TAA mutation (a stop codon that was previously reported to be more difficult to read through than TGA and TAG) and induced ATMpSer1981 foci formation after radiation (IRIF) (Figure 3a and 3b). Furthermore, both compounds were also able to induce detectable full-length ATM protein in treated A-T cells, as demonstrated by ATM-ELISA (Figure 3c).
Figure 2.
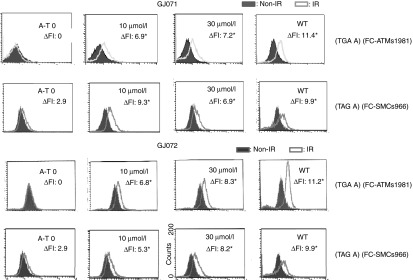
GJ071 and GJ072 induced ATM kinase activity in A-T cells carrying homozygous TGA or TAG stop codons. A-T cells were treated with each compound for 4 days before harvesting. The ATM kinase activity in AT153LA cells (TGA A) was measured by FC-based ATMs1981 autophosphorylation (FC-ATMs1981), and the ATM kinase activity in AT229LA cells (TAG A) was measured by FC-based SMC1s966 transphosphorylation (FC-SMC1s966). The experiments were repeated multiple times; representative experiments for FC-ATMs1981 and FC-SMC1s966 are shown here. A right shift of the fluorescence intensity peaks (ΔFI) indicated the restored ATM kinase activity by both compounds (top: GJ071; bottom: GJ072). *P ≤ 0.05 compared with nontreated A-T cells. A-T, ataxia telangiectasia.
Figure 3.
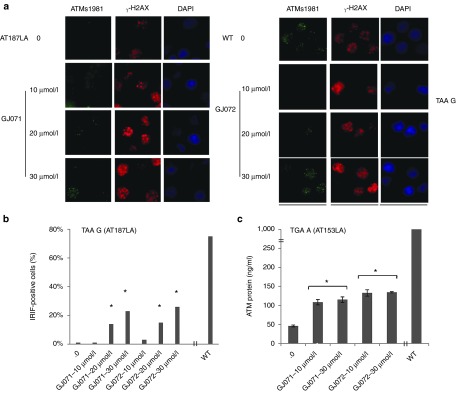
GJ071 and GJ072 induced ATMs1981 foci formation in A-T cells with a TAA mutation; full-length ATM protein levels were abrogated in A-T cells with a TGA mutation. A-T cells were treated with each compound for 4 days prior to testing for read-through effects. Wild-type cells were used as positive controls. (a) RTC-induced ATM1981 foci in AT187LA cells with a homozygous TAA mutation. The experiments were repeated multiple times; a representative experiment is used here to show ATMs1981 foci formation after 2-Gy radiation. The formation of γ-H2AX foci was included as a control for effective radiation damage; (b) Quantification of Figure 3a for IRIF-positive cells percentage; >120 cells were counted for each sample; (c) RTC-induced ATM protein in AT153LA cells with TGA mutation measured by ATM-ELISA. *P ≤ 0.05, compared with nontreated A-T cells. A-T, ataxia telangiectasia; RTC, read-through compounds.
Since a hallmark of A-T is the hypersensitivity of cells to ionizing radiation, we further assessed whether GJ071 and GJ072 could abrogate cellular radiosensitivity in A-T cells. AT242LA cells with two heterozygous stop codons, TGA (7096G>Ta, E2366X and TAG (7913G>Ab, W2638X), were treated with each compound and followed by colony survival assay. Both GJ071 and GJ072 improved AT242LA cell radiosensitivity from the “radiosensitive” range (<21 SF%) to the “intermediate” range (21–36 SF%) (Supplementary Figure S1); these ranges are currently used for A-T clinical diagnosis.30
Structure-activity relationship studies used to derive active analogs of GJ072
To establish a structure-activity relationship and improve design of novel SMRT compounds, we synthesized and tested analogs of both GJ071 and GJ072. Each analog was tested at 10 and 30 µmol/l. For GJ071, none of the six analogs of GJ071 showed significant read-through activity (data not shown). In contrast, eight of the 11 GJ072 analogs induced ATM kinase activity in A-T cells carrying different homozygous nonsense mutations; their chemical structures are shown in Figure 4. The activity of these GJ072 active analogs in TGA-containing A-T cells was demonstrated by increased FCATMpSer1981 levels, indicated by “ΔFI” (fluorescence intensity difference between paired non-irradiation (IR) and IR samples) (Figure 5), as well as by IR-induced ATMs1981 foci formation assay (IRIF) (some analogs shown in Supplementary Figure S2). Similar to the parent compound GJ072, these analogs were also active in A-T cells containing TAG or TAA stop codons. The activity of the analogs on TAA PTC (in AT187LA cells) was demonstrated by both the FCATMpSer1981 assay (Figure 6) and ATMpSer1981-IRIF assay (Supplementary Figure S3). The activity of the analogs on a TAG stop codon (AT229LA cells) is shown in Supplementary Figures S4 and S5. Notably, some GJ072 analogs (e.g., GJ103, GJ106, GJ109, and GJ111) consistently demonstrated their activities in all three PTCs by both FCATMpSer1981 (Figures 5 and 6, Supplementary Figure S4b) and IRIF assays (Supplementary Figures S2 and S5), implying their potential for further development. To date, we have developed a water soluble salt form (GJ103-salt) for one of these analogs, GJ103, which will enable us to evaluate its in vivo activity by systematic administration.
Figure 4.
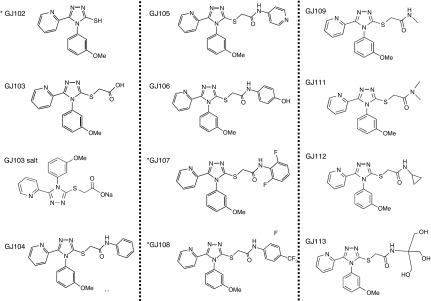
Structures of synthesized GJ072 analogs. The synthesis of the parent compounds and their analogs is described in supplemental materials (see compound preparation). All were active except GJ102, GJ107, and GJ108. *Inactive; see text and Figure 5.
Figure 5.

The effect of GJ072 analogs on TGA PTC in A-T cells measured by FC-ATMs1981. AT153LA cells with a homozygous TGA mutation were exposed to RTCs for 4 days before assaying. Restored ATM kinase activity was indicated by increased fluorescence intensity measured by flow cytometry (ΔFI). (a) Activity of GJ102, GJ103, GJ104, GJ105 and GJ106. The data shown were summarized from four independent experiments; (b) Activity of GJ109, GJ111, GJ112 and GJ113, the data shown were summarized from four independent experiments. *P ≤ 0.05 compared with nontreated A-T cells. A-T, ataxia telangiectasia; RTC, read-through compounds.
Figure 6.

The effect of GJ072 analogs on TAA in A-T cells measured by FC-ATMs1981. AT187LA cells with homozygous TAA mutation were exposed to RTCs for 4 days before assaying. Restored ATM kinase activity was indicated by increased “ΔFI.” (a) Activity of GJ103, GJ104, GJ105, and GJ106; (b) Activity of GJ109, GJ111, GJ112, and GJ113. Data shown in (a) and (b) were derived from three independent experiments, respectively. *P ≤ 0.05 as compared with nontreated A-T cells. A-T, ataxia telangiectasia.
Interestingly, we also identified two other active compounds in the primary screen, RTC204 and RTC219; both share a key structural feature with GJ072 and its analogs. In particular, RTC219 has the 3-(amidomethylthio)1,2,4-triazole unit of the earlier compounds but with different substituents at positions 4 and 5, while RTC204 has a somewhat related 5-(acylhydrazidomethylthio)tetrazole unit. The structures and activities of RTC204 and RTC219 in A-T cells are shown in Figure 7.
Figure 7.

GJ204 and GJ219 identified from primary screen shared a similar structural feature with GJ072. (a) Chemical structures of RTC204 and RTC219; (b) RTC204 and RTC219 restored ATMs1981 autophosphorylation in AT153LA cells with homozygous TGA A mutation after a 4-day treatment (indicated by the right shift of fluorescence peak). The experiments were repeated multiple times; a representative experiment is used to show histogram change of fluorescence intensity. *P ≤ 0.05 as compared with nontreated A-T cells. A-T, ataxia telangiectasia.
Comparison of GJ071 and GJ072 with other known RTCs
We next compared the efficacy of GJ071 and GJ072 with two known RTCs, RTC13, and PTC124.6,26 We also included an active analog of GJ072, GJ103, since its acid functionality could be converted to a water soluble form (GJ103-salt) for further evaluation. As shown in Figure 8, GJ072 and its analog GJ103 showed a similar activity to RTC13 and were arguably better than PTC124 in A-T cells (AT153LA, TGA A). Moreover, GJ071, GJ072, and some analogs of GJ072 (such as GJ103 and GJ105) did not show obvious cytotoxicity in A-T cells at concentration as high as 300 µmol/l (data not shown), suggesting that the new series of RTCs reported here may have greater potential for drug development.
Figure 8.

Read-through activity comparison of GJ071 and GJ072 with other known RTCs in A-T cells. AT153LA cells with homozygous TGA mutation were exposed to GJ071, GJ072, GJ103, RTC13, and PTC124 for 4 days, respectively, and ATM kinase activity was measured by FC-ATMs1981. Restored ATM kinase activity was indicated by “ΔFI.” Data shown were derived from three independent experiments. *P ≤ 0.05; **P ≤ 0.01 as compared with nontreated A-T control. A-T, ataxia telangiectasia; RTC, read-through compounds.
Discussion
In this study, we identified and characterized the read-through activity of two new groups of SMRT compounds, GJ071 and GJ072, and their analogs. This expands our experience to four parent SMRT groups: RTC13, RTC14, GJ071, and GJ072. GJ072 and RTC13 share a superficial similarity with previously described PTC124 in terms of their three-ring structures, while GJ071 and RTC14 have very different structures. Our data showed that GJ071 and GJ072 and some of their analogs (such as GJ103) had similar read-through activity as RTC13 or RTC14, but were more tolerable than RTC13 and RTC14 to A-T cells. The concentrations (10 and 30 µmol/l) we used for all analogs were based on the optimum range for the parent compounds; for the parent compounds, we did not see significant improvement of response in A-T cells at higher concentrations (data not shown). Importantly, similar to RTC13 and RTC14, these novel RTCs were active for all three premature stop codons in A-T cells. Considering the limited market potential for SMRT drugs on rare genetic diseases, the “one-drug-fits-all” model would be a distinct advantage for translational potential.
Most of the active analogs of GJ072 had low cLogP values, which is predictive of good absorption or permeation potential in vivo. In addition, the GJ103 salt form is water soluble, making it much easier to work with in in vivo experiments. Importantly, we found that two additional compounds identified in this primary screen, RTC204 and RTC219, shared a similar structural feature with GJ072 and its analogs, implying that this structural feature may be responsible for their read-through activity. Since all of the known RTCs are thought to function by interfering with parts of the ribosome,31,32 the potential ribosomal interactions of these new SMRT compounds need further attention.
The influence of nonsense-mediated decay on PTC read-through strategy should be considered since mRNAs carrying nonsense mutations are degraded by this pathway33,34 and inhibition of nonsense-mediated decay may, therefore, stabilize mutant mRNA transcripts and increase RTC-induced read-through output.35 These mutations may also be missed, if sequencing is performed on cDNA derived from patient cells without first inhibiting nonsense-mediated decay. Other major issues related to read through in A-T patients that require further studies include (i) the potential risk of triggering inappropriate “retroactive” apoptosis after restoring ATM protein to A-T cells,36 (ii) the requirement that ATM induction occurs in cerebellar cells, that is, a SMRT drug's ability to cross the blood–brain barrier,37,38 and (iii) the potential side effects of these compounds on normal stop codons. It also remains possible that substituting a “false” amino acid for a premature stop codon in ATM protein, especially in the key functional domains, may disrupt normal function. To date, we have preliminary data to suggest that many of our SMRT drugs will reach the cerebellum, a key site for reversing the neuropathology of A-T. The possibility of read through of normal stop codons by other RTCs has been investigated in numerous studies, but none have described read through of the final normal stop codon in the last exon. We are presently evaluating the activity and potential toxicities of a lead SMRT compound on AT-derived neural progenitor cells.39 Further development of these compounds may eventually contribute to the identification of a drug candidate with improved pharmacokinetic properties and minimal off-target toxicity for genetic diseases caused by nonsense mutations.
Materials and Methods
Lymphoblastoid cells. Different A-T Lymphoblastoid cells were used to assess ATM functions and cytotoxicity of RTCs. These include AT153LA, AT229LA, AT187LA, and AT242LA. All cells were derived from fresh blood samples from our laboratory, following which they were anonymized in accordance with Institutional Review Board guidance.
Chemical libraries and analogs preparation. Chemical libraries screened were provided by Molecular Shared Screening Resources, California NanoSystems Institute, UCLA. All the analogs were prepared according the protocols in Supplementary Materials.
PTT-ELISA and HTS. The HTS was based on a cell-free PTT-ELISA system that was developed in our laboratory.6,40 Approximately 36,000 compounds were screened to identify novel RTCs. Each compound was screened at a final concentration of 10 µM. Screening was performed on a fully integrated CORE System (Beckman Coulter-SAGIAN, Indianapolis, IN).
Flow cytometry analysis of ATMpSer1981 and SMC1pSer966 phosphorylation. FCpSerSMC1 and FCATMpSer1981 assay was performed as previously described.6,41 In brief, cells were resuspended in PBS and radiated for 10 Gy. After 1 hour, the cells were fixed and permeabilized using the FIX and PERM cell permeabilization kit (Invitrogen, Grand Island, NY). The cells were then incubated with ATMpSer1981 antibody or SMC1pSer966 antibody for 2 hours at room temperature. Cells were washed and stained with Alexo488-anti-mouse IgG (Invitrogen) for 45 minutes. Cells were then washed, resuspended in PBS with 0.2% paraformaldehyde, and analyzed using FACScan (BD Biosciences, San Jose, CA).
Immunofluorescence of ATMpSer1981-IRIF. Immunostaining of ATMpSer1981 nuclear foci was performed as previously described.6,42 Briefly, cells were irradiated with 2 Gy after a 4-day RTC exposure period and incubated at 37 °C for 30 minute. The cells were dropped onto coverslips, fixed with paraformaldehyde, and permeabilized. The coverslips were then blocked with PBS with 10% FBS and incubated with mouse anti-ATM pSer1981 for 1 hour at RT (Rockland Immunochemicals, Gilbertsville, PA). Cells were washed and blocked again for 1 hour and then stained with FITC-conjugated anti-mouse IgG (1:150; Jackson ImmunoResearch, West Grove, PA) and mounted onto slides.
Immunoassay for measurement of intranuclear ATM protein. Read through-induced full-length ATM protein in A-T cells was measured by ATM-ELISA immunoassay (Butch et al. 2004). Cell nuclear extracts were prepared using NE-PER protocol (Pierce, Rockford, IL). Then, ATM-ELISA was performed using 200 µg of nuclear extracts. ATM concentrations of tested samples were calculated from the standard calibration curve, using purified ATM protein.
Colony survival assay. Colony survival assay was performed as previously described.30,43 A-T cells were treated and plated in duplicate in 96-well plates at 100 and 200 cells per well, respectively. One of the duplicate plates was exposed to 1.0 Gy radiation, whereas the other one was not irradiated. The cells were incubated for 10–13 days and then stained with MTT.
Cytotoxicity by XTT assay. Cytotoxicity was measured by Cell Proliferation Kit II (Roche Applied Science, Indianapolis, IN). Briefly, cells were seeded into a flat-bottom 96-well plate, including control wells containing complete growth medium alone as blank absorbance readings. After RTC treatment, activated-XTT Solution was added into each well, and the cells were returned to the cell culture incubator for 12–14 hours. The absorbance was measured at 480 nmol/l with relevant 630 nmol/l to assess nonspecific readings.
SUPPLEMENTARY MATERIAL Figure S1. GJ071 and GJ072 abrogated the radiosensitivity of A-T LCLs. Figure S2. Active GJ072 analogs induced ATMs1981 foci in AT153LA cells with TGA PTC. Figure S3. Active GJ072 analogs induced ATMs1981 foci in AT187LA cells with TAA PTC. Figure S4. GJ072 analogs induced read through of TAG PTC in A-T cells. Figure S5. Active GJ072 analogs induced ATMs1981 foci in AT229LA cells with TAG PTC.
Acknowledgments
This work was supported by National Institutes of Health grant 1R01NS052528, A-T Ease Foundation, A-T Medical Research Foundation, A-T Medical Research Trust, and the A-T Society. FC was performed in the UCLA Jonsson Comprehensive Cancer Center and Center for AIDS Research Flow Cytometry Core Facility that is supported by NIH awards CA-16042 and AI-28697, and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA.
Supplementary Material
References
- Mendell JT, Dietz HC. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell. 2001;107:411–414. doi: 10.1016/s0092-8674(01)00583-9. [DOI] [PubMed] [Google Scholar]
- Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Sequence specificity of aminoglycoside-induced stop condon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol. 2000;48:164–169. [PubMed] [Google Scholar]
- Du M, Keeling KM, Fan L, Liu X, Bedwell DM. Poly-L-aspartic acid enhances and prolongs gentamicin-mediated suppression of the CFTR-G542X mutation in a cystic fibrosis mouse model. J Biol Chem. 2009;284:6885–6892. doi: 10.1074/jbc.M806728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingman LV, Park S, Olson TM, Alekseev AE, Terzic A. Aminoglycoside-induced translational read-through in disease: overcoming nonsense mutations by pharmacogenetic therapy. Clin Pharmacol Ther. 2007;81:99–103. doi: 10.1038/sj.clpt.6100012. [DOI] [PubMed] [Google Scholar]
- Keeling KM, Bedwell DM. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdiscip Rev RNA. 2011;2:837–852. doi: 10.1002/wrna.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Damoiseaux R, Nahas S, Gao K, Hu H, Pollard JM, et al. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J Exp Med. 2009;206:2285–2297. doi: 10.1084/jem.20081940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Kayali R, Bertoni C, Fike F, Hu H, Iversen PL, et al. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum Mol Genet. 2011;20:3151–3160. doi: 10.1093/hmg/ddr217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am J Respir Cell Mol Biol. 2002;27:619–627. doi: 10.1165/rcmb.2001-0004OC. [DOI] [PubMed] [Google Scholar]
- Gilad S, Bar-Shira A, Harnik R, Shkedy D, Ziv Y, Khosravi R, et al. Ataxia-telangiectasia: founder effect among north African Jews. Hum Mol Genet. 1996;5:2033–2037. doi: 10.1093/hmg/5.12.2033. [DOI] [PubMed] [Google Scholar]
- Kerem E. Pharmacologic therapy for stop mutations: how much CFTR activity is enough. Curr Opin Pulm Med. 2004;10:547–552. doi: 10.1097/01.mcp.0000141247.22078.46. [DOI] [PubMed] [Google Scholar]
- Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst) 2004;3:1187–1196. doi: 10.1016/j.dnarep.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Kayali R, Ku JM, Khitrov G, Jung ME, Prikhodko O, Bertoni C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum Mol Genet. 2012;21:4007–4020. doi: 10.1093/hmg/dds223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci USA. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–1284. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- Loufrani L, Dubroca C, You D, Li Z, Levy B, Paulin D, et al. Absence of dystrophin in mice reduces NO-dependent vascular function and vascular density: total recovery after a treatment with the aminoglycoside gentamicin. Arterioscler Thromb Vasc Biol. 2004;24:671–676. doi: 10.1161/01.ATV.0000118683.99628.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling KM, Brooks DA, Hopwood JJ, Li P, Thompson JN, Bedwell DM. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum Mol Genet. 2001;10:291–299. doi: 10.1093/hmg/10.3.291. [DOI] [PubMed] [Google Scholar]
- Sossi V, Giuli A, Vitali T, Tiziano F, Mirabella M, Antonelli A, et al. Premature termination mutations in exon 3 of the SMN1 gene are associated with exon skipping and a relatively mild SMA phenotype. Eur J Hum Genet. 2001;9:113–120. doi: 10.1038/sj.ejhg.5200599. [DOI] [PubMed] [Google Scholar]
- Lai CH, Chun HH, Nahas SA, Mitui M, Gamo KM, Du L, et al. Correction of ATM gene function by aminoglycoside-induced read-through of premature termination codons. Proc Natl Acad Sci USA. 2004;101:15676–15681. doi: 10.1073/pnas.0405155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politano L, Nigro G, Nigro V, Piluso G, Papparella S, Paciello O, et al. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003;22:15–21. [PubMed] [Google Scholar]
- Wilschanski M, Famini C, Blau H, Rivlin J, Augarten A, Avital A, et al. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Respir Crit Care Med. 2000;161 3 Pt 1:860–865. doi: 10.1164/ajrccm.161.3.9904116. [DOI] [PubMed] [Google Scholar]
- Clancy JP, Bebök Z, Ruiz F, King C, Jones J, Walker L, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med. 2001;163:1683–1692. doi: 10.1164/ajrccm.163.7.2004001. [DOI] [PubMed] [Google Scholar]
- Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 1999;43:1003–1012. doi: 10.1128/aac.43.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan MX, Fischel-Ghodsian N, Attardi G. A biochemical basis for the inherited susceptibility to aminoglycoside ototoxicity. Hum Mol Genet. 2000;9:1787–1793. doi: 10.1093/hmg/9.12.1787. [DOI] [PubMed] [Google Scholar]
- Zilberberg A, Lahav L, Rosin-Arbesfeld R. Restoration of APC gene function in colorectal cancer cells by aminoglycoside- and macrolide-induced read-through of premature termination codons. Gut. 2010;59:496–507. doi: 10.1136/gut.2008.169805. [DOI] [PubMed] [Google Scholar]
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007;47:430–444. doi: 10.1177/0091270006297140. [DOI] [PubMed] [Google Scholar]
- Guglieri M, Bushby K. Molecular treatments in Duchenne muscular dystrophy. Curr Opin Pharmacol. 2010;10:331–337. doi: 10.1016/j.coph.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Jung ME, Ku JM, Du L, Hu H, Gatti RA. Synthesis and evaluation of compounds that induce readthrough of premature termination codons. Bioorg Med Chem Lett. 2011;21:5842–5848. doi: 10.1016/j.bmcl.2011.07.107. [DOI] [PubMed] [Google Scholar]
- Sun X, Becker-Catania SG, Chun HH, Hwang MJ, Huo Y, Wang Z, et al. Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. J Pediatr. 2002;140:724–731. doi: 10.1067/mpd.2002.123879. [DOI] [PubMed] [Google Scholar]
- Rowe SM, Clancy JP. Pharmaceuticals targeting nonsense mutations in genetic diseases: progress in development. BioDrugs. 2009;23:165–174. doi: 10.2165/00063030-200923030-00003. [DOI] [PubMed] [Google Scholar]
- Gatti RA. SMRT compounds correct nonsense mutations in primary immunodeficiency and other genetic models. Ann N Y Acad Sci. 2012;1250:33–40. doi: 10.1111/j.1749-6632.2012.06467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MF, Shyu AB. RNA surveillance by nuclear scanning. Nat Cell Biol. 2002;4:E144–E147. doi: 10.1038/ncb0602-e144. [DOI] [PubMed] [Google Scholar]
- Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- Linde L, Boelz S, Nissim-Rafinia M, Oren YS, Wilschanski M, Yaacov Y, et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117:683–692. doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, McKinnon PJ. ATM dependent apoptosis in the nervous system. Apoptosis. 2000;5:523–529. doi: 10.1023/a:1009637512917. [DOI] [PubMed] [Google Scholar]
- Gatti RA, Vinters HV. Cerebellar pathology in ataxia-telangiectasia: the significance of basket cells. Kroc Found Ser. 1985;19:225–232. [PubMed] [Google Scholar]
- Vinters HV, Gatti RA, Rakic P. Sequence of cellular events in cerebellar ontogeny relevant to expression of neuronal abnormalities in ataxia-telangiectasia. Kroc Found Ser. 1985;19:233–255. [PubMed] [Google Scholar]
- Lee P, Martin NT, Nakamura K, Azghadi S, Amiri M, Ben-David U, et al. SMRT compounds abrogate cellular phenotypes of ataxia telangiectasia in neural derivatives of patient-specific hiPSCs. Nat Commun. 2013;4:1824. doi: 10.1038/ncomms2824. [DOI] [PubMed] [Google Scholar]
- Du L, Lai CH, Concannon P, Gatti RA. Rapid screen for truncating ATM mutations by PTT-ELISA. Mutat Res. 2008;640:139–144. doi: 10.1016/j.mrfmmm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Nahas SA, Butch AW, Du L, Gatti RA. Rapid flow cytometry-based structural maintenance of chromosomes 1 (SMC1) phosphorylation assay for identification of ataxia-telangiectasia homozygotes and heterozygotes. Clin Chem. 2009;55:463–472. doi: 10.1373/clinchem.2008.107128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Pollard JM, Gatti RA. Correction of prototypic ATM splicing mutations and aberrant ATM function with antisense morpholino oligonucleotides. Proc Natl Acad Sci USA. 2007;104:6007–6012. doi: 10.1073/pnas.0608616104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo YK, Wang Z, Hong JH, Chessa L, McBride WH, Perlman SL, et al. Radiosensitivity of ataxia-telangiectasia, X-linked agammaglobulinemia, and related syndromes using a modified colony survival assay. Cancer Res. 1994;54:2544–2547. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.