

Abstract
Immune responses directed against viral capsid proteins constitute a main safety concern in the use of adeno-associated virus (AAV) as gene transfer vectors in humans. Pharmacological immunosuppression has been proposed as a solution to the problem; however, the approach suffers from several potential limitations. Using MHC class II epitopes initially identified within human IgG, named Tregitopes, we showed that it is possible to modulate CD8+ T cell responses to several viral antigens in vitro. We showed that incubation of peripheral blood mononuclear cells with these epitopes triggers proliferation of CD4+CD25+FoxP3+ T cells that suppress killing of target cells loaded with MHC class I antigens in an antigen-specific fashion, through a mechanism that seems to require cell-to-cell contact. Expression of a construct encoding for the AAV capsid structural protein fused to Tregitopes resulted in reduction of CD8+ T cell reactivity against the AAV capsid following immunization with an adenoviral vector expressing capsid. This was accompanied by an increase in frequency of CD4+CD25+FoxP3+ T cells in spleens and lower levels of inflammatory infiltrates in injected tissues. This proof-of-concept study demonstrates modulation of CD8+ T cell reactivity to an antigen using regulatory T cell epitopes is possible.
Introduction
Clinical gene therapy has made considerable progress over the past several years. Despite the recently reported successful clinical trial results using adeno-associated virus (AAV) vectors,1 the most commonly used vector for in vivo gene transfer, host immune responses directed against the AAV capsid remain a major obstacle to achieving long-term correction of disease phenotype.2 A capsid-specific CD8+ T cell response was initially identified as the cause of short-lived transgene expression in the first clinical trial in which an AAV2 vector expressing coagulation factor IX (FIX) was introduced into the liver of severe hemophilia B subjects.3 In this trial, upon AAV gene transfer to liver, two subjects developed transient elevation of liver enzymes consequent to what appeared to be the immune rejection of transduced hepatocytes mediated by capsid-specific CD8+ T cells.4 A similar set of observations was recently made in the context of a clinical trial of hepatic gene transfer for FIX with an AAV8 vector.5 Results from these studies suggest that AAV vector administration in humans results in activation of capsid-specific CD8+ T cells in a vector dose-dependent fashion, with loss of transgene expression and increase in liver enzymes occurring above a certain vector dose threshold.
AAV vectors are nonreplicating recombinant viruses carrying a single-stranded DNA genome devoid of viral coding sequence; this genome is contained in a protein capsid comprising three structural proteins, VP1, VP2, and VP3.6 Upon infection, the viral vector capsid is present within a target cell for a defined period of time and is degraded through the proteasome pathway,7,8,9 leading to MHC class I (MHC I) presentation of AAV capsid epitopes, which ultimately flags transduced cells for destruction by capsid-specific CD8+ T cells.7,8 Humans are naturally exposed to AAV, and develop both humoral and cellular immunity to the virus early in life.10,11,12 Whereas anti-AAV antibodies can completely block vector transduction, particularly when the vector is delivered through the bloodstream,3,13 loss of therapeutic transgene expression is believed to be related to the concomitant presentation of capsid antigen and activation of capsid-specific CD8+ T cells, resulting in clearance of AAV vector-transduced cells.3,4,5,14
Immune suppression can be used to induce tolerance in a variety of settings.15 In the most recent AAV8-FIX trial, capsid T cell responses were controlled with the administration of a short course of high-dose oral prednisolone;5 however the safety and efficacy of this intervention at higher vector doses, such as the doses required to achieve therapeutic efficacy in diseases like hemophilia A or muscular dystrophy, remains unknown. Additional concerns over the risks associated with the use of immunosuppression, as well as the fact that administration of steroids in certain subsets of patients is not recommended, have prompted the exploration of alternative strategies for the modulation of capsid T cell responses.
Regulatory T cell (Treg)-mediated immunomodulation has been explored as a therapeutic strategy in transplantation16 and autoimmunity.17 Tregs play a central role in the maintenance of peripheral tolerance and the control of immune responses. Tregs have been shown to downregulate effector responses via a variety of mechanisms, which include the consumption of IL-2, secretion of suppressor cytokines, interference of antigen-presenting cell-mediated activation of effector T cells (Teff), the cytolysis of Teff, or direct cell-cell interaction mediated by surface receptor(s) at the surface of Treg and Teff.18 Whereas in some cases, the interaction between Treg and Teff results in the death or cell cycle arrest of Teff, in some cases, it results in anergy, a state of unresponsiveness that can be reversed by removing Tregs.19
De Groot and colleagues first described the use of MHC class II (MHC II) epitopes located in the Fc region of IgG to modulate immunity.20 Coadministration of these regulatory T cell epitopes (Tregitopes) with immunogenic antigen reduces immune response in vitro and in vivo.21,22,23,24 In particular, Tregitopes have been shown to downregulate CD4+ T cell responses in the setting of common immunogens20 and in models of autoimmune diseases such as experimental colitis.22 The postulated mechanism of action of Tregitopes involves the presentation of the peptides onto MHC II by antigen-presenting cells, resulting in early secretion of IL-10 and promoting proliferation of Tregs. However, no study has been published so far on the effect of Tregitopes on CD8+ T cell responses; similarly, insights on the mechanism(s) of suppression of Teff responses by Tregitope-induced Tregs are still lacking.
Here, we assessed the potential of these peptides as modulators of capsid-specific CD8+ T cell immunity and provide new insight as to their mechanism of action. In vitro experiments using human cells show that co-incubation of Tregitopes with AAV-derived epitopes completely blunts capsid-specific CD8+ T cell responses and results in robust expansion of CD4+CD25+FoxP3+ T cells. Additional experiments suggest that Tregitopes expand antigen-specific Tregs, and that these peptides can be used to modulate Th1 responses directed against several antigens in the context of multiple MHC I alleles. Finally, expression of Tregitopes in vivo also decreases CD8+ T cell responses directed against the AAV capsid, generally reducing AAV capsid immunogenicity. Therefore, our work provides proof of concept for an alternative strategy to modulate Th1-driven immunity to AAV and possibly other antigens.
Results
IgG-derived MHC II epitopes (Tregitopes) modulate AAV capsid-driven CD8+ T cell responses in vitro
To test the ability of IgG-derived MHC II epitopes (Tregitopes) to modulate AAV capsid-directed T cell responses, we used an in vitro experimental model previously described (Supplementary Figure S1).7,8 Briefly, human peripheral blood mononuclear cells (PBMCs) were restimulated in vitro with AAV capsid-derived MHC I epitopes and Tregitopes or control peptides in the presence of IL-2 before using them as effectors (Teff) in a CTL assay against HLA-matched peptide-loaded or AAV-transduced targets. Restimulation of Teff in the presence of Tregitope 167 (hTreg167), but not in the presence of three different scrambled versions of the same peptide, resulted in an almost complete loss of CTL activity against HLA-matched targets loaded with the HLA-B*0702 epitope VPQYGYLTL from AAV (Figure 1a). In this in vitro experimental system, we tested PBMCs from different donors with various MHC II haplotypes, showing a slight but not statistically significant (P = 0.1873, linear regression), correlation between the affinity of Tregitopes for one subject's MHC II alleles (iTEM score)25 and the inhibition of Teff responses (Supplementary Figure S2). Then, we tested whether Tregitope modulation of CTL responses was also effective in the context of MHC I antigens naturally processed and presented via the proteasome pathway by transducing HLA-B*0702 targets with AAV vectors at increasing multiplicity of infections, showing effective inhibition of CTL activity (Figure 1b). Restimulation of PBMCs in the presence of hTreg167 resulted in a profound downregulation of markers of T cell activation and effector functions (Figure 1c). Inhibition of Teff responses upon restimulation in the presence of hTreg167 was achieved with MHC I peptide epitopes derived from several additional HLA-B*0702-restricted viral antigens (Figure 2a–2d) but also with MHC I epitopes with different HLA restrictions (Figure 2e) and with hTreg289 (Figure 2f), a peptide with a different MHC II binding profile that was also derived from the Fc portion of human IgG.20 Suppression of CTL responses with hTreg289 was less effective than with hTreg167, which reflects the HLA haplotype of the donor (DRB1*0701 and DRB1*1501) and the fact that hTreg289 is predicted to bind only to DRB1*0701, while hTreg167 binds to both alleles.20 PBMCs labeled with CFSE prior to in vitro restimulation with AAV capsid epitopes showed that in the presence or absence of hTreg167, both CD4+ and CD8+ T cells proliferate (Supplementary Figure S3); however, CD8+ T cells restimulated in the presence of Tregitopes exhibited lower levels of expression of the activation marker CD25 (Figure 3a,b), while there was an increase in the frequency of CD25+CD4+ T cells (vide infra). ELISA assay on conditioned media from PBMC cultured in the presence of Tregitopes showed the presence of lower levels of IL-2 (Supplementary Figure S4, P = 0.0005, two-way ANOVA) and increased levels of IL-10 (Supplementary Figure S4, P = 0.0298, two-way ANOVA). No additional cytokine or chemokine analytes were changed, when measured by multiplex analysis of the conditioned media collected at multiple time points during the PBMC restimulation (not shown).
Figure 1.
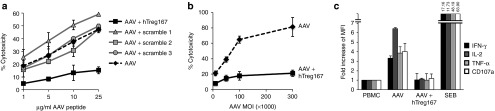
IgG-derived MHC class II epitopes (Tregitopes) mediate suppression of T cell responses in vitro. (a) CTL assay in which target cells were loaded with the AAV-derived MHC I epitope VPQYGYLTL (HLA-B*0702-restricted) at increasing concentrations and then incubated with HLA-matched Teff cells. Effectors were derived from HLA-B*0702 PBMC expanded in vitro against the same MHC I epitope alone (AAV, dashed line), or with hTreg167 (AAV+hTreg167, black line), or with three different scrambled versions of hTreg167 (Scramble 1, Scramble 2, and Scramble 3, grey lines). Teff:target ratio 10:1. Results are expressed as % cytotoxicity compared with a maximum cytotoxicity (cells treated with 10% SDS) after background subtraction. (b) CTL assay in which target cells were transduced with an AAV vector at increasing multiplicity of infections (MOIs) and then incubated with HLA-matched Teff cells restimulated with the AAV capsid MHC I peptide VPQYGYLTL alone (dashed line), or together with hTreg167 (black line). MOI, multiplicity of infection. Teff:target ratio 10:1. (c) Flow cytometry analysis of CD8+ T cell responses to the AAV MHC I epitope VPQYGYLTL. PBMC were restimulated in vitro in the presence of the AAV MHC I peptide VPQYGYLTL alone (AAV) or with hTreg167 (AAV+hTreg167) and then washed and incubated with the VPQYGYLTL peptide and stained for markers of T cell effector functions. Results are expressed as fold increase over non-restimulated PBMC incubated with the MHC I peptide VPQYGYLTL. For the analysis of flow cytometry data, after live/dead exclusion, cells were gated on lymphocytes, CD3+ T cells, and CD8+ T cells were then analyzed for IFN-γ, IL-2, TNF-α. SEB, Streptococcal enterotoxin B. Experiments shown were repeated at least twice. Error bars represent SEM. MFI, mean fluorescence intensity.
Figure 2.

Tregitopes can modulate CTL responses directed against several antigens and in the context of different HLA alleles. (a–d) Teff specific for Epstein-Barr virus (EBV), cytomegalovirus (CMV), hepatitis C virus (HCV), or influenza virus (Flu) were obtained following the protocol outlined in Figure S1. All MHC I epitopes were HLA-B*0702-restricted. Teff were used in the CTL assay at a Teff:target ratio of 1:10. Targets were peptide loaded with increasing concentrations of the relevant peptide. Results are expressed as % cytotoxicity compared to a max cytotoxicity (cells treated with 10% SDS) after background subtraction. Dashed line, Teff expanded from PBMC restimulated in vitro with indicated viral peptide only; Black line, Teff expanded in vitro with indicated viral peptide and hTreg167. (e) CTL assay in which Tregitope efficacy was tested against the AAV HLA-A*0101 restricted MHC I epitope SADNNNSEY. HLA-matched Teff were used in the CTL assay at a Teff:target ratio of 1:10. Targets were peptide loaded with increasing concentrations of the relevant peptide. Results are expressed as % cytotoxicity compared to a max cytotoxicity (cells treated with 10% SDS) after background subtraction. Dashed line, effectors obtained by restimulating PBMC in vitro with the SADNNNSEY epitope only; Black line, restimulation of PBMC was performed with the SADNNNSEY epitope from AAV and hTreg167. (f) CTL assay in which targets were loaded with the HLA-B*0702 epitope VPQYGYLTL from AAV. Teff were either obtained restimulating PBMC with the same AAV peptide only (AAV), or with the AAV peptide and Tregitope 167 (AAV+hTreg167), or with the AAV peptide and Tregitope 289 (AAV+hTreg289). Teff were used in the CTL assay at a Teff:target ratio of 1:10. Targets were peptide loaded with increasing concentrations of the relevant peptide. Results are expressed as % cytotoxicity compared to a max cytotoxicity (cells treated with 10% SDS) after background subtraction. Experiments shown were repeated at least twice. Error bars represent SEM.
Figure 3.
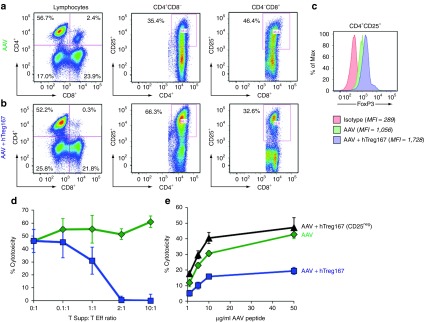
In vitro restimulation of PBMC with Tregitopes results in the expansion of a population of suppressive CD4+CD25+ T cells. Flow cytometry plots of PBMC restimulated in vitro with the AAV MHC I epitope VPQYGYLTL alone (AAV) (a) or with Tregitope 167 (AAV+hTreg167) (b). Cells were gated on lymphocytes after live/dead exclusion, CD3+ T cells, and CD4+CD8- and CD4-CD8+ T cells were analyzed for CD25+. (c) Histogram plot showing FoxP3 expression on CD4+CD25+ T cells restimulated with AAV-only from (a) or AAV+hTreg167 from (b); the isotype control is shown in red. Mean fluorescent intensity (MFI) is indicated in the figure legend. (d) Suppression experiments in which CD4+ T cells (Tsupp) negatively isolated from PBMC restimulated in vitro with AAV+hTreg167 (blue line) or with AAV only (green line) were mixed at defined ratios with CD8+ Teff negatively isolated from PBMC restimulated in vitro with the AAV only. (e) CTL assay in which Teff were derived from PBMC restimulated in vitro with AAV alone (AAV) or with Tregitope 167 (AAV+hTreg167). Alternatively, PBMC were depleted of CD25+ T cells prior to AAV+hTreg167 restimulation (AAV+hTreg167 [CD25neg]). Teff:target ratio 10:1. Results are expressed as % cytotoxicity compared to a max cytotoxicity (cells treated with 10% SDS) after background subtraction. Experiments shown were repeated at least twice. Error bars represent SEM.
These results indicate that Tregitopes added in restimulation cultures efficiently modulate CD8+ T cell responses directed against a variety of HLA class I-restricted viral epitopes in vitro.
Restimulation of PBMCs in vitro with Tregitopes expands suppressive CD4+CD25+ T cells
We then looked at the expansion of CD4+CD25+ T cells in restimulation cultures containing AAV MHC I peptide epitope only or in cultures containing Tregitopes. A population of CD4+CD25+ T cells was preferentially expanded when Tregitopes were added to the cultures (Figure 3a,b). These cells expressed FoxP3 (Figure 3c). Additional immunophenotyping of these CD4+CD25+ T cells showed that these cells were mostly CD127neg and expressed CTLA4 (not shown). Using negative bead selection, we isolated untouched CD4+ T cells (Tsupp) and used them in a suppression assay; these cells showed dose-dependent suppressor activity, with a dramatic reduction of CTL activity at a Tsupp:Teff cell ratio of 1:1 (Figure 3d). Magnetic bead depletion of the CD25+ cell fraction from PBMCs prior to in vitro restimulation resulted in the loss of modulatory activity of Tregitopes (Figure 3e), suggesting that Tregitopes exert their functions via a pool of existing T cells present in peripheral blood.
These results suggest that restimulation of PBMCs in vitro in the presence of Tregitopes expands a pool of natural CD4+CD25+FoxP3+ T cells with suppressive functions.
Tregitope-mediated modulation of CTL responses in vitro is contact dependent and associated with anergy of effector T cells
To investigate the mechanism(s) of action of Tregitopes in vitro, we carried out two experiments. In the first experiment, we tested whether cell-to-cell contact was a requirement for CTL inhibition. For this purpose, PBMCs were depleted of CD4+ or CD8+ T cells, and the CD4+CD8neg and the CD4negCD8+ fractions were separated into the two different compartments of a transwell system before restimulation in vitro (Figure 4a). When unfractionated PBMCs were cultured in both chambers of the transwell, AAV restimulation led to efficient killing of AAV peptide-loaded targets, whereas AAV+hTreg167 resulted in the inhibition of CTL activity as expected (Figure 4a). When CD4+CD8neg and CD4negCD8+ fractions of PBMCs were cultured in different chambers of the transwell, killing of target was efficient whether or not Tregitope was present for the restimulation. Suppression of CTL killing was only observed when Teff were restimulated with both AAV and hTreg167 (Figure 4a) and located in the same chamber, suggesting that contact between CD4+ and CD8+ T cells is needed to achieve suppression of CTL responses in vitro.
Figure 4.

Suppression of CTL responses to the AAV capsid in vitro mediated by Tregitopes is contact mediated and results in anergy of effector cell. (a) Transwell experiment outline. Cells were placed in a transwell chamber in which the upper and lower chambers were separated by a permeable membrane (drawing). The table shows the different conditions tested in the transwell; in each of the transwell chambers untouched PBMC or PBMC depleted of CD4+ (CD4negCD8+) or CD8+ (CD4+CD8neg) T cells were plated and restimulated with either AAV-only or AAV+hTreg167 peptides. After restimulation, PBMC or CD4negCD8+ cells were harvested from the transwell and used in a CTL assay against target cells loaded with the AAV-derived MHC I epitope VPYGYLTL at a Teff:target ratio 10:1. Results are expressed as % cytotoxicity compared to a max cytotoxicity (cells treated with 10% SDS) after background subtraction. (b) CTL assay in which target cells were transduced with an AAV vector at increasing MOIs and then incubated with HLA-matched effector cells. Teff cells were obtained by restimulating PBMC in vitro with the AAV capsid MHC I peptide VPQYGYLTL alone (AAV, black line), with AAV+Tregitope 167 (AAV+hTreg167, dashed black line), with AAV+hTreg167 followed by CD4+ T cell depletion (AAV+hTreg167 [CD4neg], gray line circles), or with AAV+hTreg167 followed by CD4+ T cell depletion and incubation with 10ng/ml IL-2 overnight (AAV+hTreg167 [CD4neg + IL-2], gray line triangles). Teff:target ratio 10:1. Results are expressed as % cytotoxicity compared to a max cytotoxicity (cells treated with 10% SDS) after background subtraction. Experiments shown were repeated at least twice. Error bars represent SEM.
We then restimulated PBMCs in the presence of Tregitopes, AAV MHC I peptide, and IL-2 (10 ng/ml) for 14 days (two rounds of restimulation) and then depleted CD4+ T cells. Following depletion, we either used the CD4negCD8+ T cell fraction in a CTL assay or incubated the cells with IL-2 (10 ng/ml) overnight before using them as source of effectors in a CTL assay. Immediately after removal of CD4+ T cells, CD4negCD8+ T cells were unable to kill the AAV peptide-loaded target (Figure 4b, CD4neg); however, a 24-hour incubation with IL-2 resulted in restoration of CTL activity to levels undistinguishable from those achieved with PBMCs restimulated with AAV MHC I epitopes only (Figure 4b, CD4neg + IL-2), suggesting that CD8+ T cells expanded in the presence of Tregitopes become anergic, possibly due to lack of costimulatory signals, and that IL-2 can restore their killing capacity.
Modulation of CTL responses in vitro is antigen specific
To test antigen specificity of modulation of CTL responses by Tregitopes, we performed the following experiment: PBMCs were restimulated in vitro in the presence of MHC I epitopes derived from AAV or EBV. Parallel cultures were established from the same PBMC donor in which cells were restimulated with the same AAV or EBV MHC I epitopes in the presence of hTreg167. At the end of the restimulation (14 days), CD4+ T cells (AAV-hTreg167 CD4+ or EBV-hTreg167 CD4+, respectively) were isolated by negative magnetic bead selection, and untouched cells were mixed with AAV or EBV Teff at a 1:1 ratio. As outlined in Figure 5a, AAV-hTreg167 CD4+ efficiently suppressed AAV Teff, whereas EBV-hTreg167 CD4+ did not. Further confirming antigen specificity, we found that AAV-hTreg167 CD4+ did not suppress EBV Teff, while EBV-hTreg167 CD4+ did (Figure 5b). These results suggest that the in vitro inhibition of Teff responses mediated by Tregitopes is antigen specific.
Figure 5.

Antigen-specificity of Tregitopes-induced suppression of CTL responses is mediated by MHC I. (a) CTL assay in which target cells were loaded with the MHC I epitope VPQYGYLTL from AAV and incubated with HLA-matched AAV-specific Teff alone (AAV, dashed line), or Teff mixed at a 1:1 ratio with negatively-selected CD4+ T cells from AAV+hTreg167 restimulated PBMC (AAV+[AAV-hTreg167 CD4+], black line), or Teff mixed at a 1:1 ratio with negatively-selected CD4+ T cells from EBV+hTreg167 restimulated PBMC (AAV+[EBV-hTreg167 CD4+], gray line). (b) CTL assay as in (a) in which target cells were loaded with the MHC I epitope RPPIFIRRL from EBV and incubated with HLA-matched EBV-specific Teff alone (EBV, dashed line), or mixed 1:1 with negatively-isolated CD4+ T cells from EBV+hTreg167 cultures (EBV+[EBV-hTreg167 CD4+], gray line) or with negatively-isolated CD4+ T cells from AAV+hTreg167 cultures (EBV+[AAV-hTreg167 CD4+], black line). Teff:target ratio 10:1. Results are expressed as % cytotoxicity compared to a max cytotoxicity (cells treated with 10% SDS) after background subtraction. (c) MHC I blockade experiment. Black bars: PBMC were restimulated in vitro with AAV+hTreg167 and CD4+ T cells were negatively selected and incubated with increasing amounts of soluble AAV-specific TCR. After washing, CD4+ T cells were mixed 1:1 with AAV-specific Teff and added to targets. Grey bar: PBMC restimulated with AAV MHC I epitope only, used as positive control (AAV only). White bar: PBMC restimulated with AAV MHC I only mixed 1:1 with CD4+ T cells derived from cell restimulated with hTreg167 only, without AAV (hTreg167 only). AAV+AAV-TCR control, CTL in which AAV-only Teff were used the CTL assay in the presence of 20 µg/ml of AAV-TCR. Shown is the % cytotoxicity. Error bars represent the standard error of the mean of quadruplicate testing. Comparison of results with the AAV-only condition gave the following p values (unpaired, two-tailed t test): AAV-TCR 0 µg/ml P = 0.00262, 1 µg/ml P = 0.00016, 5 µg/ml P = 0.00112, 20 µg/ml P = 0.03335; hTreg167 only P = 0.86133; AAV+AAV-TCR P < 0.00001. Experiments shown were repeated at least twice. Error bars represent SEM.
We further investigated the antigen specificity of Tregitope-mediated modulation by restimulating PBMCs with the HLA-B*0702-restricted AAV epitope VPQYGYLTL and hTreg167. From these cultures, CD4+ T cells isolated by negative selection were incubated with increasing amounts of soluble human T cell receptor (TCR) monomers specific for the HLA-B*0702 peptide VPQYGYLTL8 to block access to MHC I:VPQYGYLTL complexes at the surface of the CD4+ T cells. After 1 hour at room temperature, cells were washed twice, mixed at a 1:1 ratio with AAV only-restimulated Teff, and used in a CTL assay against target cells loaded with the same VPQYGYLTL peptide. Incubation of Tregitope-expanded CD4+ T cells with the soluble TCR monomers resulted in dose-dependent elimination of their suppressor activity (Figure 5c). As a control, Teff derived from PBMCs restimulated with AAV peptide and no hTreg167 were used, resulting in efficient killing of targets (Figure 5c, AAV only). As a second control, Teff restimulated with AAV peptide only were mixed 1:1 with CD4+ T cells isolated from PBMCs restimulated with hTreg167 and no AAV peptide; efficient lysis of targets was also observed in this case, indicating that Tregitopes alone without MHC I epitopes do not modulate CD8+ T cell responses. Finally, a third control in the experiment consisted of the direct addition of TCR monomers to the CTL assay in which AAV-only effectors were used. As previously described,8 this resulted in the inhibition of killing of target cells due to the blockage of MHC I by the soluble TCR monomers. These results suggest that the antigen-specific suppressive activity of Tregitope-expanded suppressor cells is mediated by the presence of antigen-specific MHC I peptide complexes which may be located on the surface of the Tregitope-specific Treg cells.
In vivo expression of Tregitopes using AAV vectors downregulates T cell responses directed against the AAV capsid
To test efficacy of modulation of T cell responses directed against the AAV capsid in vivo, we used a previously described model of adenovirus (Ad) vector-mediated immunization against AAV2, which is known to result in generation of a strong CD8+ T cell response directed against the capsid antigen.26
To deliver Tregitopes in vivo, we coexpressed Tregitopes (or a Scramble control) and the AAV2 capsid protein VP1 separated by a PACE/Furin cleavage signal27 under the transcriptional control of a CMV promoter (Figure 6a). These were packaged into AAV1 vectors and given intramuscularly to C57BL/6 mice at a dose of 2 × 1011 vector genomes (vg)/mouse. Six weeks following vector delivery, mice were challenged (intramuscularly) with 1 × 1011 viral particles of an Ad-AAV2 vector, and animals were sacrificed 9 days later (Figure 6b). Analysis of CD8+ T cell responses in splenocytes showed a significant downregulation (P = 0.0428, two-tailed unpaired t-test) of IFN-γ responses directed to the AAV capsid in animals receiving the AAV-Tregitope vector as compared with the controls (Figure 6c,d).
Figure 6.
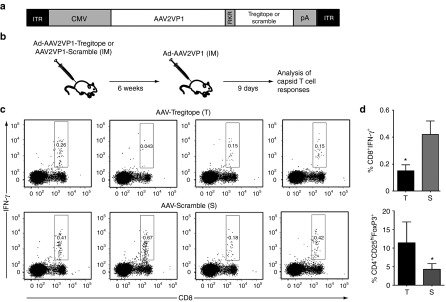
Expression of Tregitopes in vivo results in modulation of capsid-specific IFN-γ responses. (a) Diagram of the transgene expression cassette used to co-express AAV capsid antigen (AAV2VP1) and Tregitope or Scramble peptides. ITR, inverted terminal repeats; CMV, cytomegalovirus enhancer/promoter; RKR, PACE/Furin cleavage sequence; pA, poly-A signal. (b) Experimental design. At day 0, C57BL/6 mice received the Tregitope vector or the Scramble control intramuscularly, 2 × 1011 vg/mouse; six weeks later, animals were immunized with an adenoviral vector expressing the AAV2 VP1 protein injected intramuscularly, 1 × 1011 vp/mouse. Nine days after Ad immunization animals were sacrificed. (c) Intracellular cytokine staining for IFN-γ after AAV peptide restimulation of splenocytes isolated from animals 9 days after adenoviral challenge. After live/dead exclusion, cells were gated on lymphocytes, and CD8+ T cells were then analyzed for IFN-γ positivity. (d) Histogram plot of the frequency of CD8+IFN-γ+ T cells shown in panels (c) in animals receiving the AAV-Tregitope (T) or AAV-Scramble (S) vectors, the y-axis represents the percent of total lymphocytes that are CD8+IFN-γ+; *P = 0.0428. (e) Frequency of CD4+CD25hi cells that are FoxP3+ in splenocytes of animals receiving the AAV-Tregitope (T) or AAV-Scramble (S) vectors; after live/dead exclusion, cells were gated on lymphocytes, CD4+ T cells, and CD25hi cells were analyzed for FoxP3 expression; the y-axis represents the percent of CD4+ T cells that are CD25hiFoxP3+; *P = 0.0256. Shown are individual animals from one of three independent experiments. Error bars represent SEM.
An increase in frequency of CD4+CD25+FoxP3+ T cells was detected in spleens of AAV-Tregitope-injected animals (Figure 6e, P = 0.0256, two-tailed unpaired t-test), whereas a decrease in muscle CD4+ and CD8+ T cell infiltrates was observed (Supplementary Figure S5a; cells from five animals per group were pooled for the analysis). Finally, after Ad-AAV2 immunization, anti-AAV IgG levels were significantly decreased (P = 0.0031, two-tailed, unpaired t-test) in AAV-Tregitope-injected animals (Supplementary Figure S5b). RT Q-PCR confirmed transgene expression in muscle following vector delivery in mice receiving both the AAV-Tregitope and the AAV scramble vectors (Supplementary Figure S6a); similarly, vector genomes were detectable in both groups (Supplementary Figure S6b). Despite the presence of infiltrates, no loss of vector-transduced fibers was noted, a phenomenon previously associated with the apoptosis of reactive T cells.28
Although the Tregitope-mediated modulation of T cell responses in vivo was only partial, these results suggest that the proposed approach may be valuable in the modulation of capsid T cell immunity in vivo and that further studies, in which the dosing of Tregitopes is calibrated to the antigenic stimulus, may effectively modulate anti-AAV immune responses.
Discussion
Immune responses directed against the AAV capsid constitute an important obstacle to safe and effective gene transfer in humans.2 Whereas in some cases, pharmacological immunosuppression has proven efficacious in modulating these responses,5 the potential limitations of the approach prompted us to explore a more physiological strategy to achieve the goal of sustained gene transfer following in vivo gene delivery with AAV vectors.
Over the past several years, Treg-mediated modulation of immune responses has been explored in several models of autoimmunity, transplant medicine, and gene transfer.29,30 Peptide-mediated expansion of Tregs in vivo showed some degree of efficacy in preclinical31 and clinical32 models of autoimmune diseases like lupus.
The work presented here was initiated based on the observation that in vivo administration of IVIg results in modulation of immune responses and is associated with the expansion of regulatory T cells (Tregs),33 and that MHC II epitopes found in IgG (Tregitopes) have immunomodulatory properties.20,24
Here, we provide direct evidence that Tregitopes can act as potent modulators of CD8+ T cell responses against an antigen in vitro, and that expression of Tregitopes in vivo is effective in reducing T cell activation in response to adenoviral challenge.
We took advantage of an in vitro model of CD8+ T cell immunity directed against the AAV capsid,4,8 which overcomes the limitations of animal models34,35,36,37 and offers the advantage of working with effector and target cells that are of human origin and for which reagents specific to monitor AAV capsid antigen presentation and immunogenicity are well defined.4,7,8 Our data with this in vitro system indicate that Tregitopes are highly effective in modulating MHC I-driven T cell responses via the activation of a pool of pre-existing CD4+CD25+ T cells, as suggested by the following observations: (i) depletion of CD25+ T cells prior to Tregitope restimulation completely ablates the immunomodulatory effect of Tregitopes. This result is consistent with the studies that show expansion of Tregs in the thymus following Tregitope administration in vivo;22 (ii) in vitro studies also show that CD8+ Teff cells contacted by Tregitope-expanded suppressor cells lose their effector function and become anergic, as shown in suppression experiments in which negatively-selected purified CD4+ (Tsupp) and CD8+ (Teff) cells were mixed together, resulting in dose-dependent decrease in cytotoxicity; (iii) removal of CD4+ T cells and incubation of Teff with IL-2 restored cytotoxicity. This is suggestive of a model in which Tregitope-specific CD4+CD25hiFoxP3+ suppressor cells are expanded in the presence of Tregitopes, and inhibit CD8-mediated CTL activity by directly contacting the antigen-specific CD8+ Teff via MHC I and by interfering with IL-2-mediated signaling, even in the presence of IL-2-supplemented restimulation medium. Secretion of immunomodulatory cytokines does not seem to be the main mechanism of modulation of CD8+ T cell activation and function, although higher levels of IL-10 are found in cell cultures containing Tregitopes, whereas MHC I-TCR interaction seems to mediate antigen specificity of CD4+CD25+FoxP3+ T cells expanded with Tregitopes, at least in vitro. The interaction between suppressor and effector T cells results in anergy of Teff cells, which is reversible when additional IL-2 is provided.
The antigen specificity of immunomodulation mediated by Tregitopes is a desirable feature of the approach presented here, as suppression of immune responses to pathogens would otherwise constitute an important side effect of this strategy, and is a clear advantage over global suppression of immune responses with pharmacological agents. Future studies will help assess the robustness of antigen specificity and the requirement for cell-to-cell contact in vivo.
Antigen-specific tolerance via MHC I-TCR interaction has been described before in the context of veto cells.38 Since the initial description, several studies identified cells with antigen-specific MHC-restricted veto properties, including T lymphocytes, NK T cells, and dendritic cells. In the veto model, TCR-mediated recognition of the antigen presented in the context of MHC I by the veto cell results in cell death of the effector T cell.39 Furthermore, antigen presentation by veto cells has been shown to promote B cell tolerance.40 Both the in vitro and in vivo results presented here suggest that the veto cell model could be a potential mechanism of tolerance induction with Tregitopes; however, it should also be noted that MHC I presentation of antigen by CD4+ T cells in our model does not result in deletion of reactive CD8+ T cells. Rather, CD4+CD25+FoxP3+ T cells expanded with Tregitopes appear to act via deprivation of activation signals.41
For our in vivo experiments, we used a model of adenoviral vector-mediated immunization to the AAV capsid antigen.26 This vaccination model was used to overcome the limitations of several animal models proposed to mimic capsid T cell immune responses observed in humans34,35,37 and provide a stringent test to our strategy. We chose to target the muscle because expression of transgenes in this tissue is typically immunogenic42 and to colocalize the Tregitope expression and the proinflammatory signal deriving from the adenoviral vector. Expression of Tregitopes with AAV vectors allowed us to overcome the potential limitations of delivery of peptides in vivo.
The lower efficacy of effector response suppression with Tregitopes in vivo compared with in vitro may be the result of the stringency of the model. Additional factors that may account for the lower efficacy of Tregitope in vivo could include suboptimal levels of IL-2,43 or relatively low levels of expression of Tregitopes.
Despite these potential limitations, our data show that it is possible to use Tregitopes in vivo to modulate immune responses to an antigen, and the delivery of these peptides with AAV vectors could represent a tool with a wide range of applications.
Future developments will include the use of AAV serotypes with high efficiency of transduction in vivo,44 the expression of multiple Tregitopes in a single vector, the co-administration of IL-2 at the time of Tregitope expression, or the expression of Tregitopes in liver, which per se has tolerogenic properties.45 As AAV vectors are nonreplicating, recombinant viruses, the window of time in which the capsid antigen is present and immunologically detectable is limited. Direct engineering of the AAV capsid46 to carry Tregitopes at the N-terminus of the capsid protein VP2, as opposed to expressing them, could be a viable strategy to limit the duration of the protolerogenic signal deriving from Tregitopes. Finally, the recent development of a mouse model of CD8+ T cell immune responses following liver gene transfer,47 in which loss of transgene expression and increase in liver enzymes is observed like in humans, represents an important asset to the development of ad hoc tolerization protocols with Tregitopes.
In the future clinical translation of the approach presented here, one potential limitation to the use of Tregitopes is the fact that efficacy of suppression with these peptides is dependent on the HLA class II haplotype of the subject. Low affinity of Tregitope peptides for a subject's MHC II alleles (represented by the iTEM score) result, in fact, in less effective modulation of T cell reactivity. This was well exemplified in the experiments showing only partial suppression of CTL activity with hTreg289 and complete suppression with hTreg167 due to differences in binding affinity to the donor's MHC II alleles; correlation between the iTEM score and hTreg167-mediated suppression of CTLs is also suggestive of such correlation. However, the affinity of Tregitopes for several MHC II alleles20 and the coexpression of strings composed of Tregitopes that bind to multiple HLA could allow administration of the same tolerizing vector to most subjects.
An important implication of the study presented here is that our strategy could be potentially applied to a variety of diseases. AAV vectors per se are among the most efficient vectors for in vivo gene transfer,1 they have been administered both systemically and locally to humans with an excellent safety profile, and they have been shown to promote tolerance rather than immunity against the expressed transgenes.45 The availability of a wide range of AAV capsid serotypes with different tissue tropism,44 the possibility to further direct tropism of AAV vectors to a determined tissue,48 and the broad range of tissue-specific and/or inducible promoters available constitute a great advantage of the strategy presented here over the systemic administration of peptides or immunosuppressive drugs.
In conclusion, the work presented here shows that MHC II epitopes derived from IgG can be used as tolerizing agents to modulate CD8+ T cell responses in vitro and in vivo. The use of AAV vectors as delivery vehicles for Tregitopes represents a novel strategy to deliver tolerogenic peptides to modulate immune responses to viral vectors and other antigens.
Materials and Methods
Peptides. The AAV2 MHC I epitopes VPQYGYLTL and SADNNNSEY, restricted to the human HLA-B*0702 and HLA-A*0101 alleles, respectively,4 were obtained from Genemed Synthesis. The AAV2 MHC I epitope SNYNKSVNV, restricted to the mouse allele Kb,26 was also obtained from Genemed Synthesis; this epitope from the AAV2 capsid is not conserved in the AAV1 capsid. The HLA-B*0702-restricted peptide epitopes from EBV (RPPIFIRRL), CMV (TPRVTGGGAM), HCV (DPRRRSRNL), and influenza (QPEWFRNVL) were obtained from Proimmune. hTreg-167 (PAVLQSSGLYSLSSVVTVPSSSLGTQ), hTreg-289 (EEQYNSTYRVVSVLTVLHQDW), the scamble negative control peptides Scramble 1 (SPYQSVTSSVLGLLSPVSASVSQTLG), Scramble 2 (SSGVQPLLVVYSSLVTSPSASSGLTQ), and Scramble 3 (SSGSQALVLVYSPSSVLVPTSSTGQL), and the control peptide from influenza hemagglutinin (307-319) (PKYVKQNTLKLAT) were synthesized by 21st Century Biochemicals with a 5′ acylation modification and an amidylation modification at the 3′ terminus. All peptides were resuspended to a concentration of 5 mg/ml. Affinity of the Tregitopes used in this study for human class II alleles has been previously discussed.20
Vectors. The AAV serotype 1 vectors used in the study encoded the AAV2 VP1 capsid protein fused to the Tregitope sequence (AAV-Tregitope) or a scrambled version of the Tregitope sequence (AAV-Scramble). A PACE/furin cleavage sequence was inserted between the VP1 sequence and the Tregitope/Scramble sequence to ensure cleavage of the two proteins.27 Expression of transgenes was driven by a CMV promoter/enhancer sequence. All AAV vectors were administered at a dose of 2 × 1011 vector genomes (vg)/mouse. The adenoviral vector expressing the AAV2 VP1 capsid protein (Ad-AAV2) was previously described.26 The Ad-AAV2 vector was given at a dose of 1 × 1011 virus particles (vp)/mouse. All vectors were produced as previously described26 and injected intramuscularly (intramuscularly).
Cell lines, peripheral blood mononuclear cells. The human hepatocyte cell line HHL5 (carrying the human HLA-A*0101 allele) and the HHL5 cell line genetically modified to express the human HLA-B*0702 allele, used as targets in the CTL assay, were previously described.7,8 Healthy donor PBMCs were obtained from Cellular Technologies (Cleveland, OH) and selected based on their HLA haplotype; so, they were carrier of the HLA-A*0101 or HLA-B*0702 alleles as well as matching the DRB1 alleles previously reported to be high-affinity binders of Tregitopes used in the study.20 All human specimens were deidentified and handled under a protocol approved by the Children's Hospital of Philadelphia's Institutional Review Board.
In vitro expansion of T cells. Restimulation of PBMCs in vitro was performed as previously described.4 Briefly, cells were thawed, washed, counted, and resuspended at a concentration of 2 × 106 cells/ml in AIM-V lymphocyte media (Invitrogen Gibco, Carlsbad, CA) containing 3% human serum (Bioreclamation), 1% L-glutamine (Invitrogen Gibco), and 1% penicillin/streptomycin (Invitrogen Gibco). For each expansion condition, 1 × 106 cells were added to a well in a 24-well plate (BD Falcon). 1 × 106 of autologous-irradiated PBMC (3000 rad) were also added to each well together with 10 ng/ml of human recombinant IL-7 (R&D Systems, Minneapolis, MN). According to the experimental conditions for each expansion well, peptides were added at a final concentration of 10 µg/ml in 5% CO2, 10 ng/well of human IL-2 (Roche, Indianapolis, IN) was added to the cell culture and replenished every 48 hours thereafter. Cells were divided into new wells as the expansion proceeded, and antigenic stimulation (antigen and feeder cells) was repeated every 7–10 days for up to two rounds of restimulation. Expanded CD8+ T cells were characterized after each round of stimulation by IFN-γ ELISpot, MHC I pentamer staining, or intracellular cytokine staining.
Flow cytometry, intracellular cytokine staining, and polyfunctional analysis of T cell activation markers. Surface staining and IFN-γ intracellular cytokine staining was performed as previously described.4,26 Regulatory T cell staining was performed using human or mouse regulatory Treg-staining kits (eBioscience, San Diego, CA) containing antibodies for CD4, CD25, and FoxP3. For polyfunctional analysis of T cell activation markers, cells were washed and counted using an automated cell counter (Invitrogen), resuspended at a concentration of 2 × 106 cells/ml, and incubated overnight at 37 °C in 5% CO2. The next day, 1 × 106 cells were treated with 1 µg each of anti-CD28 and anti-CD49d antibodies (BD Biosciences, Franklin Lakes, NJ), and with 10 µl of anti-CD107a antibody (BD Biosciences). Cells were then stimulated with 10 µg/ml of peptide, 10 µg/ml of Staphylococcal enterotoxin B (SEB, Sigma, St Louis, MO), or left untreated as a negative control for 1 hour at 37 °C, 5% CO2 prior to adding 10 µg of brefeldin A (Sigma) and 0.7 µl of monensin (GolgiStop, BD Biosciences). After an additional 5-hour incubation, cells were washed in PBS and stained for the surface markers CD3, CD4, CD8, CD14, CD16, and CD19 (BD Biosciences or Caltag). Cells were then washed once with PBS 2% FBS, and then fixed and permeabilized with 200 µl of Cytofix/Cytoperm solution (BD Biosciences). Intracellular staining for cytokines was performed at room temperature using antibodies against IFN-γ, TNF-α, and IL-2 (BD Biosciences). Cells were fixed with 2% paraformaldehyde prior to acquisition on a FACS Canto II flow cytometer; analysis was conducted using FACSDiva (BD Biosciences) and Flowjo (Treestar) software.
CTL assay. The CTL assay was performed as previously described.7,8 Briefly, lactate dehydrogenase (LDH) release following CTL-mediated target lysis was measured with the CytoTox 96 Non Radioactive Cytotoxicity Assay (Promega, Madison, WI). Five thousand HHL5 target cells were plated in each well of a Microtest Primaria flat-bottom 96-well plate (BD Falcon) in DMEM containing no serum. Targets were either transduced at a range of multiplicity of infections (see Results) with an AAV vector, or loaded with MHC class I epitopes at indicated concentrations (see Results) and incubated for 18 hours at 37 °C, 5% CO2. Teff cells were added at a Teff:target ratio of 10:1 unless otherwise noted, and cell lysis was measured after four hours of incubation.
Cytokine profiling. Conditioned medium was sampled immediately after cells were plated (day 0) and then twice daily on day 4, 8, 13, and 18 during in vitro restimulation and prior to feeding cells with IL-2. Media withdrawn for sampling was replaced with an equal volume of medium. Cytokine analysis was performed by ELISA. The multiplex assay was performed using a Milliplex MAP Human Cytokine/Chemokine Pre-mixed 14-plex kit (Millipore, Billercia, MA) on a Luminex 200 instrument and analyzed with xPONENT software (Luminex, Austin, TX). Analytes and individual lower limit of detection were as follows: granular-macrophage colony stimulating factor (GM-CSF): 2.75 pg/ml; IFN-γ: 2.97 pg/ml; IL-1 β: 1.64 pg/ml; IL-2: 2.51 pg/ml; IL-4: 2.94 pg/ml; IL-5: 3.05 pg/ml; IL-6: 3.01 pg/ml; IL-7: 2.95 pg/ml; IL-8: 3.01 pg/ml; IL-10: 0.41 pg/ml; IL-12 (p70): 2.99 pg/ml; IL-13: 1.31 pg/ml; monocyte chemotactic protein 1 (MCP-1): 2.97 pg/ml; and TNF-α: 3.01 pg/ml. The multiplex assay did not show variation in any of the cytokines or chemokines tested (not shown).
Isolation and depletion of T cell subsets. For studies involving the selection or depletion of subpopulations of T cells, antibody-coated magnetic bead separation was performed to negatively select for untouched CD4+ and CD8+ T cells, or to deplete CD25+, CD4+, or CD8+ T cell populations (Invitrogen Dynal) according to the manufacturer's protocols. Isolated cell populations were either used directly in CTL assays or cultured until assayed. Surface staining followed by flow cytometry was used to confirm the success of cell separation or depletion.
Assay for contact-dependent inhibition of cytotoxicity. PBMCs were separated into CD4-depleted and CD8-depleted fractions and then cultured in separate chambers of a 0.4 µM Transwell permeable support (Corning). Cells were restimulated for 2 weeks in the presence of the AAV MHC I peptide VPQYGYLTL and hTreg167 as described above. After restimulation, cells were harvested from the CD4-depleted chamber of the transwell (containing the CD8+ fraction of PBMCs) and tested in a CTL assay.
MHC I blockade experiment. To demonstrate that antigen specificity of Tregitope-mediated CTL suppression is dependent on MHC I, we performed a MHC I blockade. We previously described the development of soluble TCR monomers specific for the HLA-B*0702 epitope VPQYGYLTL.4 This TCR is HLA- and epitope-specific and can mediate complete blockage of MHC I.8 In this experiment, PBMCs from the same donor were restimulated in vitro with AAV+hTreg167, AAV, or hTreg167.
Cells stimulated with AAV+hTreg167 were used to negatively select CD4+ T cells using Dynabeads (Invitrogen Dynal). These cells were then incubated with 0, 1, 5, or 20 µg/ml of soluble TCR monomers at room temperature in PBS with 1% BSA. After washing twice in DMEM (Gibco, Carlsbad, CA), cells were mixed 1:1 with Teff derived from AAV-restimulated PBMCs and used in a CTL assay.
As a control, Teff derived from AAV-restimulated PBMCs (in the absence of Tregitope) were used (Figure 5C, AAV only).
One additional control consisted of CD4+ T cells derived from PBMCs restimulated with hTreg167 only mixed at a 1:1 ratio with Teff derived from PBMC restimulated with AAV antigen only (Figure 5C, hTreg167 only). In this control, suppressor cells were never exposed to the MHC I epitope from AAV.
Finally, a third control consisted in the addition of soluble TCR monomers to the CTL assay in which effector cells were restimulated with AAV peptide only.
Animal studies. All mouse experiments were repeated at least twice, conducted using 8- to 10-week-old male C57BL/6 mice (Charles River Laboratories, Wilmington, MA), and were approved by the Children's Hospital of Philadelphia Institutional Animal Care and Use Committee. Animals received the vectors intramuscularly via direct injection into both hind limbs. Blood samples were collected into heparinized capillary tubes (Fisher Scientific, Waltham, MA) for antibody analysis as previously described;45 spleens and lymph nodes were collected and briefly stored in cold medium until processed for lymphocyte isolation. Muscle samples were snap-frozen in liquid nitrogen for RNA extraction. For cell extraction, muscle samples were collected in cold Hank's balanced salt solution (HBSS, Invitrogen Gibco) containing 0.05% of proteinase K (Pronase, Invitrogen Gibco), homogenized on a gentleMACS Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany), digested for 1 hour at 37 °C, then further homogenized, filtered through a 70 µm cell strainer, and washed twice in RPMI 1640 medium (Invitrogen Gibco).
Quantitative real-time PCR for transgene expression and vector gene copy number. RNA was extracted from snap-frozen muscle using TRIzol/chloroform and additional purification performed with RNEasy columns (QIAGEN). cDNAs were generated using the High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems, Foster City, CA). Taqman Universal Master Mix II (Applied Biosystems) was used for quantitative real-time PCR. PrimeTime Standard qPCR Assays were ordered from Integrated DNA Technologies. For AAV2 VP1 detection, the following reagents were used: forward primer: 5′-GACATTTTCACCCCTCTCCC-3′ reverse primer: 5′-CAAACTTTGCCGCACTGAAG-3′ and probe sequence: 5′-/56-FAM/ACACCCTCC/ZEN/TCCACAGATTCTCATCA/3IABkFQ/-3′. GAPDH was used as an internal control: forward primer: 5′-AATGGTGAAGGTCGGTGTG-3′ reverse primer: 5′-GTGGAGTCATACTGGAACATGTAG-3′ and probe 5′-/56-FAM/TGCAAATGG/ZEN/CAGCCCTGGTG/3IABkFQ/-3′. The same set of primers and probe were used to determine vector genome copy number on genomic DNA extracted from injected muscle. The assays were run on a Mastercycler Realplex2 (Eppendorf, Hauppauge, NY).
Bioinformatics and statistical analysis. Tregitope-binding scores (iTEM) were obtained from EpiVax (EpiVax, Providence, RI) using the EpiMatrix and ClustiMer epitope-mapping algorithms as previously described.20,25,49 Unpaired t-test was used to compare means across experimental groups, two-way ANOVA was used to compare levels of cytokines secreted in PBMC cultures. Linear regression was used to analyze the relationship between iTEM score and inhibition of CTL activity. Statistical analysis was performed using GraphPad (GraphPad Software, La Jolla, CA); P values <0.05 were considered significant.
SUPPLEMENTARY MATERIAL Figure S1. Defining an experimental design for the in vitro testing of Tregitopes efficacy. Figure S2. The binding score of Tregitopes for MHC II shows a rough correlation with the inhibition of CTL activity Figure S3. Both CD4+ and CD8+ T cells proliferate in vitro in the presence of Tregitopes. Figure S4. In vitro restimulation of PBMCs with Tregitopes leads to an antiinflammatory cytokine response in the tissue culture media. Figure S5. Expression of Tregitopes in vivo is associated with reduced T cell infltrates in muscle and lower anti-AAV antibody titers. Figure S6. After AAV vector delivery in vivo, transgene expression and vector genome copy number can be detected in injected muscle.
Acknowledgments
This work was supported by the Center for Cellular and Molecular Therapeutics at the Children's Hospital of Philadelphia. F. M. is supported by the European Union Marie Curie Career Integration Grant number 333628. The authors gratefully acknowledge Drs. K. A. High, R. W. Herzog, and J. M. Davoust for the critical discussion of the data. L.P.C. and A.S.D. are employees of EpiVax, Inc., the company that holds patents on the Tregitope technology. All other authors declare no conflict of interest.
Supplementary Material
References
- Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2011;11:321–330. doi: 10.2174/156652311796150354. [DOI] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzyczka N, Berns KI. Lippincott, Williams and Wilkins: Philadelphia; 2001. Parvoviridae: the viruses and their replication. [Google Scholar]
- Finn JD. Proteasome inhibitors decrease AAV2 capsid derived peptide epitope presentation on MHC class I following transduction. Mol Ther. 2010;18:135–142. doi: 10.1038/mt.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pien GC, Basner-Tschakarjan E, Hui DJ, Mentlik AN, Finn JD, Hasbrouck NC, et al. Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J Clin Invest. 2009;119:1688–1695. doi: 10.1172/JCI36891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA. 2008;105:7827–7832. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcedo R, Morizono H, Wang L, McCarter R, He J, Jones D, et al. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin Vaccine Immunol. 2011;18:1586–1588. doi: 10.1128/CVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther. 2011;18:288–294. doi: 10.1038/gt.2011.90. [DOI] [PubMed] [Google Scholar]
- Veron P, Leborgne C, Monteilhet V, Boutin S, Martin S, Moullier P, et al. Humoral and cellular capsid-specific immune responses to adeno-associated virus type 1 in randomized healthy donors. J Immunol. 2012;188:6418–6424. doi: 10.4049/jimmunol.1200620. [DOI] [PubMed] [Google Scholar]
- Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood. 2006;108:3321–3328. doi: 10.1182/blood-2006-04-017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, Meulenberg JJ, Hui DJ, Basner-Tschakarjan E, Hasbrouck NC, Edmonson SA, et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114:2077–2086. doi: 10.1182/blood-2008-07-167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng B, Ye P, Rawlings DJ, Ochs HD, Miao CH. Anti-CD3 antibodies modulate anti-factor VIII immune responses in hemophilia A mice after factor VIII plasmid-mediated gene therapy. Blood. 2009;114:4373–4382. doi: 10.1182/blood-2009-05-217315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safinia N, Leech J, Hernandez-Fuentes M, Lechler R, Lombardi G. Promoting transplantation tolerance; adoptive regulatory T cell therapy. Clin Exp Immunol. 2013;172:158–168. doi: 10.1111/cei.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia M, Roncarolo MG. Immune intervention with T regulatory cells: past lessons and future perspectives for type 1 diabetes. Semin Immunol. 2011;23:182–194. doi: 10.1016/j.smim.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- De Groot AS, Moise L, McMurry JA, Wambre E, Van Overtvelt L, Moingeon P, et al. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes”. Blood. 2008;112:3303–3311. doi: 10.1182/blood-2008-02-138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousens LP, Mingozzi F, van der Marel S, Su Y, Garman R, Ferreira V, et al. Teaching tolerance: New approaches to enzyme replacement therapy for Pompe disease. Hum Vaccin Immunother. 2012;8:1459–1464. doi: 10.4161/hv.21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Marel S, Majowicz A, Kwikkers K, van Logtenstein R, te Velde AA, De Groot AS, et al. Adeno-associated virus mediated delivery of Tregitope 167 ameliorates experimental colitis. World J Gastroenterol. 2012;18:4288–4299. doi: 10.3748/wjg.v18.i32.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousens LP, Najafian N, Mingozzi F, Elyaman W, Mazer B, Moise L, et al. In vitro and in vivo studies of IgG-derived Treg epitopes (Tregitopes): a promising new tool for tolerance induction and treatment of autoimmunity. J Clin Immunol. 2013;33 Suppl 1:S43–S49. doi: 10.1007/s10875-012-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousens LP, Najafian N, Mingozzi F, Elyaman W, Mazer B, Moise L, et al. In vitro and in vivo studies of IgG-derived Treg epitopes (Tregitopes): a promising new tool for tolerance induction and treatment of autoimmunity. J Clin Immunol. 2013;33 Suppl 1:S43–S49. doi: 10.1007/s10875-012-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen T, Moise L, Ardito M, Martin W, De Groot AS.2010A method for individualizing the prediction of immunogenicity of protein vaccines and biologic therapeutics: individualized T cell epitope measure (iTEM). J Biomed Biotechnol 2010doi:10.1155/2010/961752). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino DE, Mingozzi F, Hui DJ, Chen H, Colosi P, Ertl HC, et al. Identification of mouse AAV capsid-specific CD8+ T cell epitopes. Mol Ther. 2005;12:1023–1033. doi: 10.1016/j.ymthe.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Margaritis P, Arruda VR, Aljamali M, Camire RM, Schlachterman A, High KA. Novel therapeutic approach for hemophilia using gene delivery of an engineered secreted activated Factor VII. J Clin Invest. 2004;113:1025–1031. doi: 10.1172/JCI20106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez VM, Bowen DG, Walker CM. Silencing of T lymphocytes by antigen-driven programmed death in recombinant adeno-associated virus vector-mediated gene therapy. Blood. 2009;113:538–545. doi: 10.1182/blood-2008-01-131375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA, Kang SM. CD4(+)Foxp3(+) regulatory T cell therapy in transplantation. J Mol Cell Biol. 2012;4:11–21. doi: 10.1093/jmcb/mjr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DA, Leboeuf M, Gjata B, Danos O, Davoust J. CD4+CD25+ regulatory T cells inhibit immune-mediated transgene rejection. Blood. 2003;102:4326–4328. doi: 10.1182/blood-2003-05-1454. [DOI] [PubMed] [Google Scholar]
- Amarilyo G, Hahn B, La Cava A. Preclinical studies with synthetic peptides in systemic lupus erythematosus. Front Biosci. 2012;17:1940–1947. doi: 10.2741/4030. [DOI] [PubMed] [Google Scholar]
- Sthoeger ZM, Sharabi A, Molad Y, Asher I, Zinger H, Dayan M, et al. Treatment of lupus patients with a tolerogenic peptide, hCDR1 (Edratide): immunomodulation of gene expression. J Autoimmun. 2009;33:77–82. doi: 10.1016/j.jaut.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Ephrem A, Chamat S, Miquel C, Fisson S, Mouthon L, Caligiuri G, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111:715–722. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- Li C, Hirsch M, Asokan A, Zeithaml B, Ma H, Kafri T, et al. Adeno-associated virus type 2 (AAV2) capsid-specific cytotoxic T lymphocytes eliminate only vector-transduced cells coexpressing the AAV2 capsid in vivo. J Virol. 2007;81:7540–7547. doi: 10.1128/JVI.00529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Murphy SL, Giles-Davis W, Edmonson S, Xiang Z, Li Y, et al. Pre-existing AAV capsid-specific CD8+ T cells are unable to eliminate AAV-transduced hepatocytes. Mol Ther. 2007;15:792–800. doi: 10.1038/sj.mt.6300090. [DOI] [PubMed] [Google Scholar]
- Li H, Tuyishime S, Wu TL, Giles-Davis W, Zhou D, Xiao W, et al. Adeno-associated virus vectors serotype 2 induce prolonged proliferation of capsid-specific CD8+ T cells in mice. Mol Ther. 2011;19:536–546. doi: 10.1038/mt.2010.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Figueredo J, Calcedo R, Lin J, Wilson JM. Cross-presentation of adeno-associated virus serotype 2 capsids activates cytotoxic T cells but does not render hepatocytes effective cytolytic targets. Hum Gene Ther. 2007;18:185–194. doi: 10.1089/hum.2007.001. [DOI] [PubMed] [Google Scholar]
- Miller RG. An immunological suppressor cell inactivating cytotoxic T-lymphocyte precursor cells recognizing it. Nature. 1980;287:544–546. doi: 10.1038/287544a0. [DOI] [PubMed] [Google Scholar]
- Reich-Zeliger S, Eidelstein Y, Hagin D, Antebi YE, Seger R, Reisner Y. Deletion of alloreactive T cells by veto cytotoxic T lymphocytes is mediated through extracellular signal-regulated kinase phosphorylation. Transplantation. 2010;90:380–386. doi: 10.1097/TP.0b013e3181e86b28. [DOI] [PubMed] [Google Scholar]
- Nguyen P, Geiger TL. Induction of B-cell immune tolerance by antigen-modified cytotoxic T lymphocytes. Transplantation. 2010;89:667–676. doi: 10.1097/TP.0b013e3181ca9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. 2011;23:424–430. doi: 10.1016/j.smim.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Wang L, Cao O, Swalm B, Dobrzynski E, Mingozzi F, Herzog RW. Major role of local immune responses in antibody formation to factor IX in AAV gene transfer. Gene Ther. 2005;12:1453–1464. doi: 10.1038/sj.gt.3302539. [DOI] [PubMed] [Google Scholar]
- Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365:2067–2077. doi: 10.1056/NEJMoa1105143. [DOI] [PubMed] [Google Scholar]
- Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington KH, Gorbatyuk OS, Harrison JK, Opie SR, Zolotukhin S, Muzyczka N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J Virol. 2004;78:6595–6609. doi: 10.1128/JVI.78.12.6595-6609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino AT, Basner-Tschakarjan E, Markusic DM, Finn JD, Hinderer C, Zhou S, et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood. 2013;121:2224–2233. doi: 10.1182/blood-2012-10-460733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Chang M, Davidson BL. Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nat Med. 2009;15:1215–1218. doi: 10.1038/nm.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanen BC, De Groot AS, Moise L, Ardito M, McClaine E, Martin W, et al. Coupling sensitive in vitro and in silico techniques to assess cross-reactive CD4(+) T cells against the swine-origin H1N1 influenza virus. Vaccine. 2011;29:3299–3309. doi: 10.1016/j.vaccine.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.