

Abstract
The p.Ala204Thr mutation (exon 7) of the CLCNKB gene is a "founder" mutation that causes most of type III Bartter syndrome cases in Spain. We performed genetic analysis of the CLCNKB gene, which encodes for the chloride channel protein ClC-Kb, in a cohort of 26 affected patients from 23 families. The diagnostic algorithm was: first, detection of the p.Ala204Thr mutation; second, detecting large deletions or duplications by Multiplex Ligation-dependent Probe Amplification and Quantitative Multiplex PCR of Short Fluorescent Fragments; and third, sequencing of the coding and flanking regions of the whole CLCNKB gene. In our genetic diagnosis, 20 families presented with the p.Ala204Thr mutation. Of those, 15 patients (15 families) were homozygous (57.7% of overall patients). Another 8 patients (5 families) were compound heterozygous for the founder mutation together with a second one. Thus, 3 patients (2 siblings) presented with the c. -19-?_2053+? del deletion (comprising the entire gene); one patient carried the p.Val170Met mutation (exon 6); and 4 patients (3 siblings) presented with the novel p.Glu442Gly mutation (exon 14). On the other hand, another two patients carried two novel mutations in compound heterozygosis: one presented the p.Ile398_Thr401del mutation (exon 12) associated with the c. -19-?_2053+? del deletion, and the other one carried the c.1756+1G>A splice-site mutation (exon 16) as well as the already described p.Ala210Val change (exon 7). One case turned out to be negative in our genetic screening. In addition, 51 relatives were found to be heterozygous carriers of the described CLCNKB mutations. In conclusion, different mutations cause type III Bartter syndrome in Spain. The high prevalence of the p.Ala204Thr in Spanish families thus justifies an initial screen for this mutation. However, should it not be detected further investigation of the CLCNKB gene is warranted in clinically diagnosed families.
Introduction
Bartter syndrome (BS), which was first described in 1962 [1], is a heterogenic autosomal recessive disorder of the salt reabsorption at the thick ascending limb (TAL) of Henle’s loop. BS includes a group of closely related hereditary tubulopathies characterized by hypokalemia, metabolic alkalosis, hyperreninemia, hyperaldosteronism with normal or low blood pressure, renal salt loss and hyperplasia of juxtaglomerular apparatus [2]. Two phenotypic variants have been described in patients with BS, one presenting early in life with severe salt loss and marked prostaglandinuria, called “neonatal BS”, and another one with a milder phenotype or later onset of the disease, named “classic BS” [3-5]. Furthermore, patients with mutations in the same gene can present different phenotypes [6].
Loss-of-function mutations of several genes encoding those transporters involved in salt reabsorption at the TAL cause different types of BS: SLC12A1 (Na-K-2Cl cotransporter NKCC2) causes type I BS (OMIM #601678) [7], KCNJ1 (K+ channel, ROMK) causes type II BS (OMIM #241200) [8] and CLCNKB (Chloride Channel Protein ClC-Kb) causes type III BS (OMIM #607364) [9]. Loss-of-function mutations in the BSND gene, that encodes barttin, an essential beta subunit for ClC chloride channels, cause type IV-A BS with sensorineural deafness (OMIM #602522) [10]. On the other hand, gain-of-function mutations in the CASR gene (OMIM +601199) cause type V BS [11]. Finally, simultaneous mutations in both the CLCNKB and CLCNKA genes cause type IV-B BS (OMIM #613090) [12].
As mentioned above, type III BS is caused by mutations in the CLCNKB gene (OMIM *602023), on chromosome 1p36. It spans 20 exons, 19 encoding for the chloride channel protein ClC-Kb [13]. Three splice transcript variants have been found for this gene. ClC-Kb is associated with another chloride channel, ClC-Ka, which is encoded by the CLCNKA gene (OMIM *602024). CLCNKB and CLCNKA genes are separated by approximately 10 kb of genomic sequence, and they have 94% of sequence identity.
The ClC-Kb protein, which is expressed predominantly in the TAL, distal tubule and collecting duct, is required to ensure the chloride (Cl-) exit on the basolateral cellular side [14]. Thus, impaired ClC-Kb function reduces Cl- exit and decreases Na-K-Cl reabsorption through the Na-K-2Cl cotransporter (NKCC2) [7-9] by modifying the transepithelial voltage gradient and causing salt loss in urine [15].
The ClC-Kb is formed by 687 amino acids and is present as a homodimer at the membrane. Each subunit is composed of 18 α-helices (A-R), with 12 transmembrane domains and intracellular amino and carboxyl termini, the latter containing two cystathionine-β-synthase (CBS) domains [16].
At present, according to the Human Gene Mutation Database (HGMD database, Biobase-International, www.hgmd.cf.ac.uk), 75 mutations have been described in the CLCNKB gene, including gross deletions (8), small deletions (6), small insertions (4), complex rearrangements (2), nonsense or missense mutations (45) and splice site mutations (10).
The genetic diagnosis of type III BS is complex because of its large phenotypic variability. Indeed, mutations in the CLCNKB gene may cause phenotypes that overlap with other types of BS and Gitelman syndrome (OMIM #263800) as well [6,17].
Our group previously described a founder mutation in the CLCNKB gene (c.610G>A; p.Ala204Thr) that causes type III BS in Spain [18]. To date, our experience has been that almost all Spanish patients clinically diagnosed of type III BS present this mutation. However, we have seen an increasing number of patients with a clinical diagnosis of type III BS who do not carry the p.Ala204Thr mutation. Thus, genetic analysis of the entire CLCNKB gene is indicated. The aim of this study was to perform a comprehensive genetic analysis in our cohort of patients with type III BS, and to devise a strategy for gene analysis that is appropriate for the Spanish population.
Methods
Ethics Statement
The study was approved by the Comité Ético de Investigación Clínica (CEIC) from the Cruces University Hospital. The patients and their relatives provided their written informed consent to participate in this study. In case of the minors included in the cohort, we obtained written informed consent from their guardians prior to enrolment in the study.
Population
A total of 26 patients (18 females), of mean 3,4 (range: 0,3-25) years of age, belonging to 23 families with type III BS and unaffected available relatives (n=57) were analyzed. 10 patients of our cohort were included in our previous manuscript, in which we described the founder effect of the c.610G>A; p.Ala204Thr mutation in Spain (Table 1) [18]. Clinical diagnosis was performed at different hospitals, based on symptoms and biochemistry disturbances (Table 1). Most patients demonstrated a decreased distal Cl- reabsorption during hypotonic saline diuresis [19]. In 20 out of 26 cases nephrocalcinosis was not detected. One family (SOR051) was known to be consanguineous.
Table 1. Clinical and biological characteristics of BS patients.
| Patient | Age at diagnosis (years) | Gestational Age (weeks) | NC | PH | Body Height (SDS) | Body Weight(SDS) | pH blood | Na + mEq/l | K + mEq/l | Cl - mEq/l | HCO 3 - mEq/l | Mg 2+ mg/dl | Creati-Nine mg/dl | FENa + n % | FEK + % | FECl - % | Renin ng/ml/h | Aldosterone pg/ml | UCa 2+ /Cr mg/mg |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| *SOR003 | 6,8 | 36 | - | + | -0,1 | -1,3 | 7,53 | 134 | 1,4 | 100 | 35 | 0,9 | 0,7 | 1,2 | 49,9 | 2,9 | 20,9 | 945 | 0,4 |
| *SOR005 | 3,0 | 40 | + | + | -1,0 | -1,6 | 7,50 | 130 | 2,0 | 85 | 28 | 2,2 | 0,5 | 2,8 | 50,5 | 4,0 | 60,5 | 484 | 0,5 |
| *SOR008 | 2,0 | 40 | + | - | -3,2 | -4,9 | 7,47 | 134 | 2,0 | 88 | 28 | 1,5 | 0,7 | 1,5 | 62,3 | 2,1 | NA | NA | 0,3 |
| *SOR009 | 2,0 | 36 | - | + | -3,5 | -4,5 | 7,31 | 143 | 1,7 | 100 | 20 | NA | 0,7 | 1,3 | 76,8 | 2,4 | 13,6 | NA | 0,3 |
| SOR023 | 1,8 | 40 | - | - | -3,7 | -3,2 | 7,47 | 140 | 3,3 | 85 | 24 | 1,9 | 0,4 | 1,2 | 27,5 | 2,1 | 53,4 | 267 | 0,7 |
| *SOR025 | 2,3 | 40 | - | - | -2,0 | -3,7 | 7,57 | 132 | 2,3 | 96 | 27 | 1,6 | 0,5 | 1,2 | 34,2 | 2,0 | 215 | 5610 | 0,2 |
| *SOR026 | 0,7 | 40 | - | + | -1,9 | -3,7 | 7,42 | 136 | 3,3 | 90 | 37 | 2,5 | 0,2 | 0,1 | 7,20 | 0,2 | 13,6 | 1654 | 0,2 |
| SOR039 | 1,4 | 41 | - | - | -2,3 | -4,0 | 7,49 | 138 | 1,6 | 93 | 28 | 2,8 | 0,3 | 2,3 | 54,0 | 3,5 | 86,6 | 1281 | 0,2 |
| SOR045 | 0,7 | NA | - | + | -2,3 | 2,0 | 7,48 | 133 | 2,9 | 79 | 40 | 2,3 | 0,4 | 0,2 | 13,4 | 0,3 | NA | 670 | NA |
| SOR047 | 0,7 | NA | - | - | -1,9 | -3,5 | 7,65 | 137 | 2,5 | 83 | 31 | 3,1 | 0,3 | 0,1 | 17,7 | 0,3 | NA | 760 | 0,1 |
| *SOR048 | 2,0 | 42 | - | + | -1,2 | -2,3 | 7,38 | 126 | 2,7 | 82 | 27 | 3,0 | 0,2 | 0,6 | 14,2 | 0,6 | 77,4 | 282 | 0,2 |
| SOR050 | 0,7 | 41 | - | - | -5,3 | -5,3 | 7,50 | 140 | 2,2 | 99 | 31 | 2,5 | 0,3 | 0,2 | 20,8 | NA | NA | NA | 0,0 |
| SOR051 | 25 | NA | + | NA | NA | NA | 7,34 | 134 | 2,3 | 98 | 24 | 2,6 | 2,7 | NA | NA | NA | NA | NA | NA |
| SOR062 | 3,0 | 40 | + | - | -1,9 | -2,7 | NA | 130 | 1,9 | 89 | 31 | 2,4 | 0,3 | 0,9 | 42,0 | 1,4 | 80,4 | 1608 | 1,1 |
| SOR073 | 0,7 | 40 | - | - | -1,0 | -3,2 | 7,47 | 139 | 3,3 | 97 | 30 | 2,2 | 0,4 | 1,0 | 25,0 | 0,3 | 8,30 | 1370 | 0,4 |
| *SOR011 | 11 | NA | - | - | -2,3 | NA | 7,45 | 138 | 2,0 | 93 | 29 | 1,7 | 0,5 | 0,8 | 31,4 | 1,1 | 31,1 | 1015 | NA |
| *SOR024 | 0,9 | NA | - | NA | NA | -5,0 | NA | NA | 1,8 | NA | NA | NA | 0,5 | NA | NA | NA | NA | NA | NA |
| *SOR024 | 17 | 40 | - | - | 1,8 | 1,0 | NA | 132 | 2,6 | 134 | 33 | 1,3 | 0,4 | NA | NA | NA | 195 | 114 | NA |
| SOR054 | 0,3 | NA | - | - | -0,3 | -2,3 | 7,51 | 131 | 2,3 | 80 | 33 | 2,2 | 0,5 | 0,3 | 42,1 | 0,6 | NA | NA | 0,0 |
| SOR054 | 0,3 | NA | - | - | -2,1 | -2,2 | 7,46 | 134 | 2,9 | 86 | 28 | 2,2 | 0,7 | 0,5 | 25,0 | 1,2 | 21,0 | 185 | 0,0 |
| SOR054 | 0,5 | NA | - | - | -0,6 | -1,5 | 7,40 | 145 | 2,2 | 81 | 26 | NA | 0,5 | 0,2 | 30,9 | NA | 60,6 | 220 | 0,5 |
| SOR057 | 0,7 | 36 | + | + | -3,5 | -6,2 | 7,62 | 134 | 2,3 | 87 | 35 | 2,9 | 0,3 | 0,1 | 24,3 | 0,5 | 39,0 | 473 | 0,4 |
| SOR064 | 0,8 | 41 | - | + | -1,7 | -2,6 | 7,55 | 134 | 3,1 | 81 | 43 | 2,3 | 0,3 | 0,2 | 8,20 | 0,2 | 19,9 | 1584 | 0,5 |
| SOR063 | 0,4 | 41 | - | - | -1,0 | -2,9 | 7,59 | 133 | 2,7 | 94 | 33 | 2,3 | 0,3 | 0,3 | 35,2 | 1,3 | 30,0 | 384 | 0,1 |
| SOR076 | 1,0 | NA | - | - | -1,6 | 1,5 | 7,46 | 138 | 2,7 | NA | 28 | NA | 0,6 | NA | NA | NA | 50,0 | 480 | NA |
| SOR021 | 2,5 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| MEAN | 3,4 | 40 | -1,8 | -2,7 | 7,48 | 135 | 2,4 | 91 | 30 | 2,2 | 0,5 | 0,8 | 32,9 | 1,4 | 59,7 | 1020 | 0,3 | ||
| SD | 5,7 | 2 | 1,4 | 2,1 | 0,08 | 4,37 | 0,5 | 11 | 5 | 0,6 | 0,5 | 0,7 | 18,2 | 1,2 | 58,2 | 1226 | 0,3 | ||
| MEDIAN | 1,2 | 40 | -1,9 | -2,9 | 7,47 | 134 | 2,3 | 89 | 29 | 2,3 | 0,4 | 0,6 | 30,9 | 1,2 | 44,5 | 670 | 0,3 | ||
| MIN. | 0,3 | 36 | -5,3 | -6,2 | 7,31 | 126 | 1,4 | 79 | 20 | 0,9 | 0,2 | 0,1 | 7,2 | 0,2 | 8,30 | 114 | 0,0 | ||
| MAX. | 25 | 42 | 1,8 | 2 | 7,65 | 145 | 3,3 | 134 | 43 | 3,1 | 2,7 | 2,8 | 76,8 | 4 | 215 | 5610 | 1,1 |
* Patients included in our previous manuscript [18]. Abbreviations: NC, nephrocalcinosis; PH, polyhydramnios; + yes; - no; NA, not available; SDS, standard deviation score in comparison with age- and sex-matched reference population; FE, fractional excretion; U, urinary; Ca2+/Cr, calcium/creatinine ratio; SD, standard deviation; MIN, minimum; MAX, maximum.
Biological samples from the patients were received at the Molecular Genetic Laboratory at Cruces University Hospital, Bizkaia, Spain. Most samples were sent from the Pediatric Nephrology Division at Cruces University Hospital (n=14) and Renaltube team (n=7), a network tool for clinical and genetic diagnosis of primary tubulopathies (www.renaltube.com). A few samples (n=5) were received from other hospitals in Spain.
DNA analysis
Genomic DNA was extracted from peripheral blood leukocytes according to manufacturer’s instructions (QIAamp® DNA Blood Mini Kit, QIAGEN, Germany).
The exon regions and flanking intronic sequences of the CLCNKB gene (Ensembl identifiers: gene ENSG00000184908; transcript, ENST00000375679) were screened for mutations by polymerase chain reaction (PCR) followed by direct sequencing.
We used newly designed primers for non-coding exon 1 and promoter region (according to SwitchGear Genomics), and previously described primers [20-22] to amplify exons 2-20 together with their splice sites (Table 2). Due to the high (>90%) sequence similarities between CLCNKB and CLCNKA, and their close location on chromosome 1, specific primers for individual exons were used only when available, this is, when both genes’ sequence was different enough to specifically amplify the CLCNKB gene. For the cases it was not possible to directly amplify the CLCNKB gene’s individual exons, we generated by Long-range PCR three large fragments (named FD, FE and FF), specific for the CLCNKB gene. Then, we used nested PCR with internal specific primers to analyze exons 4 (within product FD), 8, 9-10 (within product FE) and 13-14 (within product FF) (Table 2). All PCR reactions were performed in a total volume of 50µl, in presence of 10% DMSO (dimethyl sulfoxide). Annealing temperatures and amplicons’ sizes are shown in Table 2.
Table 2. PCR and sequencing primers for the analysis of the human CLCNKB gene.
| Exon/ Promoter | Forward primer (5´- 3´) | Reverse primer (5´- 3´) | Annealing T (°C) | Amplicon Length (bp) |
|---|---|---|---|---|
| Promoter | * CTCTCCCCATTCACAGGGTG | * TGGACAGGTGTGTGTTCCAA | 56,3 | 966 |
| † CGGCCTCCGTGATCTTAGAC | † AGCATGACCACAGCCTCC | |||
| 1 | * CTCTGTGCAGCTATGGTGGG | * CTGTCCACCTATGAGCACCC | 56.3 | 429 |
| 2 | ‡ ACTGGAAGGGCCTAGAGGCAGT | ‡ GATGTCCTGAGTGGTCCTCCAG | 60 | 231 |
| 3 | § CACTGTGTCACCACTGTCACC | § AGGAGTAAAGCCAGGACCAGA | 60 | 460 |
| FD | || ¶ TGCCCCACCCTGTGCCGTGAC | || ¶ GGGTGGTTGGGATGCCCTCAC | 66,8 | 2558 |
| 4 | || # GAGGCTGTGGGTGCCTCCCTG | || # AGTGGGGACTGGCGTAGCGAC | 61,4 | 200 |
| 5-7 | § AGATCTTGTCCCCAAAGGAAA | § GGCTGAAGTGAGAACTAGAATGA | 60 | 950 |
| † AGTGGTATGGGCAGGGGT | † AGGTAGGCAGCCATCATCAC | |||
| † CACCTGTCTGTGATGATGG | † GGGTAGGGTGGTTGGGA | |||
| FE | || ¶ CCCTCCTGGCCCTGCCCAC | || ¶AGCTCGCTGAGAGGTCCCCAG | 66,8 | 2082 |
| 8 | || # GGAGGGCCCACCTGAGATCAG | || # GCAGGGCCAGGGTCAGGCAG | 61,4 | 193 |
| 9-10 | || # CGCCATCTTGGCTCCCCACTG | || # AGCTCGCTGAGAGGTCCCCAG | 61,4 | 374 |
| 11-12 | § CTGACCCCACAGGTTCTGT | § ** CCAGGGCAGAGGTTAGAGGC | 60 | 1099 |
| † GCCTCCTTTGCGTGTAT | † TAACCAGGAAGAAGGCAAGG | |||
| † GGTTCACCATCTTTGGGA | † CTGACCTCCCGAAGCTGTAG | |||
| FF | || ¶ GTCGGGCTCTGGGCTCATGTC | || ¶ CAGTCAGCCTGAGGTGGGCAC | 66,8 | 2660 |
| 13-14 | || # TCTAGGACACTCCCCTGTCCC | || # CCCTGGGGAACCACCAGCCAA | 61,4 | 519 |
| 15 | †† CATCACTCCCTCGTGGCTCCTG | †† CTACGGTGGCGTTTCTTTTTCG | 60 | 518 |
| 16 | †† GCTAAAGTGGAGCTGGTCTG | †† GCAACAAGGATTTGGAGG | 60 | 750 |
| † CCACAGCATCACCACACT | † CACAACCTTGACCACCTCCT | |||
| 17-18 | § CACAATAGCCCCATAGGAACA | § CTCTCCCACTTCCCTCATCTC | 60 | 672 |
| † GGTCAGAGAGAGGCATCCTG | † ATGTCCTGGAGACACTGCTG | |||
| 19 | || GGGCACCTTCTACCCTCCAGTG | || GTCTTCTCAGGCATAGGTTCCCTG | 60 | 187 |
| 20 | || CTACATCCCCCCGCACCACCAC | || AGGGTCTCAGCCCAACCTC | 60 | 153 |
According to DNA sequence (Ensembl: ENST00000375679); Bases that differs from the CLCNKA gene (Ensembl: ENST00000331433) are marked in bold; * Newly designed primers; † Newly designed internal primers for sequencing; ‡ Previously designed primers [20]; § Previously designed primers [21]; || Primers kindly provided by the Hôpital Européen Georges Pompidou, Service de Génétique, Paris; ¶ Specific primers for long-range PCR; # Internal specific primers for nested PCR; ** Reverse primer modified (last base removed because it is a SNP); †† Previously designed primers [22].
PCR products were then purified by extracting them from agarose gels using QIAquick Gel Extraction Kit (QIAGEN), according to the manufacturer’s specifications.
Purified amplified products were directly sequenced with fluorescent dideoxynucleotides (BigDye Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems, Foster City, CA). We used newly designed internal primers for direct sequencing of fragments 5-7, 11-12, 16, 17-18 and promoter (Table 2). In the rest of exons, the same primers as in PCR were used for direct sequencing.
Excess dye terminators were removed (ethanol-ethylenediaminetetraacetic acid-sodium acetate) and samples were denatured and loaded onto an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA). Pathogenic effect of specific DNA variants was assessed using Ensembl (www.ensembl.org, EMBL-EBI) and HGMD database. Missense DNA variants, not listed either in the Ensembl database or in the HGMD database, were assessed using prediction pathogenic software: Mutation t@sting (www.mutationtaster.org), PolyPhen-2 (genetics.bwh.harvard.edu), SIFT (sift.jcvi.org), SNPs & GO (snps-and-go.biocomp.unibo.it) and NetGene2 Server (www.cbs.dtu.dk) for variations in the splice sites. DNA mutations were named according to the Human Genomic Variation Society guidelines (HGVS, www.hgvs.org).
For the purpose of detecting large deletions or duplications a commercially available kit, SALSA MLPA (Multiplex Ligation dependent Probe Amplification) probemix P266-B1 (MRC, Holland, Amsterdam, The Netherlands) was used. This kit contains probes for 14 out of 20 exons of the CLCNKB gene and more telomeric probes, including 2 probes for the CLCNKA gene, 1 probe for the CASP9 gene and 1 probe for the PRDM2 gene.
In order to analyse the remaining exons without a MLPA probe (exons 4, 7, 9, 12, 16 and 20) we performed Quantitative Multiplex PCR of Short Fluorescent Fragments (QMPSF) assays. QMPSF is a simple method based on the simultaneous amplification of short genomic sequences under quantitative conditions, using dye-labelled primers, and on the comparison of profiles generated from tested and control DNA, and is considered a very sensitive method for the detection of genomic deletions and duplications. Briefly, we simultaneously amplified our exons of interest together with exon 7 of the HNF1β gene (used as a control fragment), stopping the reaction at the exponential phase of the amplification in order to get a semiquantitative estimation of each PCR product. Fragments were then separated by capillary electrophoresis (ABI 3130xl Genetic Analyzer) and analysed. Primer sequences, annealing temperatures and amplicons’ sizes are shown in Table 3. Two QMPSF assays were performed for each sample to easily discriminate between all amplicons.
Table 3. PCR Primers for QMPSF.
| QMPSF1 Exon | Forward primer (5´- 3´) | Reverse primer (5´- 3´) | AnnealingT (°C) | Amplicon Length (bp) |
|---|---|---|---|---|
| 4 | *TGTACCCTGTGGCCCTCGTC | */5’6-FAM/ CGCAGAAGATCCTCCCACCA | 50 | 323 |
| 12 | *TCGCTGTTCGACAACCACTC | */5’6-FAM/CCACCAGCTGCGATGAGGT | 50 | 251 |
| 16 | */5’6-FAM/GGGTCT CACATCCCTGACTGT | *CCTTGACCACCTCCTCCAGT | 50 | 157 |
| 17-18 | */5’6-FAM/CCTCCTTCC TGGGCTCCT | *GCAGCCTGCAGCCAAGAT | 50 | 231 |
| †7 (HNF1 β gene) | ‡/5’6-FAM/TCAACACCTCCCAAGCACA | ‡TGAGTCACAGCTGCCATGA | 50 | 197 |
| QMPSF2 Exon | Forward primer (5´- 3´) | Reverse primer (5´- 3´) | AnnealingT (°C) | Amplicon Length (bp) |
| 6-7 | */5’6-FAM/CGTGCACCTGTCTGT GATGATG | *GGAGCTGCAAAGACTGTGGC | 50 | 244 |
| 9-10 | *CAGTTTCCGGGTGGACGTT | */5’6-FAM/ AACCTATTGTTCCTGATGAAGCC | 50 | 270 |
| 20 | */5’6-FAM/GCTCTACTATT TACCCAGAAACCAC | *CTGGCGGATTTGTCAGGTT | 50 | 166 |
| †7 (HNF1 β gene) | ‡/5’6-FAM/TCAACACCTCCCAAGCACA | ‡TGAGTCACAGCTGCCATGA | 50 | 197 |
According to DNA sequence (Ensembl: ENST00000375679); Bases that differ from the CLCNKA gene (Ensembl: ENST00000331433) are marked in bold; * primers kindly provided by the Hôpital Européen Georges Pompidou, Service de Génétique, Paris; † Control for the QMPSF technique. ‡ Previously designed primers [30].
In order to improve the genetic diagnostic the algorithm was as follows: first, detection of the previously described Spanish founder mutation (c.610G>A; p.Ala204Thr) [18]; second, detecting large deletions or duplications by MLPA and QMPSF; and third, sequencing of the coding and flanking regions of the whole CLCNKB gene.
Statistics
Reported data correspond to the mean ± SD unless otherwise specified.
Results
Clinical results
Clinical symptoms consisted of polyuria, polydipsia, vomiting, constipation, salt craving, dehydration, hypotonia, and failure to thrive. In 8 cases (30,7%) a history of hydramnios was also recorded. Characteristically, all patients had hypokalemia (K+ 2,4 ± 0,5 mEq/l) of renal origin (FEK mean 32,9%), commonly associated with hypochloremia (Cl- 91 ± 11 mEq/l), and metabolic alkalosis (plasma pH 7,48 ± 0,08, bicarbonate 30 ± 5 mEq/l). Increased plasma renin activity (mean 59,7 ng/ml/h), and aldosterone levels (mean 1020 pg/ml) were observed despite normal blood pressure, a marker of BS. At the time of the study two cases (7,7%) presented hypomagnesemia, and 5 (19%) nephrocalcinosis. Further, growth retardation and poor weight gain (mean Z-height -1,8, and Z-weight -2,7) were very common in the study cohort (Table 1).
Mutation analysis in the CLCNKB gene
Spanish founder mutation detection (c. 610G>A; p.Ala204Thr)
We found that 23 patients (88%), belonging to 20 families (Table 4), presented with the Spanish founder mutation c.610G>A; p.Ala204Thr (exon 7) [18]. From those, 15 patients (15 families) were homozygous for the mutation (58%). Another 8 patients (5 families) were compound heterozygous (31%). We observed a high frequency (38/52 of the alleles) of the p.Ala204Thr founder mutation in our population.
Table 4. Molecular results for the CLCNKB gene in the studied cohort.
| Patient | Sex | Ethnic origin | Exon | Mutation‡ 1 | Mutation‡ 2 | Father‡ | Mother‡ |
|---|---|---|---|---|---|---|---|
| SOR003 | F | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR005 | F | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR008 | F | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR009 | F | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR023 | M | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR025 | F | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR026 | F | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR039 | F | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR045 | M | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR047 | M | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR048 | F | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR050 | M | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR051 | M | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR062 | M | Spain | 7 | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | c.610G>A p.Ala204Thr | |
| SOR073 | F | Spain | 7 | c.610G>A p.Ala204Thr | |||
| SOR011 | F | Spain | 7 1-20 | c.610G>A p.Ala204Thr | c. -19-? _2053+?del | c.610G>A p.Ala204Thr | c. -19-? _2053+?del |
| SOR024* | F | Spain | 7 1-20 | c.610G>A p.Ala204Thr | c. -19-? _2053+?del | c. -19-? _2053+?del | c.610G>A p.Ala204Thr |
| SOR024* | F | Spain | 7 1-20 | c.610G>A p.Ala204Thr | c. -19-? _2053+?del | c. -19-? _2053+?del | c.610G>A p.Ala204Thr |
| SOR054* | F | Spain | 7 14 | c.610G>A p.Ala204Thr | c.1325A>G† p.Glu442Gly | c.610G>A p.Ala204Thr | c.1325A>G† p.Glu442Gly |
| SOR054* | F | Spain | 7 14 | c.610G>A p.Ala204Thr | c.1325A>G† p.Glu442Gly | c.610G>A p.Ala204Thr | c.1325A>G† p.Glu442Gly |
| SOR054* | F | Spain | 7 14 | c.610G>A p.Ala204Thr | c.1325A>G† p.Glu442Gly | c.610G>A p.Ala204Thr | c.1325A>G† p.Glu442Gly |
| SOR057 | F | Spain | 7 14 | c.610G>A p.Ala204Thr | c.1325A>G† p.Glu442Gly | c.1325A>G† p.Glu442Gly | c.610G>A p.Ala204Thr |
| SOR064 | M | Spain | 6 7 | c.610G>A p.Ala204Thr | c.508G>A p.Val170Met | c.610G>A p.Ala204Thr | c.508G>A p.Val170Met |
| SOR063 | M | Spain African | 12 1-20 | c.1192_1203del12† p.Ile398_Thr401del | c. -19-? _2053+?del | c.1192_1203del12† p.Ile398_Thr401del | c. -19-? _2053+?del |
| SOR076 | F | Latin America | 7 16 | c.629C>T p.Ala210Val | c.1756+1G>A† Splice defect | ||
| SOR021 | F | Spain |
Siblings
† These mutations (also marked in bold) have not been reported to date
Numbering is according to DNA sequence (Ensembl: ENST00000375679)
(All patients were homozygous unless a second mutation is given; all parents were heterozygous for the given mutation)
MLPA and QMPSF analysis
Among those 8 compound heterozygous patients for the founder mutation, MLPA and QMPSF techniques demonstrated that 3 patients (SOR011 and the 2 siblings of SOR024) had an entire gene deletion in one allele (c. -19-?_2053+? del). Further, we found another patient (SOR063) that had an entire gene deletion in one allele in combination with a small deletion of 12 base pairs (c.1192_1203del12) in the other, detected by direct sequencing. This small deletion is confirmed using the QMPSF technique; we observed an amplified fragment 12 base pairs smaller than the one corresponding to exon 12 (Figure 1). In all patients with the whole CLCNKB deletion, we confirmed that the probes for the nearly CLCNKA, CASP9 and PRDM2 genes included in the MLPA were present.
Figure 1. Detection of CLCNKB deletions by the QMPSF and MLPA techniques.
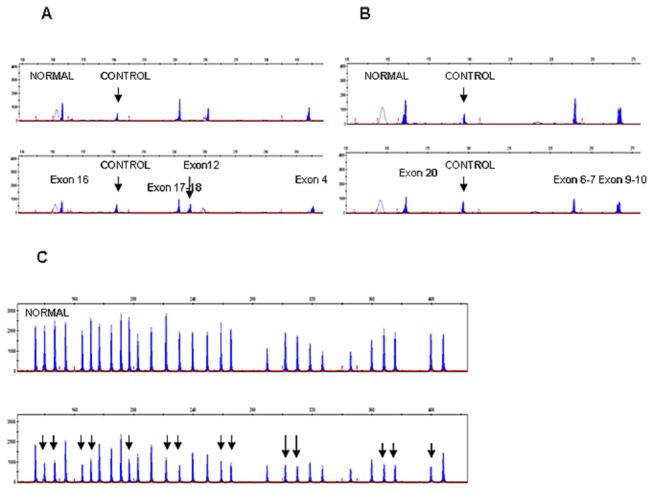
QMPSF and MLPA electropherograms for the CLCNKB gene from controls (upper panels) and patients (lower panels). (A) QMPSF half doses for exons 16, 17-18, 12 and 4. The arrow shows the peak for exon 12 with the small deletion c.1192_1203del12 (patient SOR0063). Exon 7 of the HNF1B gene is the internal control. (B) QMPSF half doses for exons 20, 6-7 and 9-10. (C) For MLPA, each peak represents the exons for the CLCNKB gene and 15 control probes. The arrows show half doses for all the CLCNKB exons (patient SOR0011, family SOR0024 and SOR0063).
CLCNKB gene analysis
Complete structural CLCNKB gene analysis found missense mutations in the remaining 5 patients with the founder mutation: one case (SOR064) carried the described mutation c.508G>A; p.Val170Met (exon 6) [23], and 4 cases (SOR057 and the 3 siblings of SOR054) carried the novel mutation c.1325A>G; p.Glu442Gly (exon 14) (Figure 2).
Figure 2. Detection of novel CLCNKB mutations by direct sequencing.
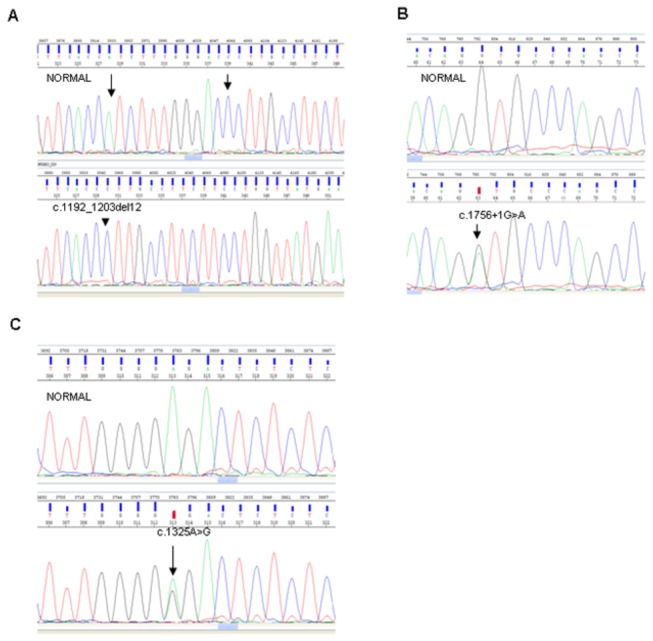
Figures represent the sequencing chromatograms from controls (upper panels) and patients (lower panels). (A) Heterozygous c.1192_1203del12 mutation in exon 12; arrows show the extension of the deletion. (B) Heterozygous c.1756+1G>A mutation at the splicing site in exon 16. (C) Heterozygous missense mutation c.1325A>G in exon 14.
Furthermore, we found two patients having other mutations in the CLCNKB gene. We observed one patient (SOR063) with a novel small deletion of 12 base pairs (c.1192_1203del12; p.Ile398_Thr401del) (Figure 2) in combination with the entire CLCNKB gene deletion (altogether, we found that 15% of cases -4/26- carried an entire gene heterozygous deletion within the cohort). The second patient (SOR076) presented the described mutation c.629C>T; p.Ala210Val (exon 7) [22] in combination with a novel splice-donor mutation c.1756+1G>A, a substitution of the 5´-donor splice site of intron 16 (Figure 2). Finally, in one case (SOR021), the molecular analysis of CLCNKB turned out to be negative.
We also detected 51 healthy relatives with heterozygous mutations in CLCNKB (43 presented the founder mutation p.Ala204Thr in heterozygosis), who were thus considered carriers in our genetic screening.
We tested 95 control subjects of Spanish ancestry in order to additionally confirm the pathogenicity of the novel mutations found in this study. None presented any of the mutations in either allele.
The complete description of the genetic results in our series is detailed in Table 4.
Discussion
In the present study, we demonstrated that the Spanish p.Ala204Thr mutation, despite being very prevalent, is not the unique cause of type III BS in Spain, with other distinct CLCNKB gene mutations being observed. Importantly, the complete genetic study revealed three not previously reported CLCNKB mutations: p.Glu442Gly, c.1756+1G>A and p.Ile398_Thr401del.
We identified the genetic defect in almost all cases by polymerase chain reaction (PCR) and direct CLCNKB gene sequencing. Most of our cohort of BS patients presented the substitution of the hydrophobic non-polar amino acid alanine to the polar amino acid threonine at codon 204 in the fifth transmembrane domain, thus confirming our previous report regarding a founder mutation in most type III Bartter patients among Spanish population [18]. It has also been described that point mutations in the fifth transmembrane domain of the protein are associated with altered channel activity [24]. In previous studies realized by our group, 300 control subjects of Spanish ancestry were sequenced, none was homozygous for the p.Ala204Thr allele and it was identified in only 2 of 600 control chromosomes [18]. Therefore, that amino acid change represents a true loss of function mutation and not a common polymorphism.
It has been described that homozygous deletion encompassing the whole CLCNKB gene represents the most common molecular finding in type III BS [9]. Accordingly, we decided to screen for large rearrangements at the CLCNKB gene. We emphasize that in our study we have only found heterozygous deletions for the entire gene. CLCNKA and CLCNKB genes are separated only by 9,727 kb of genomic sequence, sharing a 94% of sequence identity and, therefore, it has been stated that there is a predisposition to a high rate of rearrangements by unequal crossing over [13], which would explain the high frequency of the whole gene deletion. Thus, some authors have suggested a complete gene analysis also in cases of an apparently homozygous mutation, due to the high possibility of being indeed hemizygous [25].
Among heterozygous cases with the founder mutation, we found two different mutations: the previously described mutation c.508G>A; p.Val170Met [23] and the novel mutation c.1325A>G; p.Glu442Gly. In the first one, the change of a hydrophobic valine to a nonpolar methionine (with a thioether group) causes a defective protein. In the second case, the exchange of a negative charged hydrophilic glutamic acid to a small uncharged glycine predicts a disruption of the charge distribution of the domain and the alteration of the conformation of ClC-Kb.
Other mutations were found in compound heterozygosis: one patient (SOR063) presented a not previously described small deletion of four amino acids in the paternal allele (c.1192_1203del12; p.Ile398_Thr401del) in combination with the entire gene deletion in the maternal allele. Lack of these four amino acids might lead to a defective chloride channel. The program Mutation t@sting predicts loss of one transmembrane helical domain and additional activations of splice sites; another patient (SOR076) presented the described mutation c.629C>T; p.Ala210Val [22]. Alanine 210 is a residue located within the transmembrane G helix. Mutations in many residues within the G helix have been described to cause functional alterations of chloride channels [26-28]. This mutation is present in combination with a novel heterozygous splice-donor mutation c.1756+1G>A. This guanine to adenine substitution might lead to the exon skipping and generation of an aberrant chloride channel ClC-Kb.
One case turned out to be negative in our genetic screening. Despite we cannot discard the possible presence of a deep intronic mutation or any complex rearrangement (i.e., intragenic inversions) not detected with our strategy, it is more plausible that this patient might have mutations in other transporter-codifying genes (KCNJ1, SLC12A1), or in the SLC12A3 gene (OMIM *600968). Indeed, SLC12A3 encodes for the thiazide-sensitive Na+-Cl- cotransporter localized in the distal tubule. Mutations in this gene cause Gitelman syndrome [29], which is characterized by hypokalaemic metabolic alkalosis in combination with hypomagnesemia and hypocalciuria and may cause phenotypes that overlap with BS [17].
In conclusion, based on our results, we propose this algorithm as a way to reduce costs and accelerate the genetic study of the type III BS in the Spanish population: because the p.Ala204Thr mutation in CLCNKB gene is the most prevalent mutation in Spain, we propose to first look for this mutation in exon 7. Nevertheless, we have also found other mutations, including gross deletions. Therefore, should the p.Ala204Thr mutation not be detected in clinically diagnosed type III BS patients, whole gene sequencing, MLPA and QMPSF analyses must be carried out.
Acknowledgments
We thank patients and families, and their pediatric nephrologists who collaborated with the genetic study. We also thank Dra. Rosa Vargas-Poussou from the genetic department of the Hôpital Européen Georges Pompidou who has helped us assembling the techniques for genetic analysis.
The members of the RenalTube Group are: Alejandro García Castaño, Research Unit, Ciberer, Cruces University Hospital, Bizkaia, Spain;
Elizabeth Córdoba, Pediatric Nephrology, Nuestra Señora de Candelaria University Hospital, Tenerife, Canarias, Spain;
Eliecer Coto, Pediatric Nephrology, Asturias Central University Hospital, Oviedo, Asturias, Spain;
Enrique García, Pediatric Nephrology, Asturias Central University Hospital, Oviedo, Asturias, Spain;
Elena Ramos, Pediatric Nephrology, Nuestra Señora de Candelaria University Hospital, Tenerife, Canarias, Spain;
Flor Ángel Ordóñez, Pediatric Nephrology, Asturias Central University Hospital, Oviedo, Asturias, Spain;
Félix Claverie, Pediatric Nephrology, Nuestra Señora de Candelaria University Hospital, Tenerife, Canarias, Spain;
Francisco Javier González, Pediatric Nephrology, Nuestra Señora de Candelaria University Hospital, Tenerife, Canarias, Spain;
Fernando Santos, Pediatric Nephrology, Asturias Central University Hospital, Oviedo, Asturias, Spain:
Gema Ariceta, Pediatric Nephrology, Materno Infantil Vall d’Hebron Hospital, Barcelona, Spain;
Gustavo Pérez de Nanclares, Research Unit, Ciberer, Cruces University Hospital, Bizkaia, Spain;
Helena Gil, Pediatric Nephrology, Asturias Central University Hospital, Oviedo, Asturias, Spain;
Hilaria González, Pediatric Nephrology, Nuestra Señora de Candelaria University Hospital, Tenerife, Canarias, Spain;
Julián Rodríguez, Pediatric Nephrology, Asturias Central University Hospital, Oviedo, Asturias, Spain;
Luis Castaño, Research Unit, Ciberer, Cruces University Hospital, Bizkaia, Spain; Department of Pediatrics, School of Medicine and Odontology, University of Basque Country UPV/EHU, Bizkaia, Spain;
Leire Madariaga, Pediatric Nephrology, Cruces University Hospital, Bizkaia., Spain; Department of Pediatrics, School of Medicine and Odontology, University of Basque Country UPV/EHU, Bizkaia, Spain;
Mireia Aguirre, Pediatric Nephrology, Cruces University Hospital, Bizkaia., Spain;
María Isabel Luis Yanes, Pediatric Nephrology, Nuestra Señora de Candelaria University Hospital, Tenerife, Canarias, Spain;
Natalia Mejía, Pediatric Nephrology, Asturias Central University Hospital, Oviedo, Asturias, Spain;
Victoria Álvarez, Pediatric Nephrology, Asturias Central University Hospital, Oviedo, Asturias, Spain;
Victor Manuel García, Pediatric Nephrology, Nuestra Señora de Candelaria University Hospital, Tenerife, Canarias, Spain;
Vanessa Loredo, Pediatric Nephrology, Asturias Central University Hospital, Oviedo, Asturias, Spain
Funding Statement
This study was supported by two grants (PI09/90888 and PI11/01412) from the FIS of the Instituto de Salud Carlos III, Madrid, Spain, and the Eitb Maratoia-Bioef (BIO08/ER/020) the Basque Foundation for Health Innovation and Research (BIOEF, from the Basque Berrikuntza + Ikerketa + Osasuna Eusko Fundazioa). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bartter FC, Pronove P, Gill JR, MacCardle RC (1962) Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med 33: 811-828. doi:10.1016/0002-9343(62)90214-0. PubMed: 13969763. [DOI] [PubMed] [Google Scholar]
- 2. Rodríguez Soriano J (1998) Bartter and related syndromes: the puzzle is almost solved. Pediatr Nephrol 12: 315-327. doi:10.1007/s004670050461. PubMed: 9655365. [DOI] [PubMed] [Google Scholar]
- 3. Seyberth HW, Rascher W, Schweer W, Kühl PG, Mehls O et al. (1985) Congenital hypokalemia with hypercalciuria in preterm infants: a hyperprostaglandinuric tubular syndrome different from Bartter syndrome. J Pediatr 107: 694-701. doi:10.1016/S0022-3476(85)80395-4. PubMed: 3863906. [DOI] [PubMed] [Google Scholar]
- 4. Proesmans W (1997) Bartter syndrome and its neonatal variant. Eur J Pediatr 156: 669-679. doi:10.1007/s004310050688. PubMed: 9296528. [DOI] [PubMed] [Google Scholar]
- 5. Rodríguez Soriano J (1999) Bartter’s syndrome comes of age. Pediatrics 103: 663-664. doi:10.1542/peds.103.3.663. PubMed: 10049972. [DOI] [PubMed] [Google Scholar]
- 6. Jeck N, Konrad M, Peters M, Weber S, Bonzel KE et al. (2000) Mutations in the chloride channel gene, CLCNKB, leading to a mixed Bartter-Gitelman phenotype. Pediatr Res 48: 754-758. doi:10.1203/00006450-200012000-00009. PubMed: 11102542. [DOI] [PubMed] [Google Scholar]
- 7. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA et al. (1996) Bartter’s syndrome, hypokalemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13: 183-188. doi:10.1038/ng0696-183. PubMed: 8640224. [DOI] [PubMed] [Google Scholar]
- 8. Simon DB, Karet FE, Rodríguez Soriano J, Hamdan JH, DiPietro A et al. (1996) Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet 14: 152-156. doi:10.1038/ng1096-152. PubMed: 8841184. [DOI] [PubMed] [Google Scholar]
- 9. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E et al. (1997) Mutations in the chloride channel gene CLCNKB cause Bartter’syndrome type III. Nat Genet 17: 171-178. doi:10.1038/ng1097-171. PubMed: 9326936. [DOI] [PubMed] [Google Scholar]
- 10. Birkenhäger R, Otto E, Schürmann MJ, Vollmer M, Ruf EM et al. (2001) Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet 29: 310-314. doi:10.1038/ng752. PubMed: 11687798. [DOI] [PubMed] [Google Scholar]
- 11. Vargas-Poussou R, Huang C, Hulin P, Houillier P, Jeunemaître X et al. (2002) Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J Am Soc Nephrol 13: 2259-2266. doi:10.1097/01.ASN.0000025781.16723.68. PubMed: 12191970. [DOI] [PubMed] [Google Scholar]
- 12. Schlingmann KP, Konrad M, Jeck K, Waldegger P, Reinalter SC et al. (2004) Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med 350: 1314-1319. doi:10.1056/NEJMoa032843. PubMed: 15044642. [DOI] [PubMed] [Google Scholar]
- 13. Konrad M, Vollmer M, Lemmink HH, Van den Heuvel LP, Jeck N et al. (2000) Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol 11: 1449-1459. PubMed: 10906158. [DOI] [PubMed] [Google Scholar]
- 14. Takeuchi Y, Uchida S, Marumo F, Sasaki S (1995) Cloning, tissue distribution, and intrarenal localization of ClC chloride channels in human kidney. Kidney Int 48: 1497-1503. doi:10.1038/ki.1995.439. PubMed: 8544406. [DOI] [PubMed] [Google Scholar]
- 15. Ellison DH (2000) Divalent cation transport by the distal nephron: insights from Bartter´s and Gitelman’s syndromes. Am J Physiol Renal Physiol 279: F616-F625. PubMed: 10997911. [DOI] [PubMed] [Google Scholar]
- 16. Dutzler R, Campbell EB, Cadene M, Chait BT, Mackinnon R (2002) X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 415: 287-294. doi:10.1038/415287a. PubMed: 11796999. [DOI] [PubMed] [Google Scholar]
- 17. Zelikovic I, Szargel R, Hawash A, Labay V, Hatib I et al. (2003) A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int 63: 24-32. doi:10.1046/j.1523-1755.2003.00730.x. PubMed: 12472765. [DOI] [PubMed] [Google Scholar]
- 18. Rodríguez Soriano J, Vallo A, Pérez de Nanclares G, Bilbao JR, Castaño L (2005) A founder mutation in the CLCNKB gene causes Bartter syndrome type III in Spain. Pediatr Nephrol 20: 891-896. doi:10.1007/s00467-005-1867-z. PubMed: 15875219. [DOI] [PubMed] [Google Scholar]
- 19. Rodríguez Soriano J, Vallo A, Castillo G, Oliveros R (1981) Renal handling of water and sodium in infancy and childhood: a study using clearance methods during hypotonic saline diuresis. Kidney Int 20: 700-704. doi:10.1038/ki.1981.199. PubMed: 7334744. [DOI] [PubMed] [Google Scholar]
- 20. Fukuyama S, Okudaira S, Yamazato S, Yamazato M, Ohta T (2003) Analysis of renal tubular electrolyte transporter genes in seven patients with hypokalemic metabolic alkalosis. Kidney Int 64: 808-816. doi:10.1046/j.1523-1755.2003.00163.x. PubMed: 12911530. [DOI] [PubMed] [Google Scholar]
- 21. Dong Y, Ji QW, Zeng XT, Jiang GR (2010) A novel splicing mutation in CLCNKB in a Chinese patient with Bartter syndrome type III. Chin Med J (Engl) 123: 3151-3153. [PubMed] [Google Scholar]
- 22. Yu Y, Xu C, Pan X, Ren H, Wang W et al. (2010) Identification and functional analysis of novel mutations of the CLCNKB gene in Chinese patients with classic Bartter syndrome. Clin Genet 77: 155-162. doi:10.1111/j.1399-0004.2009.01288.x. PubMed: 19807735. [DOI] [PubMed] [Google Scholar]
- 23. Vargas-Poussou R, Dahan K, Kahila D, Venisse A, Riveira-Munoz E et al. (2011) Spectrum of mutations in Gitelman Syndrome. J Am Soc Nephrol 22: 693-703. doi:10.1681/ASN.2010090907. PubMed: 21415153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waldegger S, Jentsch TJ (2000) Functional and structural analysis of ClC-Kb channel involved in renal disease. J Biol Chem 275: 24527-24533. doi:10.1074/jbc.M001987200. PubMed: 10831588. [DOI] [PubMed] [Google Scholar]
- 25. Nozu K, Fu XJ, Nakanishi K, Yoshikawa N, Kaito H et al. (2007) Molecular analysis of patients with type III Bartter syndrome: picking up large heterozygous deletions with semiquantitative PCR. Pediatr Res 62: 364-369. doi:10.1203/PDR.0b013e318123fb90. PubMed: 17622951. [DOI] [PubMed] [Google Scholar]
- 26. Lin YW, Lin CW, Chen TY (1999) Elimination of the slow gating of ClC-0 chloride channel by a point mutation. J Gen Physiol 114: 1-12. doi:10.1085/jgp.114.1.1. PubMed: 10398688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duffield M, Rychkov G, Bretag A, Roberts M (2003) Involvement of helices at the dimer interface in ClC-1 common gating. J Gen Physiol 121: 149-161. doi:10.1085/jgp.20028741. PubMed: 12566541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Accardi A, Ferrera L, Pusch M (2001) Drastic reduction of the slow gate of human muscle chloride channel (ClC-1) by mutation C277S. J Physiol 534: 745-752. doi:10.1111/j.1469-7793.2001.00745.x. PubMed: 11483705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gitelman HJ, Graham JB, Welt LG (1966) A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians 79: 221-235. PubMed: 5929460. [PubMed] [Google Scholar]
- 30. Bellanné-Chantelot C, Clauin S, Chauveau D (2005) Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 54: 3126–3132. doi:10.2337/diabetes.54.11.3126. PubMed: 16249435. [DOI] [PubMed] [Google Scholar]