

Abstract
Lipocalin 2 (Lcn2) has previously been characterized as an adipokine/cytokine playing a role in glucose and lipid homeostasis. In this study, we investigate the role of Lcn2 in adipose tissue remodeling during high-fat diet (HFD)-induced obesity. We find that Lcn2 protein is highly abundant selectively in inguinal adipose tissue. During 16 weeks of HFD feeding, the inguinal fat depot expanded continuously, whereas the expansion of the epididymal fat depot was reduced in both wild-type (WT) and Lcn2−/− mice. Interestingly, the depot-specific effect of HFD on fat mass was exacerbated and appeared more pronounced and faster in Lcn2−/− mice than in WT mice. In Lcn2−/− mice, adipocyte hypertrophy in both inguinal and epididymal adipose tissue was more profoundly induced by age and HFD when compared with WT mice. The expression of peroxisome proliferator-activated receptor-γ protein was significantly down-regulated, whereas the gene expression of extracellular matrix proteins was up-regulated selectively in epididymal adipocytes of Lcn2−/− mice. Consistent with these observations, collagen deposition was selectively higher in the epididymal, but not in the inguinal adipose depot of Lcn2−/− mice. Administration of the peroxisome proliferator-activated receptor-γ agonist rosiglitazone (Rosi) restored adipogenic gene expression. However, Lcn2 deficiency did not alter the responsiveness of adipose tissue to Rosi effects on the extracellular matrix expression. Rosi treatment led to the further enlargement of adipocytes with improved metabolic activity in Lcn2−/− mice, which may be associated with a more pronounced effect of Rosi treatment in reducing TGF-β in Lcn2−/− adipose tissue. Consistent with these in vivo observations, Lcn2 deficiency reduces the adipocyte differentiation capacity of stromal-vascular cells isolated from HFD-fed mice in these cells. Herein Rosi treatment was again able to stimulate adipocyte differentiation to a similar extent in WT and Lcn2−/− inguinal and epididymal stromal-vascular cells. Thus, combined, our data indicate that Lcn2 has a depot-specific role in HFD-induced adipose tissue remodeling.
Adipose tissue plays a crucial role as a lipid-storing and endocrine organ in the regulation of energy metabolism, body weight homeostasis, inflammation, and insulin resistance (1, 2). Loss of function within adipose tissue characterized by abnormal production of adipokines/cytokines as a result of defective adipogenesis and/or lipid metabolism has been linked to obesity and associated inflammation, insulin resistance, and metabolic disorders. Multiple types of cells constitute adipose tissue, including lipid-filled adipocytes, preadipocytes, fibroblasts, endothelial cells, immune cells (macrophages and T cells), and other unknown cell types. Adipose tissue composition is compromised in response to metabolic stress, excessive caloric intake, and aging. Specifically, adipose tissue undergoes infiltration of proinflammatory macrophages and T cells, adipocyte hyperplasia and hypertrophy, and extracellular matrix (ECM) remodeling during obesity, leading to adipose tissue dysfunction and insulin resistance (3, 4).
Adipose tissue is located in several different anatomical depots, and different fat depots display differential remodeling in pathological states (5–7). For instance, sc adipose tissue has a higher hyperplastic capacity (7) but is less inflamed in response to a high-fat diet (HFD) than visceral adipose tissue (5). As such, adipose tissue expansion and remodeling in distinct depots could lead to different metabolic consequences. Numerous studies indicate that an increased risk of insulin resistance and metabolic syndrome is closely associated with increased visceral but not sc adipose tissue mass. In contrast, sc fat expansion is considered to be beneficial for increasing lipid buffering capacity and prevents the development of insulin resistance (8, 9). However, the molecular mechanisms underlying depot differences in adipose tissue development and remodeling, particularly in the disease state, remains elusive. It is also unclear whether development of abnormal fat mass and adipose tissue remodeling are causally related to the development of insulin resistance or whether they are the result of lipid derangement.
We have recently identified lipocalin 2 (Lcn2), also known as neutrophil gelatinase-associated lipocalin (NGAL), as a novel adipose-derived cytokine belonging to the lipocalin subfamily of small secreted proteins that bind hydrophobic molecules including retinoids, fatty acids, and various steroids (10–13). A number of studies have shown that the Lcn2 gene promoter region contains CCAAT-enhancer-binding protein and nuclear factor-κB binding sites (14) and a glucocorticoid response element (15), suggesting that Lcn2 may have a function in adipose tissue remodeling. In our previous studies, we demonstrated that Lcn2 is a critical regulator of energy metabolism, glucose and lipid homeostasis, and insulin resistance in Lcn2-deficient mice (16–18). Lcn2-deficient mice were cold-sensitive and developed significantly increased body fat mass as well as exacerbated dyslipidemia, fatty liver, and insulin resistance upon HFD feeding compared with wild-type (WT) mice (16). We also demonstrated that thiazolidinedione (TZD) treatment, which acts as a peroxisome proliferator-activated receptor-γ (PPARγ) agonist, was able to markedly improve HFD-induced insulin resistance and dyslipidemia in Lcn2−/− mice (17). However, TZD treatment failed to induce sc white fat mass expansion in Lcn2−/− mice (17). In this study, we investigated in more detail the potential role and mechanism for Lcn2 activity in the regulation of adipogenesis and adipose tissue remodeling in Lcn2-deficient mice.
We observe that Lcn2 is expressed in white adipose tissue (WAT) in a depot-selective and age-dependent manner. HFD-induced fat mass expansion occurs selectively in the sc depot of Lcn2−/− mice. Furthermore, Lcn2 deficiency reduces the adipocyte differentiation capacity of stromal-vascular (SV) cells isolated from HFD-fed mice and increases ECM gene expression and collagen deposition selectively in epididymal adipose tissue. In the absence of Lcn2, adipocyte hypertrophy induced by age and HFD was markedly exacerbated. Lcn2 deficiency interferes with the TZD effect on adipose ECM remodeling but does not interfere with adipogenesis. Meanwhile, further activation of the adipogenic transcription process by providing TZD treatment induces further enlargement of these adipocytes, presumably due to an improved metabolic function of these cells. Based on our data, we conclude that Lcn2 has a critical role in regulating adipogenesis and adipose ECM remodeling in a depot-specific manner.
Materials and Methods
Animals
The mice used in this study were C57BL/6 WT and Lcn2-null (Lcn2−/−) mice as described previously (16). Mice were housed in a specific pathogen-free facility at the University of Minnesota. Animal handling was performed in adherence to National Institutes of Health guidelines, and experimental procedures were approved by the University of Minnesota animal care and use committee. Mice were allocated into groups (3–4 mice per cage) and fed an HFD (fat calories, 60%) obtained from Bio-Serv Inc (F3282) or a regular chow diet (RCD), with free access to water for all studies.
During the time-course study, age-matched male WT and Lcn2−/− mice (6–7 mice per group) at 4 weeks of age were fed either an HFD or RCD for 4 weeks, 10 to 12 weeks, 16 weeks, and 21 weeks. At the end of experiments, mice were killed for blood and tissue collection after 18 hours fasting.
In the study of TZD treatment, age-matched male WT and Lcn2−/− mice at 3 to 4 weeks of age were fed an HFD and subjected to oral gavage of rosiglitazone (Rosi) (10 mg/kg body weight per day) for 25 days after the development of insulin resistance and obesity in response to HFD preloading for 14 weeks. At 21 weeks of age, mice were killed and blood and tissues were collected after 18 hours fasting.
Isolation of primary adipocytes and SV cells
Primary adipocytes and SV cells were isolated from WT and Lcn2−/− mice as described previously (16, 19). After mincing, epididymal and inguinal fat pads were digested with collagenase (2 mg/mL solution) in digestion vials containing Krebs-Ringer bicarbonate HEPES buffer (pH 7.4), 200 nM adenosine, and 3.5% BSA. After a 2-hour digestion, adipocytes and SV cells were separated by centrifugation at 1200 rpm for 10 minutes and washed twice with Krebs-Ringer bicarbonate HEPES buffer. After the final wash, adipocytes and SV cells were collected for RNA extraction.
Adipose cell size analysis
Adipose tissue was obtained from epididymal and inguinal fat pad of WT and LCN2−/− mice fed on RCD or HFD with or without TZD treatment. Tissue samples (20–30 mg) were immediately fixed in 12 mL of the 2% osmium tetroxide solution in collidine buffer in a Wheaton vial (SPI-Chem no. 986704) and incubated in a water bath at 37°C for 48 hours as described previously (20, 21). Collidine buffer was prepared from a 4× stock collidine buffer of 0.2M 2,4,6-trimethyl-pyridine (C-0505; Sigma Chemical Co) dissolved in distilled water. Subsequently, the contents of the vial were washed out using a 25-μm filter to catch the fixed cells. The filter containing the cells was washed extensively under running tap water at least until the run-through had become clear. After a final rinse using 0.9% saline, the material was transferred into a 250-μm filter using a squirt bottle filled with 0.9% saline. A gentle rub on the 250-μm mesh was applied to crush any large chunks while rinsing with 0.9% saline. The procedure was repeated to collect the cells. The end volume should not exceed more than 1 conical tube (50 mL). Samples were analyzed with a Multisizer III Coulter counter (Beckman Coulter Inc) set at 256 linear bins, 6000 particle count, using a 400-μm aperture under continuous stirring. The range of cell sizes that can effectively be measured using this aperture is 20 to 240 μm. Each sample is measured at least in duplicate, thus generating a minimum of 4 measurements per adipose tissue sample. After collection of pulse sizes, the data are expressed in particle diameters and displayed as histograms of counts against diameter using linear bins and a linear scale for the x-axis. Figures were generated using GraphPad Prism version 5.01 for Windows (GraphPad Software Inc).
Statistical analysis
Results are expressed as mean ± SEM. Group means for genotypes and TZD treatment were analyzed by two-way ANOVA using genotype and treatment as factor. Genotype by treatment interaction was also inspected. If the interaction term was significant, mean differences for treatment and genotype were analyzed separately using Student's t test. Similar statistical approaches were used for analyzing body weight and fat mass at different time points.
Results
Lcn2 deficiency exacerbates the depot-different effect of HFD on body fat mass
Depot-different response to HFD in fat mass changes has been reported previously (5, 22). In these studies, sc (inguinal) fat mass of C57 mice was found to be consistently increased under HFD-induced weight gain. However, epididymal fat mass was increased after short-term (7 weeks) HFD, but reduced during the additional 12 weeks of HFD feeding (22). The molecular mechanism behind this interesting phenomenon remains elusive. We have previously reported that Lcn2 expression in WAT is gender-, depot-, and age-dependent (17); Lcn2 protein is highly abundant in inguinal WAT (Ing-WAT) (Figure 1A). This depot-specific abundance of Lcn2 protein suggests that Lcn2 may have depot-specific local effects on adipose tissue remodeling and metabolism. To address this hypothesis, we determined whether Lcn2 protein expression is different in response to HFD feeding in different fat depots. As shown in Figure 1, B and C, 13 weeks of HFD feeding significantly increases Lcn2 protein expression in both epididymal WAT (Epi-WAT) and Ing-WAT of normal mice. Additionally, serum Lcn2 levels are significantly increased by HFD as well (Figure 1D).
Figure 1.

A, Lcn2 protein is more abundant in inguinal adipose tissue. B–D, Lcn2 protein expression in Epi-WAT (B), Ing-WAT (C), and serum Lcn2 (D) in normal mice under RCD and HFD conditions. E–J, Time course of RCD or HFD effect on body weight gain (E and F) and inguinal (G and I), and epididymal (H and J) fat mass increase in WT and Lcn2−/− mice. Results of E–J represent mean ± SE of 7 to 10 animals. *, P < .05; **, P < .01; WT vs Lcn2−/−.
We then performed a time-course analysis of body weight and fat mass gain in Lcn2−/− mice under RCD and HFD conditions. The results show that the difference in body weight and fat mass gain of Epi-WAT and Ing-WAT by HFD were statistically significant between WT and Lcn2−/− mice at different time points (Supplemental Table 1, published on The Endocrine Society's Journals Online website at http://endo.endojournals.org). However, in RCD-fed mice, the differences in fat mass gain between the 2 genotypes were not statistically significant (Supplemental Table 1 and Figure 1, G and H). Nor is the difference in body weight by 30 weeks of age on an RCD statistically significant between the 2 genotypes, as is shown in Figure 1E. However, as mice age or are fed an HFD, body weight increased, and this age- or HFD-related increase was enhanced significantly in Lcn2−/− mice as compared with WT mice (Figure 1F). Moreover, HFD consistently increased the expansion of the inguinal fat depot as demonstrated by normalizing fat mass to body weight (Figure 1I), but it did not induce the expansion of epididymal fat depots during 16 weeks of HFD feeding in both WT and Lcn2−/− mice (Figure 1J). Most intriguingly, in the absence of Lcn2, the HFD-induced expansion of the inguinal fat depot was dramatically exacerbated. We observe that HFD feeding induces both more rapid and more extensive expansion of the inguinal fat depots in Lcn2−/− mice when compared with WT mice. Significant expansion of inguinal depots appeared after 15 weeks on HFD in WT mice (at 19 or 20 weeks of age), whereas only 9 weeks of HFD feeding was required to achieve a similar effect in Lcn2−/− mice (Figure 1I). Similar to the observations by Jo et al (22), the expansion of epididymal fat mass was significantly reduced after 15 weeks of HFD in WT mice; in Lcn2−/− mice, this HFD-induced reduction of fat mass expansion occurred earlier after 9 weeks of HFD feeding and was more profound (Figure 1J).
Lcn2 deficiency alters adipose tissue remodeling induced by HFD and age
Adipose tissue is enlarged through mechanisms involving both hyperplasia and hypertrophy to store excess energy intake during periods of caloric excess. Studies on adipose tissue growth during HFD-induced obesity have shown that both adipocyte hyperplasia and hypertrophy occur during the initial stages of HFD feeding (22, 23). However, as HFD feeding continues, the adipocyte size continues to increase, whereas the total adipocyte numbers are actually decreased in all the fat depots (22), suggesting that long-term HFD feeding impairs adipogenesis. Thus, in other words, late-stage fat mass expansion appears a process driven by hypertrophy over hyperplasia. To better understand whether Lcn2 deficiency alters adipose tissue remodeling, we investigated adipose tissue cellularity of Lcn2−/− mice. Osmium tetroxide fixation was used to analyze fat cell size distribution. Figure 2, A–H, shows the distribution of adipocytes with different mean diameters in epididymal and inguinal adipose tissue from the curve-fitting analysis. As shown in Figure 2, A–D, the nadir is slightly shifted to a larger peak diameter in adipocytes from Lcn2−/− mice as compared with WT mice at the age of 15 weeks under RCD condition. This nadir is shifted significantly as mice aged 30 weeks. These data indicate that Lcn2−/− adipose tissue consists of a population of larger adipocytes in both fat depots compared with WT adipose tissue. Age amplifies this difference significantly. HFD feeding for 4 weeks increases the peak diameter of the large fat cell population in both epididymal and inguinal adipose tissues of both WT and Lcn2−/− mice (Figure 2, E and F). Interestingly however, the differential response between WT and Lcn2−/− mice to HFD-induced increases in the large fat cell population appears sooner in the sc fat depot than in the epididymal fat depot (Figure 2, E and F). After 4 weeks of HFD, the difference in fat cell size distribution profiles between WT and Lcn2−/− mice was indistinguishable in epididymal adipose tissue (Figure 2E) but is already clearly evident in inguinal adipose tissue (Figure 2F). After 9 weeks of HFD feeding, the fat cell size distribution profile is clearly distinguished in both epididymal (Figure 2G) and inguinal adipose tissue between the 2 genotypes (Figure 2H). These results suggest that the inguinal fat depot is more responsive to HFD-induced adipocyte hypertrophy than the epididymal fat depot in the absence of Lcn2, which may be associated with the above observation that the expansion of the epididymal fat depot is more restricted in Lcn2−/− mice.
Figure 2.

A–D, Age effect on fat cell size distribution of epididymal (A and C) and inguinal (B and D) adipose tissue of WT and Lcn2−/− mice fed RCD. E–H, HFD effect on fat cell size distribution of epididymal (E and G) and inguinal (F and H) fat depots of WT and Lcn2−/− mice. Results represent mean of 3 animals.
LCN2 deficiency reduces adipogenic gene expression but increases ECM gene expression and collagen deposition selectively in epididymal adipose tissue
It has been proposed that reduced adipogenesis, adipose tissue fibrosis, and adipocyte hypertrophy are tied together, linking to obesity-related insulin resistance. The ECM is understood to primarily play a role as a static mechanic support for tissues. Recently, attention has been focused on their functional relevance in adipose tissue physiology, inflammation, and insulin resistance. In a recent study, the reduction of collagen VI expression was shown to have a significant impact on adipose tissue fibrosis and adipocyte hypertrophy affecting systemic insulin sensitivity and glucose homeostasis (24). Therefore, we set out to determine whether Lcn2 deficiency alters the adipogenic capacity and ECM remodeling of adipose tissue. As shown in Figure 3, A and B, the mRNA expression levels of PPARγ, glucose transporter 4 (Glut4, also known as Slc2a4), and lipoprotein lipase were significantly lower in primary inguinal adipocytes (Figure 3A) and trended to a decrease in epididymal adipocytes (Figure 3B) isolated from HFD-fed Lcn2−/− mice as compared with those from WT mice. Moreover, to obtain a better indication of the changes in adipogenesis in Lcn2−/− mice, we examined PPARγ protein expression in adipose tissue of HFD-fed mice. Interestingly, PPARγ protein expression was markedly decreased in Epi-WAT (Figure 3D) but remained unchanged in Ing-WAT of Lcn2−/− mice (Figure 3C).
Figure 3.

A and B, The mRNA expression of adipogenic genes in primary inguinal (A) and epididymal (B) adipocytes isolated from WT and Lcn2−/− mice fed an HFD for 16 weeks. C and D, PPARγ protein expression in Epi-WAT (C) and Ing-WAT (D) of HFD-fed WT and Lcn2−/− mice. E–H, The mRNA expression of ECM molecules in epididymal adipocytes (E) and SV cells (F) and inguinal adipocytes (G) and SV cells (H) isolated from WT and Lcn2−/− mice fed an HFD. I, Picrosirius staining of epididymal and inguinal adipose tissue from HFD-fed WT and Lcn2−/− mice; collagen fibers are stained with red. Results (A and B and E–H) represent mean ± SE of 4 to 6 animals. *, P < .05; **, P < .01; WT vs Lcn2−/−; #, P < .05; ##, P < .01, H2O vs Rosi.
Subsequently, we examined the expression of ECM in primary adipocytes and SV cells isolated from adipose tissue of WT and Lcn2−/− mice. As indicated in Supplemental Tables 2 and 3, there was an overall statistically significant difference in ECM gene expression between two genotypes in epididymal adipocytes and SV cells but not in inguinal adipocytes and SV cells. Specifically, as shown in Figure 3, E and F, the mRNA expression levels of collagen I and III, fibronectin 1, and TGF-β1 were significantly higher in epididymal adipocytes isolated from Lcn2−/− mice fed HFD as compared with WT mice (Figure 3E), whereas, in the SV fraction, collagen III and fibronectin 1 were down-regulated in Lcn2−/− mice compared with WT mice (Figure 3F). Interestingly, such alterations in ECM expression were not observed in the inguinal depot, with the notable exception of a decrease in collagen IV expression in Lcn2−/− inguinal adipocytes (Figure 3G) and down-regulation of TGF-β1 in SV cells (Figure 3H). Consistent with the increased ECM gene expression, collagen staining shows that collagen deposition is significantly higher in epididymal, but not inguinal, adipose tissue of Lcn2−/− mice as compared with WT mice (Figure 3I). These data suggest that the influence of Lcn2 deficiency on HFD regulation of adipogenesis, ECM gene expression and adipose tissue fibrosis is cell type- and epididymal depot-specific.
Lcn2 deficiency alters TZD effect on ECM gene expression but not its effects on adipogenesis
In our previous study, we have shown that TZD, a PPARγ ligand, is able to reverse HFD-induced insulin resistance and dyslipidemia in Lcn2−/− mice as effectively as it does in WT mice (17), but fails to increase sc fat mass (17). Herein, we assessed the effect of TZD on adipogenesis and ECM remodeling. As shown in Figure 3, A and B, Rosi administration significantly increased the expression of PPARγ, GLUT4, and LPL in primary epididymal and inguinal adipocytes from both WT and Lcn2−/− mice to a similar extent. As indicated in Supplemental Tables 2 and 3, the inguinal fat depot appears to be generally more responsive to Rosi treatment in ECM gene expression as compared with the epididymal depot. In WT mice, Rosi treatment led to the down-regulation of mRNA expression of all the ECM genes examined in both inguinal SV cells (Figure 4A) and adipocytes (Figure 4B). In Lcn2−/− mice, only 2 ECM genes, collagen type III, α 1 (Col3a1) and fibronectin 1, were significantly down-regulated. Although the expression of collagen type IV, α 1 (Col4a1), collagen type I, α 1 (Col1a1), and matrix metallopeptidase 9 were also reduced by Rosi, these did not reach a statistically significant difference in inguinal adipocytes (Figure 4B) and SV cells (Figure 4A). In epididymal SV cells, all of the examined ECM genes were significantly down-regulated by Rosi in WT mice (Figure 4C) but had only a trend toward a decrease in Lcn2−/− mice (with an exception for FN1) (Figure 4C). In epididymal adipocytes, Rosi caused the up-regulation of TGF-β, COL3A1, and FN1 expression in WT mice (Figure 4D), whereas none of the ECM genes examined in Lcn2−/− mice had a significant change in their expression by Rosi treatment (Figure 4D).
Figure 4.

The mRNA expression of ECM molecules in primary epididymal and inguinal adipocytes and SV cells isolated from control and TZD-treated WT and Lcn2−/− mice. A–D, Primary inguinal SV cells (A), primary inguinal adipocytes (B), primary epididymal SV cells (C), and primary epididymal adipocytes (D) from HFD WT and Lcn2−/− mice with or without TZD treatment. E and F, TGF-β protein expression in Epi-WAT (E) and Ing-WAT (F) of HFD WT and Lcn2−/− with or without TZD treatment. Results (A–D) represent mean ± SE of 4 to 6 animals. *, P < .05; **, P < .01; WT vs Lcn2−/− (KO); #, P < .05; ##, P < .01, H2O vs Rosi.
In agreement with the gene expression data, Western blotting on adipose tissue showed that TGF-β protein expression levels were higher (especially TGF-β precursor) in Epi-WAT (Figure 4E) but markedly lower (mature TGF-β) in Ing-WAT of HFD-fed Lcn2−/− mice (Figure 4F) when compared with HFD-fed WT mice. Rosi treatment significantly reduced TGF-β protein expression in Epi-WAT (Figure 4E) but did not further reduce TGF-β (precursor and mature) in Ing-WAT of HFD-fed Lcn2−/− mice (Figure 4F). Interestingly, after Rosi treatment, TGF-β protein expression levels were significantly lower in both Epi-WAT and Ing-WAT of Lcn2−/− mice compared with WT mice. Together with the PPARγ protein expression levels (Figure 3, C and D) and immunohistochemical staining of collagen (Figure 3I), our data suggest that Lcn2 deficiency increases Epi-WAT fibrosis but does not significantly affect Ing-WAT ECM remodeling. In addition, in ECM expression, Lcn2 deficiency alters the responsiveness of adipose tissue to Rosi treatment.
SV cells from adipose tissue of HFD-fed Lcn2−/− mice show reduced adipogenesis capacity in vitro
To determine whether Lcn2 locally and directly affects adipose tissue remodeling and adipocyte function, we assessed the adipogenic capacity in SV cell cultures. SV cells were isolated from inguinal and epididymal adipose tissue of WT and Lcn2−/− mice fed RCD and HFD, respectively, and then cultured and induced to differentiate into adipocytes. Morphological analysis of oil red O staining showed that about 90% to 95% of SV cells isolated from inguinal adipose tissue of WT mice were differentiated into lipid-filled adipocytes. The rate of Ing-SV cell differentiation into adipocytes is not significantly changed in Lcn2−/− mice fed an RCD (data not shown) but was noticeably decreased in Lcn2−/− mice when fed an HFD (Figure 5A) as compared with WT mice. Unlike the results of fat cell sizing analysis of the adipose tissue in vivo, the difference in the degree of the hypertrophy of differentiated adipocytes in SV cell cultures was not evident between WT and Lcn2−/− genotypes in vitro (Figure 5A). This suggests that Lcn2−/− SV cells are functionally capable of differentiating into adipocytes with the normal size; therefore, the enlarged adipocytes observed in Lcn2−/− adipose tissue in vivo are likely secondary to the dysregulation of lipid homeostasis or some other extrinsic factor, rather than the nature of Lcn2−/− adipocytes per se. These observations support the concept that adipocyte hypertrophy is a response to lipid overload and overflow in the pathological state in conjunction with other extrinsic factors (such as, for instance, the ECM).
Figure 5.
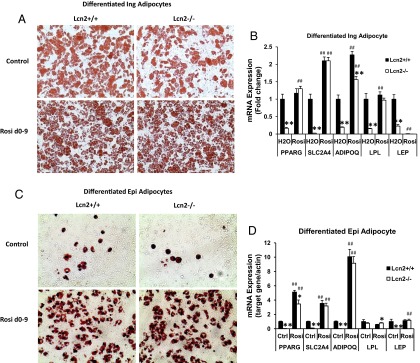
A, Oil-red O staining of inguinal SV cell cultures with or without Rosi treatment during the 9-day differentiation process. B, The mRNA expression of adipogenic genes in differentiated inguinal adipocytes with or without TZD treatment. C, The morphology of epididymal SV cell cultures with or without TZD treatment. D, The mRNA expression of adipogenic genes in differentiated epididymal adipocytes with or without TZD treatment. The morphological results of adipocytes (A and C) represents 4 independent experiments. Results of gene expression represent mean ± SE of 2 independent SV cell cultures from 4 animals per experiment. *, P < .05; **, P < .01, WT vs Lcn2−/−; #, P < .05; ##, P < .01, H2O vs Rosi. Abbreviation: Ctrl, control.
We proceeded to examine the expression of genes involved in adipogenesis and lipid metabolism in the SV cell cultures. As expected, the mRNA expression levels of adipogenic genes PPARγ, GLUT4, and LPL are markedly reduced in Lcn2−/− as compared with WT inguinal adipocytes in inguinal SV cell cultures from HFD-induced obese mice (Figure 5B), and so was the expression of both adiponectin and leptin (Figure 5B). In addition, we evaluated the effect of TZD on adipogenesis in inguinal SV cell culture. In line with the in vivo results, treatment with Rosi during the 9-day differentiation period was able to restore the adipogenic capacity of Lcn2−/− inguinal SV cells almost completely, as determined by both adipocyte morphology (Figure 5A) and adipogenic gene expression (Figure 5B).
However, SV cells from epididymal adipose tissue had a very poor capacity for differentiation in vitro. Consistent with the report by Macotela et al (25), our results showed that about 10% to 15% of epididymal SV cells from both WT and Lcn2−/− mice were differentiated into adipocytes using the standard adipocyte differentiation cocktail; it was difficult to distinguish the morphological difference in adipocyte differentiation between two genotypes of cells at this low rate of differentiation (Figure 5C). The presence of Rosi during the differentiation process was able to significantly increase the differentiation rate of WT and Lcn2−/− epididymal SV cells isolated from HFD-fed mice up to 50% to 60% (Figure 5C). Similar to what we perceived in inguinal SV cell cultures, the effect of Rosi on epididymal adipocyte differentiation was not significantly different between WT and Lcn2−/− epididymal SV cell cultures (Figure 5C). The analysis of adipogenic gene expression demonstrated that the expression of PPARγ, GLUT4, adiponectin, and leptin was significantly decreased in Lcn2−/− epididymal SV cell cultures without Rosi treatment (Figure 5D); this decrease was significantly restored by Rosi treatment (Figure 5D). All these results together suggest that Lcn2 deficiency does not impair the TZD effect on adipogenesis in Ing-WAT and Epi-WAT.
TZD further enhances hypertrophy but improves metabolic activity of adipocytes in Lcn2−/− mice
Previous investigations in human studies have shown that TZD therapy leads to increased size of fat cells in sc adipose tissue (26, 27). In this study, we showed that TZD administration could also amplify adipocyte hypertrophy in both Epi-WAT (Figure 6A) and Ing-WAT (Figure 6C) in WT mice. In the absence of Lcn2, this effect of TZD was much more pronounced (Figure 6, B and D), without leading to an overall fat mass increase (data not shown). Unlike HFD, TZD-induced adipocyte hypertrophy was not accompanied by a decreased cell number as evidenced by a slight increase in the peak height of the fat cell size distribution curve in both epididymal and inguinal fat depots (Figure 6, B and D), suggesting that TZD is able to increase the recruitment of new adipocytes in Lcn2−/− mice and that the enlarged adipocytes may be developed from these newly recruited cells.
Figure 6.

A–D, TZD effect on fat cell size distribution of epididymal (A and B) and inguinal (C and D) adipose tissue of WT and Lcn2−/− mice. Results represent mean of 3 animals. D–H, Comparison of mRNA expression of adipogenic genes and genes involved in lipid metabolism in epididymal and inguinal adipocytes between HFD-fed WT and Lcn2−/− mice (E and F) and between HFD-fed Lcn2−/− mice with and without TZD treatment (G and H). Results represent mean ± SE of 4 to 6 animals. *, P < .05; **, P < .01, WT vs Lcn2−/−; #, P < .05; ##, P < .01, H2O vs Rosi.
We then further characterized the metabolic activity of TZD-hypertrophied adipocytes by comparing the metabolic gene expression of these cells with that of HFD-hypertrophied adipocytes in Lcn2−/− mice. The expression profiles of genes involved in multiple metabolic pathways including adipogenesis, lipid metabolism, and adipose tissue remodeling was determined and compared between WT and Lcn2−/− primary adipocytes isolated from mice with or without Rosi treatment. Intriguingly, we found that the expression patterns of genes in the above-mentioned pathways between WT and Lcn2−/− adipocytes are completely mirrored between Lcn2−/− control and Rosi-treated adipocytes. As illustrated in Figure 6, E and F, adipocytes from HFD-fed Lcn2−/− mice express significantly lower levels of mRNA of adipogenic genes and genes involved in lipid homeostasis than those from HFD-fed WT mice. When compared with control Lcn2−/− adipocytes, Lcn2−/− adipocytes from mice with Rosi treatment express significantly higher levels of PPARγ, GLUT4, and SREBP-1c (Figure 6, G and H). Similar phenomena are observed in the expression of genes involved in lipid metabolism and homeostasis such as LPL, HSL, DGAT1, and PEPCK1 (Figure 6, E–H). Moreover, as shown in Supplemental Figure 1, Rosi treatment also significantly increased serum adiponectin levels in WT mice and in Lcn2−/− mice, although in the latter, the trend was marginal, not reaching statistical significance (P = .07). Combined, these results suggest that TZD-hypertrophied adipocytes have improved metabolic activity in Lcn2−/− mice.
Discussion
The recently discovered adipokine Lcn2 has a critical role in regulation of whole-body energy metabolism and lipid and glucose homeostasis (16). We further explored the role of Lcn2 in adipogenesis and adipose tissue remodeling to gain a better understanding of the function of Lcn2 in adipocyte biology. In Lcn2−/− mice, the HFD effect on increasing inguinal (sc) fat mass and decreasing epididymal fat mass is significantly exacerbated; both depots were more responsive when compared with WT mice. Moreover, the adipogenic capacity of SV cells from the inguinal depot of HFD-fed Lcn2−/− mice is reduced compared with that from WT mice. The down-regulation of PPARγ protein expression and up-regulation of ECM genes and collagen deposition are selectively observed in epididymal adipose tissue and not significantly changed in inguinal adipose tissue from Lcn2−/− mice when compared with WT mice. This suggests a more profound alteration in adipogenesis and ECM remodeling/fibrosis in epididymal adipose tissue. In the absence of Lcn2, the PPARγ agonist TZD is able to reverse impaired adipogenesis and lipid metabolism in adipose tissue in vivo and in SV cell cultures in vitro. However, Lcn2 deficiency alters the responsiveness of adipose tissue to the TZD effect in ECM expression. Concomitantly, TZD treatment led to a further enlargement of adipocyte, and an improvement of their metabolic activity in Lcn2−/− mice.
Adipose tissue has several location-dependent stereotypical depots, and white adipocytes from different depots display differential metabolic and physiological characteristics (28–31). Thus, identifying depot-specific factors and understanding their roles in controlling the development and function of specific fat depots in metabolism in pathophysiological states is of great importance and interest. However, not many such adipose depot-specific adipokines have been identified and characterized. We demonstrate here that Lcn2 is preferentially expressed in inguinal adipose tissue in mice. Lcn2 protein expression in both Epi-WAT and Ing-WAT is significantly increased in normal mice in response to 13 weeks of HFD feeding. However, Lcn2 deficiency affects the HFD effect on fat mass expansion most profoundly in the inguinal fat depot. For instance, HFD selectively induces sc depot but reduces epididymal depot expansion; the magnitude of this effect of HFD appeared sooner and was larger in Lcn2-deficient mice as compared with WT mice in our time-course studies of HFD feeding. HFD feeding also induced a greater degree of adipocyte hypertrophy in both epididymal and inguinal adipose tissue in Lcn2−/− mice as compared with WT mice. However, Lcn2 deficiency affects neither fat mass distribution nor adipogenesis or ECM expression under RCD (control) conditions. These data suggest that Lcn2 functions as an important depot-selective regulator of adipose tissue remodeling under metabolic stress conditions such as HFD feeding, rather than a regulator of adipose tissue development per se.
The adipose tissue expandability has been connected to ectopic lipid accumulation, lipotoxicity, and insulin resistance (32, 33). Adipose tissue is assumed to have a limited lipid storage capacity. Once the maximum capacity of storing lipids is reached, excessive lipids flux to ectopic sites leads to lipotoxicity and the development of insulin resistance (32, 33). According to this hypothesis, ectopic lipid deposition should therefore occur later than the maximum expansion of adipose tissue. The results from our time-course studies of HFD-induced adipose tissue mass expansion demonstrate that epididymal and inguinal adipose tissue have a different response to HFD in depot expansion. Epididymal adipose tissue seems to be more rigid than inguinal adipose tissue and reaches its maximum expansion earlier (after 4 weeks of HFD feeding); the epididymal fat mass was not increased further but was actually reduced with continued HFD feeding. However, the inguinal fat mass continued to increase in both WT and Lcn2−/− mice during the entire period of the HFD feeding study. Interestingly, Lcn2 deficiency significantly alters the degree and timing of HFD effect on epididymal fat mass decrease and inguinal fat mass increase.
The expansion of the sc fat depot has been considered to be beneficial for preventing/reducing ectopic fat deposition and improving metabolic deterioration (8). During the development of obesity, the expansion of adipose tissue mass can be accomplished through adipocyte hyperplasia (the recruitment of new adipocytes) and/or hypertrophy (the enlargement of existing adipocytes). Adipocyte hypertrophy has been known to reflect the situation of lipid overflow and to be closely associated with insulin resistance (34, 35). There are two hypotheses for the development of adipocyte hypertrophy and ectopic lipid deposition in obesity-induced insulin resistance: an inflammation-related adipogenic defect, which is attributed more to the reduced recruitment of new adipocytes, and adipose tissue fibrosis, which is related to adipocyte hypertrophy. In addition to increased inflammation and cytokine production, up-regulation of ECM molecules has been suggested to provide a direct link between adipose tissue fibrosis and adipocyte hypertrophy in obesity. Others have also reported that an array of ECM proteins was up-regulated in obese adipose tissue (36–39) and that PPARγ agonists down-regulate the expression of a majority of ECM components (39), thus corroborating our observations. Thus, adipocyte hypertrophy might actually be a compensatory mechanism in an attempt to prevent lipids overflow to ectopic storage sites.
In obesity, adipose tissue fibrosis is increased, restricting the further expansion of adipocytes, ultimately leading to lipid overflow and ectopic lipid accumulation. In this study, we found that the mRNA expression levels of collagens, fibronectin, and TGF-β was up-regulated in Lcn2−/− epididymal adipocytes as compared with WT adipocytes. Moreover, collagen deposition was increased in the Lcn2−/− epididymal depot, suggesting an increase in epididymal adipose tissue fibrosis in Lcn2−/− mice. In addition, Lcn2−/− epididymal adipose tissue expressed lower levels of PPARγ protein. Thus, it is reasonable to suggest that the combination of increased fibrosis and decreased adipogenesis may largely contribute to a reduction in the maximal level of epididymal adipose tissue expansion observed in Lcn2−/− mice. It is noteworthy that even though Lcn2−/− epididymal adipocytes are larger in size than WT adipocytes, the total number of cells capable of storing lipids may actually be reduced in Epi-WAT of Lcn2−/− mice due to decreased adipogenesis (decreased PPARγ) as compared with WT mice. Interestingly, the up-regulation of ECM genes and down-regulation of PPARγ protein expression were not observed in Ing-WAT of Lcn2−/− mice. In contrast, TGF-β protein expression was markedly reduced in Ing-WAT of Lcn2−/− mice. These results suggest that Lcn2 has a depot-specific role in regulating adipose tissue remodeling. It is likely that the decrease in TGF-β levels we observed may be one of the more important contributors to the inguinal adipocyte hypertrophy and increased expansion of inguinal fat depot induced by HFD in Lcn2−/− mice. The increased expansion of inguinal fat depot meanwhile may be a compensatory mechanism to overcome the decreased lipid storage capacity of Epi-WAT in Lcn2−/− mice.
It is also worth mentioning that sc fat mass increase doesn't always reflect the increased lipid storage capacity of the depot. Adipocyte hypertrophy or fat mass increase in obesity could just be the result of adipocyte dysfunction or reflect a reduced lipid-handling capacity in other fat depots such as visceral depots. The results shown in Figure 6 and Supplemental Figure 1 support that HFD-induced Lcn2−/− adipocytes are metabolically dysfunctional, whereas TZD treatment improves metabolic activity and promotes further hypertrophy of adipocytes. Thus, Lcn2−/− inguinal and epididymal adipocytes have significantly lower expression levels of LPL, DGAT1, and PEPCK1 genes under the HFD conditions; whereas TZD treatment significantly restores the expression pattern of these genes.
PPARγ is well-known as a key transcription factor that controls adipogenesis and antagonizes the activation of the proinflammatory nuclear factor-κB pathway. Recent studies have reported that PPARγ has a role in the regulation of fibrosis in multiple organs and tissues including adipose tissue (40–43). Several PPARγ agonists are able to reduce collagen deposition in kidney (42) and reduce the expression levels of most collagens in adipose tissue (39). In a previous study, we demonstrated that TZD administration was unable to increase fat mass and lipogenesis in Lcn2−/− mice regardless of a marked improvement of insulin resistance and dyslipidemia (17). These data suggest that Lcn2 is required for the activity of PPARγ ligands on adipose tissue remodeling and expansion. In this study, we supplement these earlier findings with our observation that TZD treatment did lead to a more significant enlargement of adipocytes in both epididymal and inguinal fat depots in Lcn2−/− mice compared with WT mice. In other words, the process appears driven more by hypertrophy (albeit without an alteration in the lipogenic rate) and less by hyperplasia. Moreover, TZD treatment was able to restore adipogenesis in both depots in vivo and in vitro in SV cell cultures. Interestingly, we found that the responsiveness of adipose tissue to TZD treatment is changed in Lcn2−/− mice. Lcn2 deficiency slightly blunts TZD-induced reduction in collagens and FN1, while enhancing the TZD effect on reducing TGF-β protein expression in Epi-WAT. More strikingly, after TZD treatment, TGF-β protein expression becomes significantly lower in Epi-WAT and Ing-WAT in Lcn2−/− mice as compared with WT mice. TGF-β is a key factor that controls local concentrations of various ECM proteins by stimulating the production and inhibiting the degradation of these matrix proteins (44). Because of these actions, TGF-β plays an important role in tissue fibrosis, and increased TGF-β may lead to increased tissue fibrosis (44). Thus, the most likely explanation for our results is that lower levels of TGF-β in Epi-WAT and Ing-WAT attribute to TZD-induced adipocyte hypertrophy in Lcn2−/− mice. More importantly, we observed that TZD-hypertrophied adipocytes express higher levels of adipogenic genes and genes involved in lipid metabolism than control HFD-hypertrophied adipocytes in Lcn2−/− mice, suggesting that TZD-enlarged Lcn2−/− adipocytes are indeed more metabolically active. This conclusion is further supported by increased serum adiponectin levels in Lcn2−/− mice with TZD treatment.
In summary, in this study, we demonstrate that Lcn2 is preferentially expressed in inguinal fat depot in mice. Lcn2 deficiency significantly alters the effect of HFD on depot-specific body fat mass expansion and distribution. Our time-course analysis of HFD feeding shows that HFD induces an increase in inguinal fat mass but a decrease in epididymal fat mass. It does so more rapidly and to a greater degree in Lcn2−/− mice. Additional studies show that Lcn2 deficiency reduces the adipogenic capacity and increases ECM remodeling and fibrosis selectively in epididymal adipose tissue. Adipocyte hypertrophy was more profound in both inguinal and epididymal tissues in response to HFD feeding. The PPARγ agonist TZD is able to stimulate adipogenesis independent of Lcn2. However, Lcn2 deficiency alters the TZD effect on adipose ECM remodeling suggesting these pathways are regulated by different cofactors (one of which is tied into Lcn2-mediated signaling). An increased effect of TZD on the reduction of TGF-β may further contribute to the enlargement of adipocytes in Lcn2−/− mice; these enlarged adipocytes display an improved metabolic gene expression profile. Combined, our findings demonstrate that Lcn2 plays a significant role in HFD-induced adipose tissue remodeling in a fat depot-specific manner.
Acknowledgments
This research and the manuscript was supported by Grant R01DK080743 (to X.C.) from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and Minnesota Obesity Center (Grant 2P30DK050456 from the NIDDK).
Disclosure Summary: The authors have nothing to declare. No potential conflicts of interest relevant to this article were reported.
Footnotes
- ECM
- extracellular matrix
- Epi-WAT
- epididymal WAT
- HFD
- high-fat diet
- Ing-WAT
- inguinal WAT
- Lcn2
- lipocalin 2
- PPARγ
- peroxisome proliferator-activated receptor-γ
- RCD
- regular chow diet
- Rosi
- rosiglitazone
- SV
- stromal-vascular
- TZD
- thiazolidinedione
- WAT
- white adipose tissue
- WT
- wild-type.
References
- 1. Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87:507–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867 [DOI] [PubMed] [Google Scholar]
- 3. Lee MJ, Wu Y, Fried SK. Adipose tissue remodeling in pathophysiology of obesity. Curr Opin Clin Nutr Metab Care. 2010;13:371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Strissel KJ, Stancheva Z, Miyoshi H, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56:2910–2918 [DOI] [PubMed] [Google Scholar]
- 6. Nishimura S, Manabe I, Nagasaki M, et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest. 2008;118:710–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DiGirolamo M, Fine JB, Tagra K, Rossmanith R. Qualitative regional differences in adipose tissue growth and cellularity in male Wistar rats fed ad libitum. Am J Physiol. 1998;274:R1460–R1467 [DOI] [PubMed] [Google Scholar]
- 8. Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008;7:410–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Medina-Gomez G, Gray SL, Yetukuri L, et al. A PPARγ2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet. 2007;3:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. LaLonde JM, Bernlohr DA, Banaszak LJ. The up-and-down β-barrel proteins. FASEB J. 1994;8:1240–1247 [DOI] [PubMed] [Google Scholar]
- 11. Flower DR. Beyond the superfamily: the lipocalin receptors. Biochim Biophys Acta. 2000;1482:327–336 [DOI] [PubMed] [Google Scholar]
- 12. Zhang J, Wu Y, Zhang Y, Leroith D, Bernlohr DA, Chen X. The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol Endocrinol. 2008;22:1416–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yan QW, Yang Q, Mody N, et al. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes. 2007;56:2533–2540 [DOI] [PubMed] [Google Scholar]
- 14. Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–24148 [DOI] [PubMed] [Google Scholar]
- 15. Garay-Rojas E, Harper M, Hraba-Renevey S, Kress M. An apparent autocrine mechanism amplifies the dexamethasone- and retinoic acid-induced expression of mouse lipocalin-encoding gene 24p3. Gene. 1996;170:173–180 [DOI] [PubMed] [Google Scholar]
- 16. Guo H, Jin D, Zhang Y, et al. Lipocalin-2 deficiency impairs thermogenesis and potentiates diet-induced insulin resistance in mice. Diabetes. 2010;59:1376–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin D, Guo H, Bu SY, et al. Lipocalin 2 is a selective modulator of peroxisome proliferator-activated receptor-γ activation and function in lipid homeostasis and energy expenditure. FASEB J. 2011;25:754–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo H, Zhang Y, Brockman DA, Hahn W, Bernlohr DA, Chen X. Lipocalin 2 deficiency alters estradiol production and estrogen receptor signaling in female mice. Endocrinology. 2012;153:1183–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen X, Al-Hasani H, Olausson T, Wenthzel AM, Smith U, Cushman SW. Activity, phosphorylation state and subcellular distribution of GLUT4-targeted Akt2 in rat adipose cells. J Cell Sci. 2003;116:3511–3518 [DOI] [PubMed] [Google Scholar]
- 20. McLaughlin T, Sherman A, Tsao P, et al. Enhanced proportion of small adipose cells in insulin-resistant vs insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia. 2007;50:1707–1715 [DOI] [PubMed] [Google Scholar]
- 21. Hirsch J, Knittle JL. Cellularity of obese and non-obese human adipose tissue. Fed Proc. 1970;29:1516–1521 [PubMed] [Google Scholar]
- 22. Jo J, Guo J, Liu T, et al. Hypertrophy-driven adipocyte death overwhelms recruitment under prolonged weight gain. Biophys J. 2010;99:3535–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jo J, Gavrilova O, Pack S, et al. Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLoS Comput Biol. 2009;5:e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khan T, Muise ES, Iyengar P, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29:1575–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Macotela Y, Emanuelli B, Mori MA, et al. Intrinsic differences in adipocyte precursor cells from different white fat depots. Diabetes. 2012;61:1691–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McLaughlin TM, Liu T, Yee G, et al. Pioglitazone increases the proportion of small cells in human abdominal subcutaneous adipose tissue. Obesity (Silver Spring). 2010;18:926–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ciaraldi TP, Kong AP, Chu NV, et al. Regulation of glucose transport and insulin signaling by troglitazone or metformin in adipose tissue of type 2 diabetic subjects. Diabetes. 2002;51:30–36 [DOI] [PubMed] [Google Scholar]
- 28. Giorgino F, Laviola L, Eriksson JW. Regional differences of insulin action in adipose tissue: insights from in vivo and in vitro studies. Acta Physiol Scand. 2005;183:13–30 [DOI] [PubMed] [Google Scholar]
- 29. Gesta S, Bluher M, Yamamoto Y, et al. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci U S A. 2006;103:6676–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–896 [DOI] [PubMed] [Google Scholar]
- 31. Macotela Y, Boucher J, Tran TT, Kahn CR. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes. 2009;58:803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann N Y Acad Sci. 2002;967:363–378 [DOI] [PubMed] [Google Scholar]
- 33. Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome: an allostatic perspective. Biochim Biophys Acta. 2010;1801:338–349 [DOI] [PubMed] [Google Scholar]
- 34. Stefan N, Kantartzis K, Machann J, et al. Identification and characterization of metabolically benign obesity in humans. Arch Intern Med. 2008;168:1609–1616 [DOI] [PubMed] [Google Scholar]
- 35. Marini MA, Succurro E, Frontoni S, et al. Metabolically healthy but obese women have an intermediate cardiovascular risk profile between healthy nonobese women and obese insulin-resistant women. Diabetes Care. 2007;30:2145–2147 [DOI] [PubMed] [Google Scholar]
- 36. Alessi MC, Bastelica D, Morange P, et al. Plasminogen activator inhibitor 1, transforming growth factor-beta1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes. 2000;49:1374–1380 [DOI] [PubMed] [Google Scholar]
- 37. Chavey C, Mari B, Monthouel MN, et al. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J Biol Chem. 2003;278:11888–11896 [DOI] [PubMed] [Google Scholar]
- 38. Maquoi E, Munaut C, Colige A, Collen D, Lijnen HR. Modulation of adipose tissue expression of murine matrix metalloproteinases and their tissue inhibitors with obesity. Diabetes. 2002;51:1093–1101 [DOI] [PubMed] [Google Scholar]
- 39. Chen X, Hunt D, Cushman SW, Hess S. Proteomic characterization of thiazolidinedione regulation of obese adipose secretome in Zucker obese rats. Proteomics Clin Appl. 2009;3:1099–1111 [DOI] [PubMed] [Google Scholar]
- 40. Galli A, Crabb DW, Ceni E, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–1940 [DOI] [PubMed] [Google Scholar]
- 41. Shiomi T, Tsutsui H, Hayashidani S, et al. Pioglitazone, a peroxisome proliferator-activated receptor-γ agonist, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2002;106:3126–3132 [DOI] [PubMed] [Google Scholar]
- 42. Kawai T, Masaki T, Doi S, et al. PPAR-γ agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-β. Lab Invest. 2009;89:47–58 [DOI] [PubMed] [Google Scholar]
- 43. Wei J, Ghosh AK, Sargent JL, et al. PPARγ downregulation by TGFβ in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis. PLoS One. 2010;5:e13778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leask A, Abraham DJ. TGF-β signaling and the fibrotic response. FASEB J. 2004;18:816–827 [DOI] [PubMed] [Google Scholar]