

Abstract
Background:
Squamous cell lung cancer (SqCC) is the second most common type of lung cancer in the United States. Previous studies have used gene-expression data to classify SqCC samples into four subtypes, including the primitive, classical, secretory and basal subtypes. These subtypes have different survival outcomes, although it is unknown whether these molecular subtypes predict response to therapy.
Methods:
Here, we analysed RNAseq data of 178 SqCC tumour samples and characterised the features of the different SqCC subtypes to define signature genes and pathway alterations specific to each subtype. Further, we compared the gene-expression features of each molecular subtype to specific time points in models of airway development. We also classified SqCC-derived cell lines and their reported therapeutic vulnerabilities.
Results:
We found that the primitive subtype may come from a later stage of differentiation, whereas the basal subtype may be from an early time. Most SqCC cell lines responded to one of five anticancer drugs (Panobinostat, 17-AAG, Irinotecan, Topotecan and Paclitaxel), whereas the basal-type cell line EBC-1 was sensitive to three other drugs (PF2341066, AZD6244 and PD-0325901).
Conclusion:
Compared with the other three subtypes of cell lines, the secretory-type cell lines were significantly less sensitive to the five most effective drugs, possibly because of their low proliferation activity. We provide a bioinformatics framework to explore drug repurposing for cancer subtypes based on the available genomic profiles of tumour samples, normal cell types, cancer cell lines and data of drug sensitivity in cell lines.
Keywords: squamous cell lung cancer subtypes, gene expression, RNAseq, microarray, signature genes, cells of origin, representative cell line, drug sensitivity, classification
Lung cancer now accounts for 13% of new cancer cases and 29% of all cancer deaths each year in the United States. Lung cancer is the leading cause of cancer-related mortality worldwide, leading to an estimated 1.4 million deaths in 2010. Lung cancer is a heterogeneous disease with multiple histological and molecular subtypes. Lung cancers are usually classified according to the histological types because the histopathological type of lung cancer correlates with tumour behaviour and prognosis (Beadsmoore and Screaton, 2003). The vast majority of lung cancer types are carcinoma malignancies that arise from epithelial cells. The two most common subtypes of lung cancer are adenocarcinoma and squamous cell lung cancer types (SqCC).
Squamous cell lung cancer is the second most common type of lung cancer, usually originating in the large airways in the central part of the lungs. Even for patients with SqCCs, outcomes are variable, suggesting that heterogeneity exists within this subtype. Further classification of SqCC into subtypes may help to understand the disease mechanism better, define the pathways relevant in disease origin and pathogenesis, and help guide treatment.
Gene-expression-profiling studies have been undertaken in an attempt to identify the cell-of-origin for a variety of malignancies, including lung cancer, by comparing expression profiles of cancer cells to those obtained from sorted cell populations or cell lines obtained from non-neoplastic lung tissue. These studies have not yet clearly identified a cell-of-origin for SqCCs but have correlated lung SqCC expression subtypes with specific time points in lung development in the mouse (Mariani et al, 2002; Chen et al, 2012).
In our study, we aim to characterise the features of the different SqCC subtypes, gain deeper understanding about how subtypes are correlated with the developmental stages of airways, and further explore the subtypes within SqCCs and correlation with response to therapeutic agents. We leveraged a large RNAseq data set (Hammerman et al, 2012) of 178 SqCC individuals classified into four previously defined dominant SqCC subtypes. We found the primitive subtype to be related to differentiation stages of normal bronchial epithelial cells and characterised SqCC cell lines by identifying their similarities to the four SqCC subtypes. The cell line response to anticancer therapies in vitro was generalised to SqCC subtypes.
Materials and methods
SqCC subtype data and the analysis
The 178 samples include 43 basal, 65 classical, 27 primitive and 43 secretory samples (Wilkerson et al, 2010). Raw count data of each gene were obtained. There are 22 283 genes. R function calcNormFactors in edgeR package (Robinson et al, 2010) was used on the raw count data to calculate normalisation factors to scale the raw library size in RNAseq. R function voom in limma package (Smyth, 2005), using the above normalisation factor and the design matrix, converted the read counts to log2 counts per million, with associated weights ready for linear modelling.
The log2 scale data were fitted with a model that has the subtype variable of four categories. Using ‘contrast.matrix', one type of comparison is the six pairwise comparisons of the four SqCC subtypes, and another type is ‘1 vs other'. The function eBayes was used. The false discovery rate (FDR) was controlled globally using the Benjamini and Hochberg algorithm. Probes with FDR <0.05 and fold change >2 were judged to be differentially expressed. These packages are from Bioconductor (Gentleman et al, 2004) in R (R Core Team, 2012).
On the basis of the criterion for a DE gene in a comparison, the signature gene set for a subtype was further defined. For one SqCC subtype, when comparing with all other three subtypes, the genes that are significantly upregulated in each of the three comparisons are defined as positive-signature genes of this SqCC subtype. The genes that are significantly downregulated in each of the three comparisons are defined as negative-signature genes of this SqCC subtype.
Time-course data of human bronchial epithelial cells
From GEO website, the normalised data of GSE 5264 for human bronchial epithelial cells were downloaded. Affymetrix Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA, USA) was used with 54 675 probes. We used the GPL570 as the annotation file for the platform. The data set contains 30 samples at 11 time points of cell culture from 0 to 28 days. There are biological duplicates or triplicates at each time point from two or three different donors.
Data of the 28 SqCC cell lines
The microarray data of the 28 SqCC cell line data (Table 2) were downloaded from www.broadinstitute.org/ccle. The array platform was HG-U133Plus2 with 54 675 probe sets. To just obtain SqCC data, we restricted firstly filtered data sets by primary the site in lung and then by ‘Hist.Subtype1' as squamous cell carcinoma. 28 cell lines were obtained. We also downloaded the annotation of samples. Quality control using an affycoretool package (MacDonald, 2008) has been carried out, including the examination of degradation levels of each sample, boxplot of log-expression data, density plot of the expression data and so on. To carry out log transformation of the expression value, we replaced the value 0 by the minimum non-zero value 0.000216. The R function gcrma (Wu et al, 2004) was used for normalisation. The DE analysis was performed using the limma package (Smyth, 2005).
Multidimensional scaling (MDS) plot and heatmap
The functions in limma – for example, plotMDS – have been used to look at the sample relationship as a step of quality control. In plotMDS, the default 500 genes were used for each pair of samples. We mostly present the dimension 1 and dimension 2 of the MDS plots. The R function heatmap2 in the gplots (Warnes et al, 2012) package has been used. In this function, both the sample and gene dimensions were clustered. The top 500 probe sets with the largest variability in the data were chosen.
Gene sets
The human gene sets in Category 2 (C2) were from http://bioinf.wehi.edu.au/software/MSigDB, which is based on MSigDB v3.0 (Subramanian et al, 2005) downloaded on 28 September 2010. C2 includes 3272 curated gene sets.
Correlation Adjusted MEan Rank (CAMERA)
CAMERA (Wu and Smyth, 2012) is a competitive gene set test. Here, we use the default setting that allows negative gene–gene correlation and does not use rank. The multiple testing adjusting method for P-values is the Benjamini and Hochberg algorithm. It outputs P-values, set sizes and the estimated average correlation.
After fitting the variable of four subtypes in the linear model, the four contrasts were tested in CAMERA, including the basal subtype vs the average of the other three subtypes and the primitive subtype vs the average of the other three subtypes.
ROAST
The test statistic we have used in ROAST is the average of moderated t statistics. The contrasts are among the three stages in the time-course data. The donor information (three donors) was considered as the block variable in the model. The correlation within a subject was estimated using R function duplicateCorrelation in limma package. For the bronchial epithelial cell data, the highest expressed probe set was kept when there were multiple probe sets for the same gene. Then the number of probe sets dropped from 54 675 to 20 723 by keeping the unique gene name in the data prepared for ROAST. Gene weights of 1 or −1 have been used to represent the direction of differential expression (DE) in the SqCC subtypes.
ROAST is a self-contained gene set test for complex designed microarray experiments. ROAST is usually used for the direct testing of a few focused gene sets. Other genes in the platform are not considered in self-contained tests.
Active genes in a gene set were defined as the moderated t statistics from the empirical Bayesian model larger than 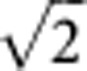 or smaller than
or smaller than 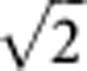 (Wu et al, 2010).
(Wu et al, 2010).
Signature scores
The probe set with the largest average value was kept when there were multiple probe sets for one gene. The detail to calculate the signature scores for a time-course sample and SqCC subtype was previously published as the Supplementary Method in (Lim et al, 2009). Here, g is for all the signature genes of the SqCC subtype. Signature score was calculated as 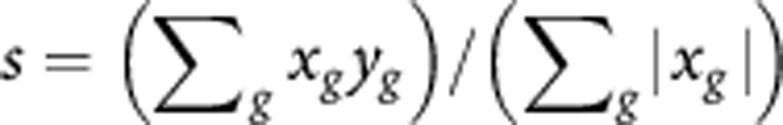 . In this calculation, the sum is over genes in the signature set, xg is the average log2-fold change while comparing one SqCC subtype to others for that gene from SqCC data, and yg is log2 expression for the same gene in a time-course sample.
. In this calculation, the sum is over genes in the signature set, xg is the average log2-fold change while comparing one SqCC subtype to others for that gene from SqCC data, and yg is log2 expression for the same gene in a time-course sample.
ClaNC
Among the total 1463 signature probe sets, 53 probe sets are in more than one subtype signatures. After removing the duplicated probe sets, 1383 probe sets represent all the signature genes of the SqCC subtypes corresponding to 1349 genes.
R code of ClaNC (Dabney, 2006) was obtained from the author. ClaNC is to classify each of the samples into one of the subtypes, whereas signature scores are to find the similarity of a group of samples to the subtype.
For the 20 cell-line data, the probe set with the largest average value was kept when there were multiple probe sets for one gene. This resulted in with 20 027 unique genes. Then gene names were matched with the 1349 subtype signature genes, which ended up with 1176 matched gene names.
To centralise subtype data of the 1176 genes, the expression value of a gene minus average of that gene across samples was calculated. The generated value was then divided by the square root of variance of that gene. Twenty cell line data of the 1176 genes were centralised using the same procedure.
To train, 1–100 genes were tried. With the selected genes, the five-fold cross-validation error rate of subtypes is <0.1. These selected genes were used to classify the cell-line samples into one of the four SqCC subtypes. On the basis of the SqCC subtype expression data of the signature genes, 40 genes for each subtype were finally selected by ClaNC for classification.
Drug database
Pharmacological profiles for 24 anticancer drugs across 504 cell lines were downloaded from www.broadinstitute.org/ccle/data. Eight dosages are included. A new score, the so-called activity area, was defined and presented in this data set. Only 17 SqCC cell lines overlapped between the cell-line expression data and the drug-sensitivity data.
The annotation of drug profiling can be found in the same CCLE website.
Proliferation scores
Proliferation gene set includes BIRC5, AURKB, CDC6, CKS2, TRAIP, CHEK1, PTTG1, DNMT1, NASP, UNG, FEN1, MCM3, MCM4, MCM5, MCM6, ORC1L, PCNA, PRIM1, RFC1, RRM1, RRM2, TOP2A, MAD2L1, CENPE, BUB1, CTPS, DHFR, TYMS, CCNA1, CCNB1, CCNE1, CCNF, CDC20, DDX11, E2F3MKI67, PKMYT1, PLK1, TIMP1, CDC25C, CENPF, MAPK13, EXOSC9 and MYB. Proliferation score for a sample is computed as the average log expression of these 43 genes in that sample.
Results
Signature genes for SqCC subtypes
The 178 samples include 43 basal, 65 classical, 27 primitive and 43 secretory samples with robust separation among the subtypes. The sample relationship in the SqCC subtype data is shown in the MDS plot (Figure 1A).
Figure 1.

Differential expression analysis results of the 178 SqCC tumour samples. (A) Multidimensional scaling plot of the normalised data for the 178 SqCC subtype samples. (B) Venn diagram of the DE analysis of the comparisons among six groups. The numbers represent the number of DE genes in each comparison (red for up and blue for down). The total genes in each diagram is the number of DE genes including up and down in any of the three comparisons involving that subtype. Regarding the subtypes, b for basal, c for classical, p for primitive and s for secretory. The overlap of the three comparison results for that subtype defines the signature gene set for each of the four SqCC subtypes. (C) Heatmap to shown the expression pattern of the signature gene sets (including up and down diretions) of the four SqCC subtypes in the 178 tumour samples. No clustering method was used in this heatmap. Rows for genes, columns for samples.
To define a specific gene signature for each subtype, we performed the DE analysis of the RNAseq data (Smyth, 2004, 2005). We compare the gene-expression profiles among the SqCC subtypes in the samples examined above. After normalisation, the data set was fitted to the linear model in which the subtype is a covariate. Two types of comparisons were made, the pairwise comparison among the four SqCC subtypes (six pairwise comparisons among four subtypes, Figure 1B) and the comparison of each single subtype to the average of the other three subtypes termed as ‘1 vs others' (four comparisons for four subtypes). More detail of the DE results, including the heatmaps of the significant DE genes in each of the six pairs, and these gene lists in the pairwise comparisons are shown in Supplementary Figures 1–6 and Supplementary Tables 8–13. The significant DE gene sets in the six comparisons (among four subtypes) may be overlapped.
On the basis of the DE results of the first type of six pairwise comparisons, we defined a signature gene set for each of the subtypes. A signature gene set of a subtype captures the uniqueness of the subtype and has a very important role in relating different data sets. In brief, signature genes were chosen if they were consistently up- or downregulated in that subtype vs each of the other subtypes (Figure 1B, Table 1). This procedure (Lim et al, 2009) selects a set of signature genes that strongly characterise each subtype by their high or low transcriptional activity (heatmap in Figure 1C). The signature genes in each of the four subtypes are shown in Supplementary Tables 4–7. It is worth noting that there are more positive-signature genes than negative-signature genes in each of the subtypes. As shown in Figure 1C, the signature gene sets are uniquely upregulated or downregulated in a subtype so that the genes in signature gene sets are unique regarding regulation directions.
Table 1. The number of signature/DE genes based on SqCC data.
| |
Signature genes from pairwise comparison |
DE genes from ‘1
vs
others' comparison |
||||||
|---|---|---|---|---|---|---|---|---|
| Direction | Basal | Classical | Primitive | Secretory | Basal | Classical | Primitive | Secretory |
| Up |
164 |
278 |
217 |
378 |
618 |
702 |
626 |
1360 |
| Down | 18 | 145 | 160 | 39 | 305 | 866 | 785 | 436 |
Abbreviations: DE=differential expression; FDR=false discovery rate.
FDR <0.05, fold change >2.
For the second type of four comparisons, we identified 618 upregulated and 305 downregulated genes while comparing the basal subtype to other three subtypes. There are 626 upregulated and 785 downregulated genes in the primitive subtype, 702 up- and 866 downregulated genes in the classical subtype, and 1360 up- and 436 downregulated in the secretory subtype (Table 1).
Pathway analysis for SqCC subtypes
To better understand the four subtypes in terms of pathways, we performed a CAMERA gene set test (Wu and Smyth, 2012), which considers the expression of gene sets, such as pathways, instead of individual genes. We used the curated gene sets in a publicly available gene set database, the Category 2 in MsigDB (human version) (Subramanian et al, 2005), which comprises 3272 sets. We aimed to identify sets of genes that are differentially expressed among the SqCC subtypes.
Here, we focused on the basal and primitive subtypes using the ‘1 vs others' comparison. On the basis of the FDR cutoff 0.05 from CAMERA in those four comparisons, the basal subtype has seven significant gene sets in the up direction but no gene sets in the down direction (Supplementary Table 1), and the primitive subtype has 628 significant gene sets in the down direction but no gene sets in the up direction (all these seven gene sets are also among the 628 gene sets but in the reverse directions in the two SqCC subtypes).
Next, for the 628 gene sets that are down in the primitive subtype, we adjusted the multiple testing P-values generated by CAMERA in the comparison of basal vs others. This is to focus on the significance of the primitive signature gene set in the comparison of basal vs others. On the basis of this procedure, 26 among the 628 are significant in basal vs others, and all of them are upregulated in basal (Supplementary Table 2). The significance of the gene set CHARAFE-BREAST-CANCER-BASAL-VS-MESENCHYMAL-UP (adjusted P 0.01 in primitive down, 0.03 in basal up) with 115 genes suggests some shared genes in the breast cancer basal subtype and the SqCC basal subtype.
Both of the gene sets RICKMAN-TUMOR-DIFFERENTIATED-WELL-VS-POORLY-DN (with 361 genes) and RICKMAN-TUMOR-DIFFERENTIATED-WELL-VS-MODERATELY-DN (with 108 genes) are significantly downregulated in primitive, and significantly upregulated in basal at adjusted P 0.006 and 0.011, respectively. On the other hand, the RICKMAN-TUMOR-DIFFERENTIATED-WELL-VS-POORLY-UP gene set (with 226 genes) is the most upregulated gene set in primitive, with nominal P-value 0.0015.
This gene set is also the second most downregulated gene set in basal vs others, with the nominal P-value 0.0001. In brief, genes overexpressed in the well-differentiated tumours are upregulated in primitive and genes underexpressed in the well-differentiated tumours are downregulated in primitive subtype. Therefore, the primitive subtype may originate from a later differentiation stage than the basal subtype.
Our pathway analysis results indicate that the basal and primitive subtypes are very different subtypes, as they do not share significant pathways in the same direction. The fact that the basal SqCC subtype and the primitive subtype display the most discordant overall survival in patients (Wilkerson et al, 2010) may be explained by the differences in signature genes and signature gene sets of the two subtypes.
Relationship between human bronchial epithelial normal cells at different culture time points and SqCC subtypes
SqCC has been considered to initiate in human bronchial epithelial cells but it is not clear when, how and in which subpopulation of the cells (Wilkerson et al, 2010). Investigators have compared the SqCC subtypes to several model systems of normal lung cell compartments. Here, we sought to evaluate this subtype to cell-type relationship using the TCGA cohort and alternative statistical methodology to that reported by Wilkerson et al (2010). We used a previously reported gene-expression data set from the HBEC-ALIC cell line in a time series of cultured normal, healthy, human bronchial epithelial cells (Ross et al, 2007; Wilkerson et al, 2010). The cells were collected at 11 different time points from day 0 to day 28.
The order of the time points of the samples can almost be recovered in the first dimension of the MDS plot (Figure 2). This suggests that the samples can be clustered according to the time points. The heatmap in this data set further confirms that there are three clusters among the samples (Figure 2). To be convenient, three clusters were defined by us as days 0, 1, 2, 4 for the early stage, days 8, 10, 12 for the middle stage and days 14, 17, 21, 28 for the late stage.
Figure 2.

Sample relationship in the normal airway time-course data. (A) Multidimensional scaling plot of the normal airway time-course data. The first dimension represents the HBEC-ALIC time points well. It suggests that the samples can be clustered into three stages of early, middle and late. (B) Heatmap of the bronchial time-course data based on the hierarchical clustering. Five hundred genes with the largest variability across samples were used. Columns are for samples and rows are for genes. The label for x axis is the days in culture. This plot further supports to cluster the 11 tHBEC-ALIC time points into three clusters. Days 0, 1, 2, 4 are the early stage; days 8, 10, 12 are the middle stage and days 14, 17, 21, 28 are the late stage.
We used the linear models and empirical Bayes methods (Smyth, 2005) to perform the DE analysis of the bronchial time-course data. In the linear model, the difference among time points was considered. We also included the donor ID as a factor variable in a random effect model, in which the correlation among samples from the same donor was computed first before fitting the model. In fact, the variable of time points can be either taken as a continuous variable or as a categorical variable into three stages of early, middle and late (Figure 2).
Here, we only showed the DE results from the three clusters of samples as follows. The FDR was controlled globally using the Benjamini and Hochberg algorithm. Probes with FDR <0.05 and fold change >2 were judged to be differentially expressed. Comparing the middle stage to the early stage, there are 605 upregulated probe sets and 302 downregulated probe sets. Comparing the late stage to the middle stage, using the same criteria, there are 843 upregulated probe sets and 534 downregulated probe sets.
Following logic similar to that of prior work (Lim et al, 2009), signature scores of the subtypes and the bronchial epithelial culture time points were computed. The signature scores include two pieces of information – the average logFC of signature genes in SqCC subtypes and the expression level at bronchial epithelial culture time points. A signature score is defined for each SqCC subtype and each bronchial sample. The higher the scores the more similar the subtype and the bronchial samples are. The signature scores were plotted according to subtypes and bronchial culture time points in Figure 3. A linear model was run to test the trend of the signature scores with the corresponding actual time points. The P-values obtained from the linear model are as follows: 4.13e-05 for basal with slope −0.014, 0.205 for classical with slope −0.001, 1.74e-05 for primitive with slope 0.010733 and 0.0519 for secretory with slope 0.0017. This suggests that there is a significant association between SqCC subtype signature scores and time points in basal and primitive subtypes. There is marginal significance in the secretory subtype and no significance in the classical subtype. Regarding the slope, classical and secretory subtypes also have much smaller slopes that are 10–20% of the slopes of the other two subtypes. In general, our results confirm what was previously published using different tumour cohorts (Wilkerson et al, 2010). The basal signature scores are highest in the early bronchial samples, whereas the primitive signature scores are highest in the late stage. Therefore, the basal SqCC subtype is most similar to early bronchial samples in which there are predominantly basal cells, and the primitive subtype is most similar to late bronchial samples in which there are many cell types and greater proliferation. The classical signature score is highest at 2d and 4d but lower in other early and late time points. The classical subtype may come from early time points, but later than the stage from which the basal subtype comes. The secretory subtype is similar to the middle stage and the late stage culture in which there are more secretory cells, although this association is not as extreme as the primitive trend in our signature score approach. This analysis discriminated the primitive and secretory subtypes clearly, further indicating that these subtypes have distinct biological properties.
Figure 3.

Signature scores of the SqCC subtype signature genes in the normal bronchial time-course data. x-axis represents the days. The higher the scores are, the more similar the subtype and the time-course samples become.
To statistically confirm the conclusion we draw from the signature scores, we performed a self-contained gene set test called Rotation gene set test (ROAST) (Wu et al, 2010) to each subtype signature gene set in the comparisons between the middle and early stages, and between the late and middle stages. A self-contained gene set test has high power to relate the two data sets of the subtype data and the bronchial time series data by giving the significance level of P-values. We used the average of moderated t value as the summary statistics in ROAST. The results (ROAST P-values 0.001–0.004 in different comparisons, detail not shown here) confirmed the above conclusion and further suggested that the order of similarity to the early bronchial epithelial culture time from high to low is basal, classical, secretory and primitive, whereas the order of similarity to the late culture is reversed as being highest in the primitive subtype and lowest in the basal subtype.
Relationship between 20 SqCC human cell lines and the SqCC subtypes
To help direct efforts for the discovery of therapeutic targets in lung SqCCs, we classified 28 lung SqCC lines by expression subtypes. In our study, a microarray data set of 28 SqCC cell lines (Table 2) was obtained from the Broad-Novartis Cancer Cell Line Encyclopedia (CCLE) downloaded from www.broadinstitute.org/ccle in May 2013. In Wilkerson et al (2010), four SqCC cell lines, HCC-15, HCC-95, HCC-2450 and H-157, have been previously classified into one of the four SqCC subtypes. Among the four cell lines, HCC-15 and HCC-95 are the only two cell lines in the 28 SqCC cell lines in CCLE.
Table 2. Classification results of the 28 SqCC cell lines to SqCC subtypes.
| |
Subtype rank |
ClaNC |
|
|||||
|---|---|---|---|---|---|---|---|---|
| Cell line | First subtype | Second subtype | First subtype | Permutaion P | Second subtype | Permutation P | Predicted | With drug data |
| NCI-H226 |
Secretory |
Primitive |
Secretory |
0.16 |
Basal |
0.457 |
Secretory |
y |
| EBC-1 |
Secretory |
Basal |
Basal |
0.426 |
Primitive |
0.277 |
Basal |
y |
| VMRC-LCP |
Basal |
Classical |
Basal |
0.033 |
Classical |
0.32 |
Basal |
|
| HCC-1588 |
Basal |
Primitive |
Basal |
0.082 |
Secretory |
0.595 |
Basal |
|
| RERF-LC-AI |
Primitive |
Secretory |
Primitive |
0.07 |
Secretory |
0.311 |
Primitive |
y |
| SK-MES-1 |
Primitive |
Secretory |
Basal |
0.293 |
Secretory |
0.318 |
Secretory |
y |
| NCI-H520 |
Classical |
Primitive |
Primitive |
0.076 |
Classical |
0.678 |
Primitive |
y |
| HCC-15 |
Classical |
Secretory |
Classical |
0.061 |
Basal |
0.528 |
Classical |
y |
| SW 900 |
Primitive |
Basal |
Secretory |
0.109 |
Basal |
0.25 |
Secretory |
y |
| LUDLU-1 |
Basal |
Classical |
Classical |
0.024 |
Basal |
0.077 |
Classical |
y |
| HARA |
Secretory |
Classical |
Basal |
0.308 |
Secretory |
0.557 |
Secretory |
y |
| KNS-62 |
Basal |
Classical |
Basal |
0.118 |
Classical |
0.551 |
Basal |
y |
| EPLC-272H |
Basal |
Primitive |
Basal |
0.114 |
Secretory |
0.339 |
Basal |
|
| HCC-95 |
Classical |
Secretory |
Classical |
0.009 |
Basal |
0.667 |
Classical |
|
| LC-1/sq-SF |
Classical |
NA |
Classical |
0.013 |
Basal |
0.688 |
Classical |
y |
| SW 1573 |
Secretory |
Primitive |
Primitive |
0.108 |
Secretory |
0.277 |
Primitive |
y |
| LC-1F |
Classical |
Basal |
Classical |
0.012 |
Primitive |
0.539 |
Classical |
|
| LOU-NH91 |
Secretory |
Primitive |
Secretory |
0.19 |
Primitive |
0.231 |
Secretory |
y |
| LK-2 |
Classical |
Primitive |
Primitive |
0.074 |
Classical |
0.585 |
Primitive |
|
| RERF-LC-Sq-1 |
Primitive |
Classical |
Secretory |
0.293 |
Primitive |
0.209 |
Primitive |
|
| HLF-a |
Primitive |
Classical |
Secretory |
0.063 |
Primitive |
0.233 |
Secretory |
|
| Calu-1 |
Primitive |
Secretory |
Secretory |
0.08 |
Basal |
0.583 |
Secretory |
y |
| NCI-H2170 |
Basal |
Classical |
Classical |
0.114 |
Primitive |
0.085 |
Classical |
y |
| Sq-1 |
Primitive |
NA |
Secretory |
0.129 |
Basal |
0.166 |
Secretory |
y |
| HCC-2814 |
Classical |
Basal |
Classical |
0.048 |
Basal |
0.369 |
Classical |
|
| HCC-1897 |
Secretory |
Primitive |
Secretory |
0.07 |
Basal |
0.39 |
Secretory |
|
| NCI-H1385 |
Classical |
Primitive |
Primitive |
0.069 |
Classical |
0.352 |
Primitive |
|
| NCI-H1869 | Basal | NA | Secretory | 0.244 | Basal | 0.303 | Basal | y |
Abbreviations: ClaNC=classification method to nearest centroids; SqCC=squamous cell lung cancer.
The signature scores of the four SqCC subtypes in each of the cell lines were computed (Figure 4). Here, these signature scores were calculated based on the general cutoff as fold-change 2 and FDR 0.05 to obtain the signature gene sets. The cell line ranks of the scores in the four subtypes remain similar even if a less stringent cutoff (fold-change 1.5 and FDR 0.1, detail unpublished) is used. Higher scores represent higher similarities between cell lines and SqCC subtypes.
Figure 4.

Cell-line ranks of signature scores of the SqCC subtype signature genes in the 28 SqCC cell lines. x-axis represents the different SqCC cell lines. y-axisrepresents the ranks of the signature scores in each SqCC subtype.
A procedure was developed to generate reproducible classification results using signature scores. We rank the 28 cell lines based on their signature scores in a subtype – for example the basal subtype. The rank is from 1 to 28. If each row is for a cell line in a data matrix, we have four columns (cell line rank per subtype) of the ranks as seen in Supplementary Table 14 and the ranks are plotted in Figure 3, shown per cell line. The top ranks have smaller rank numbers. Therefore, for each cell line, the subtype with the smallest rank number was considered as the ‘1st subtype' of that cell line, followed by the second smallest rank number for the ‘2nd subtype' of that cell line as in ‘subtype rank' as seen in Supplementary Table 14 and the corresponding brief Table 2. Here, we use this procedure to determine the most similar subtype to a cell line based on signature scores. We also include the ‘2nd subtype' to represent variability.
The cell line LUDLU-1 is similar to both basal and classical subtypes. The cell line LC-1/sq-SF is most similar to the classical subtype, and the similarity was ranked quite low in all the other three subtypes. HCC-95 is most similar to the classical subtype, the same as suggested in Wilkerson et al (2010). Although Wilkerson et al (2010) suggested HCC-15 to be a primitive subtype, it may have some mixed features of other subtypes for two reasons. First, the range of signature scores of the cell lines to the primitive subtypes is small; therefore, the difference among cell lines may be subtle. Second, the signatures of HCC-15 in other three subtypes are not very low. The brief results are shown in Table 2.
We assigned cell lines by two methods. Signature scores using signature genes of subtypes provide a general relationship between cell lines and subtypes. Some cell lines do not fit easily into a subtype – for example, sq-1, Calu-1 and LUDLU-1 (Figure 4). The classification method we next used gave a more clear indication of subtypes.
As a complementary subtype assignment, a classification method to nearest centroids (ClaNC), classified the 28 cell lines into the four SqCC subtypes, with seven basal, seven classical, five primitive and nine secretory cell lines (first subtype of ClaNC in Table 2).
As ClaNC uses distance between 28 cell lines and 4 subtype centroids, we output the distance matrix of 28 × 4. The nearest distance is used to define the ‘1st subtype' – that is, the classification results. We also obtained the ‘2nd subtype' for a cell line – that is, the second nearest centroid. To evaluate the uncertainty of this classification method, we first made an MDS plot (Supplementary Figure 7) with the centralised tumour samples together with the centralised cell line samples. This shows that the cell line samples tend to be on the centre of all samples. We permute the subtype labels randomly to generate four random centroids for 1000 times. The distance between cell lines and centroids is mostly in a much smaller scale (data not shown) compared with the observed distance. This is because of the fact that the cell-line samples tend to be on the centre of all samples. To correct this scaling bias, for each sample, we convert the distance to percentage of the distance to the sum of the four distances. This was carried out in both permutation-based samples and the observed cell-line samples. P-values were computed, as the probability of the observed percentage–distance is larger than the percentage–distance from the permutations. Smaller P-value represents how significant the distance from the cell-line sample to the nearest centroid is. Same procedure was performed for the ‘2nd subtype' that shares exactly the same permutation. Here we explained how to access the uncertainty of the classification method ClaNC.
With the results from signature scores (first and second subtypes) and ClaNC (first and second subtypes and P-values), we determine the predicted subtype of a cell line. The criteria are as follows: the permutation-based P-value for ClaNC first subtype <0.2. If not, the majority vote is used among four columns regarding first and second subtypes, and the prediction of these samples is highlighted as blue in Supplementary Table 14, with less certainty (Table 2).
In the ClaNC results, both cell lines HCC-95 and HCC-15 were classified as classical samples. Therefore, HCC-95 is highly likely to be a classical cell line, being consistent with the previous classification (Wilkerson et al, 2010) and the results of signature scores. HCC-15 has a mixed background.
Drug target of cancer cell lines
To determine whether expression subtype may predict drug response in SqCC cell lines, we obtained publicly available drug-sensitivity data for SqCC cell lines (www.broadinstitute.org/ccle/data).
In the context of the pharmacological profiles for 24 anticancer drugs across 504 cancer cell lines (Barretina et al, 2012), we located the drug sensitivities for the different SqCC subtypes. Only 17 cell lines among the 28 SqCC cell lines were treated in this drug-response experiment (Barretina et al, 2012) (Table 2), with 24 drugs at eight dosages. According to ClaNC, the 17 cell lines comprise four basal, four classical, three primitive and six secretory SqCC cell lines. In Barretina et al (2012), a novel score termed ‘activity area' was created to combine the information of the half maximal inhibitory concentration (IC50), the half maximal effective concentration (EC50) and maximum inhibited percentage (MIP). A large activity area comprising small IC50, small EC50 and larger MIP indicates high sensitivity of a cell line to a drug (Supplementary Table 3, previously published (Barretina et al, 2012)).
The response sensitivities, represented by activity area, for the 17 cell lines as shown in Figures 5A and B, are varied across drugs. Most cell lines responded to five of the drugs (Panobinostat, 17-AAG, Irinotecan, Topotecan and Paclitaxel). The drug targets of these five drugs are HDAC, HSP90, Topoisomerase-I, Topoisomerase-I and beta-tubulin, respectively. A basal cell line EBC-1 is also sensitive to three additional drugs (PF2341066 with target c-MET, AZD6244 with target MEK and PD-0325901 with target MEK).
Figure 5.

Drug sensitivity to SqCC subtypes through CCLE. (A) Heatmap of the activity area score for 17 SqCC cell lines (four basal, four classical, three primitive, six secretory and SqCC cell lines based on ClaNC) and the 24 drugs. The white block in the plot is for missing data due to the lack of some drug treatments to the cell lines. The rows are for the SqCC cell lines and the columns are for the 24 drugs. Both dimensions have been clustered by hierarchical clustering. The ClaNC results of SqCC subtype classification were shown for each cell line. (B) Scatter plot of the activity area score for 17 cell lines and 24 drugs. Colours represent the four subtypes. (C) On the left panel, proliferation scores for secretory SqCC samples or other SqCC samples (P-value 7.5e-05). On the right panel, the activity area score of all 24 drugs for secretory cell lines or others (P-value 0.014). Secretory SqCC subtype has lower proliferation scores and lower activity area scores of drug treatment. Two-sided P-value was obtained by Wilcoxon Rank sum test (P-value 7.5e-05 on the left, 0.068 on the right). (D) Focusing on the five drugs (Panobinostat, 17-AAG, Irinotecan, Topotecan and Paclitaxel), this shows the area scores for secretory cell lines are significantly different to the scores in each of the other three SqCC types of cell lines. Two-sided Wilcoxon mean rank test was used (secretory vs basal P-value 0.002, vs classical P-value 0.006 and vs primitive P-value 0.071).
Compared with most of the basal and classical cell lines, the secretory cell line NCIH-226 is least sensitive to the drugs Panobinostat, 17-AAG, Irinotecan, Topotecan, Paclitaxel, AZD6244 and PF2341066. For each cell line, we can calculate an average activity area across drugs by averaging the columns in the heatmap. The global mean of the average activity areas across the drugs for the 17 cell lines is 1.34 (s.d.=0.41).
On the basis of Figure 5B, there is a trend that the secretory cell lines (orange colour) have lower activity area scores across drugs. Particularly, all cell lines have at least moderate response to Paclitaxel, whereas secretory cell lines have lower drug response to Paclitaxel. As the secretory SqCC tumours have lower proliferation activity, we investigate a proliferation score for each tumour sample. We used a proliferation signature set of 43 genes (Whitfield et al, 2006) to get a proliferation score for a tumour sample that is the average log expression of these 43 genes in that sample. Higher proliferation score represents higher proliferation activity. Figure 5C (left) shows that the secretory SqCC samples have significant (P 7.5e-05) lower proliferation scores than other subtypes. This might explain why the cell lines of the secretory SqCC subtype have lower activity area scores of drug treatment at P-value 0.014 (Figure 5C-right). Focusing on the five drugs (Panobinostat, 17-AAG, Irinotecan, Topotecan and Paclitaxel), we used two-sided Wilcoxon mean rank test to test whether the average area scores for secretory cell lines are significantly different to the scores in each of the other three SqCC types of cell lines (Figure 5D). The secretory cell lines have significant lower scores than basal (P-value 0.002), classical (P-value 0.006) and primitive cell lines (P-value 0.071).
Overall, the SqCC cell lines are not sensitive to any drug with EGFR or FGFR as the target. We reported what we observed and the results seem reasonable in terms of the prior knowledge that EGFR-targeted drugs are not for SqCC patients. Our results may be generalised without the current limitation of the number of cell lines and number of drugs. Generally speaking, a larger number of SqCC cell lines and an increased number of profiled compounds may be required to make more robust conclusions about the drug repurpose for SqCC subtypes.
Discussion
In this study, we describe the comprehensive data integration of the relevant four data sets. They are the SqCC subtype data, the human bronchial epithelial cell air–liquid interface culture time-course data, the CCLE data and the drug-sensitivity data of SqCC cell lines. This article not only shows a pipeline from hypothesis-generating, DE data analysis to relating data sets across experiments but also finds the difference of primitive and secretory subtypes in terms of cells of origin and reveals a fundamental question of the available representative cell lines for each of the SqCC molecular subtypes. To be relevant to the clinic, drug repurposing of SqCC has been explored based on the CCLE data and is shown to have promising results.
Four SqCC subtypes have been identified (Wilkerson et al, 2010), including basal, primitive, classical and secretory subtypes. Among them, the primitive subtype has the worst prognosis and the basal subtype has better prognosis than the other subtypes. We checked the sample relationship in the SqCC-subtype gene-expression data and the four subtypes were roughly clustered by themselves. To characterise functionally the different subtypes, particularly the basal and the primitive subtypes, a competitive gene set test that takes care of the gene–gene correlation (CAMERA) has been used against the C2 gene sets in MsigDB. We found that 26 significant gene sets overlap in the basal vs others and the primitive vs others, but in opposite directions (upregulated in the basal subtype and downregulated in the primitive subtype).
We defined signature gene sets for each SqCC subtype based on the DE analysis of array data and calculated the signature scores for each subtype and each bronchial epithelial sample. Signature scores were used to represent the similarities between the subtype data with the bronchial epithelial sample data. We have used the signature scores to show that the early-stage bronchial epithelial cells are most similar to the basal subtype, and the late-stage bronchial epithelial cells are most similar to the primitive subtype. Compared with the results in Wilkerson et al (2010), we have clearly shown the difference between primitive and secretory subtypes regarding the similarities to the bronchial epithelial cells at different culturing days, as well as the stage of cells that needs to be targeted. Potentially, we also aim to integrate the data of the available cell types, stem cells among others, from normal lungs to find which normal cell type tends to be the cell-of-origin of which of the SqCC subtypes.
Lung cancer has been classified into two large categories and a few smaller categories. New molecularly targeted therapies for lung cancer (Sun et al, 2007) are of great interest. One piece of evidence is that Gefitinib (IRESSA)-sensitive lung cancer cell lines show phosphorylation of Akt without ligand stimulation (Noro et al, 2006). Perez-Moreno et al (2012) has reviewed the therapeutic opportunities to SqCC based on known genetic alternations. Hammerman et al (2011) among others investigated the novel therapeutic target in SqCC. Wilkerson et al (2010) provided lists of 2—8 genes for the different SqCC subtypes.
The ultimate goal to study SqCC subtypes is to find a better treatment for the patients who carry these subtypes. To potentially use the genomic findings to guide drug repurposing (Collins, 2011), we also integrate publicly available drug-titration data with the classification results of SqCC tumour cell lines to the SqCC subtypes. Ideally, drug repurposing can be performed by knowing the drug target (or a critical causal gene) of a disease and the drug-targeted gene of a drug. Overlapping between the two targets indicates that the drug can be repurposed as treatment of the disease (Sanseau et al, 2012). Subtype-specific sensitivity in breast cancer subtypes has been investigated (Heiser et al, 2012). For lung cancer, previously Wistuba et al (1999) compared the features of human NSCLC cell lines and their parental tumours using immunohistochemical expression of 37 biomarkers and microsatellite markers, and confirmed that the NSCLC cell lines generally retain the properties of their parental tumours for quite long culture periods. Very recently, Barretina et al (2012) have investigated the possibility to predict anticancer drug sensitivity for hundreds of cell lines in the CCLE project, including lung cancer cell lines. To investigate the drug sensitivity in different SqCC subtypes, we have used the drug-titration data for SqCC cell lines in CCLE.
Among this data set, 28 cell lines are annotated as SqCC cell lines. Using ClaNC (Dabney, 2006), a novel classification method, we were able to assign each of the 17 cell lines that have drug-titration data to one of the SqCC subtypes. Although the primitive SqCC subtype tends to have worse prognosis results and requires effective drugs, it is not clear which drug can be proposed to be for this subtype, as there is only one primitive SqCC cell line in the drug data. To fully study the aggressive primitive subtype, more primitive SqCC cell lines need to be developed. We also have investigated the association between the proliferation activity in secretory tumour samples and the activity score of drugs in secretory cell lines, which suggests that the lower drug activity may be because of the lower proliferation activity in the secretory subtype.
With more and more available data, our data-integration procedure can be extended to many other heterogeneous cancer types.
Acknowledgments
We thank Drs Neil Hayes at the University of North Carolina at Chapel Hill for providing the SqCC subtype information of the samples in Raponi et al (2006), Alan Dabney at Department of Statistics, Texas A&M University for providing the R code of ClaNC, Heidi Greulich for helping to find the CCLE data sets, Ke Deng and Simeng Han at Harvard Statistics for their discussion, Catherine Wu for proof reading, Laurent Jacob and Terry Speed at the Statistics Department UC Berkely for their suggestion about classification methods, Gordon Smyth and Charity Law at the Walter and Eliza Hall Institute for providing suggestions on how to use ‘voom', Robert Plenge at Brigham Women Hospital, Tianxi Cai at Harvard Biostatistics, Bryan Williams at Monash Institute of Medical Research and Matthew Meyerson at Dana Farber Cancer Institute for being supportive. This work was supported by grants from the Australian National Health and Medical Research Council (1036541 to Di Wu), and National Science Foundation (NSF IIS-1017967 for publication).
Footnotes
Supplementary Information accompanies this paper on British Journal of Cancer website (http://www.nature.com/bjc)
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Material
References
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P., Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The cancer cell line encyclopaedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadsmoore CJ, Screaton NJ. Classification, staging and prognosis of lung cancer. Eur J Radiol. 2003;45:8–17. doi: 10.1016/s0720-048x(02)00287-5. [DOI] [PubMed] [Google Scholar]
- Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, Liu Y, Tupper T, Ouyang J, Li J, Gao P, Woo MS, Xu C, Yanagita M, Altabef A, Wang S, Lee C, Nakada Y, Pena CG, Sun Y, Franchetti Y, Yao C, Saur A, Cameron MD, Nishino M, Hayes DN, Wilkerson MD, Roberts PJ, Lee CB, Bardeesy N, Butaney M, Chirieac LR, Costa DB, Jackman D, Sharpless NE, Castrillon DH, Demetri GD, Janne PA, Pandolfi PP, Cantley LC, Kung AL, Engelman JA, Wong KK. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483:613–617. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS. Reengineering translational science: the time is right. Sci Transl Med. 2011;3:90cm17. doi: 10.1126/scitranslmed.3002747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney AR. Clanc: point-and-click software for classifying microarrays to nearest centroids. Bioinformatics. 2006;22:122–123. doi: 10.1093/bioinformatics/bti756. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerman PS, Hayes DN, Wilkerson MD, Schultz N, Bose R, Chu A, Collisson EA, Cope L, Creighton CJ, Getz G, Herman JG, Johnson BE, Kucherlapati R, Ladanyi M, Maher CA, Robertson G, Sander C, Shen R, Sinha R, Sivachenko A, Thomas RK, Travis WD, Tsao MS, Weinstein JN, Wigle DA, Baylin SB, Govindan R, Meyerson M. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, Zhou W, Brace LE, Woods BA, Lin W, Zhang J, Deng X, Lim SM, Heynck S, Peifer M, Simard JR, Lawrence MS, Onofrio RC, Salvesen HB, Seidel D, Zander T, Heuckmann JM, Soltermann A, Moch H, Koker M, Leenders F, Gabler F, Querings S, Ansen S, Brambilla E, Brambilla C, Lorimier P, Brustugun OT, Helland A, Petersen I, Clement JH, Groen H, Timens W, Sietsma H, Stoelben E, Wolf J, Beer DG, Tsao MS, Hanna M, Hatton C, Eck MJ, Janne PA, Johnson BE, Winckler W, Greulich H, Bass AJ, Cho J, Rauh D, Gray NS, Wong KK, Haura EB, Thomas RK, Meyerson M. Mutations in the ddr2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011;1:78–89. doi: 10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiser LM, Sadanandam A, Kuo WL, Benz SC, Goldstein TC, Ng S, Gibb WJ, Wang NJ, Ziyad S, Tong F, Bayani N, Hu Z, Billig JI, Dueregger A, Lewis S, Jakkula L, Korkola JE, Durinck S, Pepin F, Guan Y, Purdom E, Neuvial P, Bengtsson H, Wood KW, Smith PG, Vassilev LT, Hennessy BT, Greshock J, Bachman KE, Hardwicke MA, Park JW, Marton LJ, Wolf DM, Collisson EA, Neve RM, Mills GB, Speed TP, Feiler HS, Wooster RF, Haussler D, Stuart JM, Gray JW, Spellman PT. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc Natl Acad Sci USA. 2012;109:2724–2729. doi: 10.1073/pnas.1018854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, Feleppa F, Huschtscha LI, Thorne HJ, Fox SB, Yan M, French JD, Brown MA, Smyth GK, Visvader JE, Lindeman GJ. Aberrant luminal progenitors as the candidate target population for basal tumour development in brca1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- MacDonald JW. affycoretools: Functions useful for those doing repetitive analyses with Affymetrix GeneChips. R package version 1.28.0. 2008.
- Mariani TJ, Reed JJ, Shapiro SD. Expression profiling of the developing mouse lung: insights into the establishment of the extracellular matrix. Am J Respir Cell Mol Biol. 2002;26:541–548. doi: 10.1165/ajrcmb.26.5.2001-00080c. [DOI] [PubMed] [Google Scholar]
- Noro R, Gemma A, Kosaihira S, Kokubo Y, Chen M, Seike M, Kataoka K, Matsuda K, Okano T, Minegishi Y, Yoshimura A, Kudoh S. Gefitinib (iressa) sensitive lung cancer cell lines show phosphorylation of akt without ligand stimulation. BMC Cancer. 2006;6:277. doi: 10.1186/1471-2407-6-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Moreno P, Brambilla E, Thomas R, Soria JC. Squamous cell carcinoma of the lung: molecular subtypes and therapeutic opportunities. Clin Cancer Res. 2012;18:2443–2451. doi: 10.1158/1078-0432.CCR-11-2370. [DOI] [PubMed] [Google Scholar]
- R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria; 2012. [Google Scholar]
- Raponi M, Zhang Y, Yu J, Chen G, Lee G, Taylor JM, Macdonald J, Thomas D, Moskaluk C, Wang Y, Beer DG. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006;66:7466–7472. doi: 10.1158/0008-5472.CAN-06-1191. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edger: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AJ, Dailey LA, Brighton LE, Devlin RB. Transcriptional profiling of mucociliary differentiation in human airway epithelial cells. Am J Respir Cell Mol Biol. 2007;37:169–185. doi: 10.1165/rcmb.2006-0466OC. [DOI] [PubMed] [Google Scholar]
- Sanseau P, Agarwal P, Barnes MR, Pastinen T, Richards JB, Cardon LR, Mooser V. Use of genome-wide association studies for drug repositioning. Nat Biotechnol. 2012;30:317–320. doi: 10.1038/nbt.2151. [DOI] [PubMed] [Google Scholar]
- Smyth GK.2004Linear models and empirical bayes methods for assessing differential expression in microarray experiments Stat Appl Genet Mol Biol 3Article3. [DOI] [PubMed] [Google Scholar]
- Smyth GK.2005Limma: linear models for microarray dataIn: Bioinformatics and Computational Biology Solutions using R and Bioconductor Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, (eds). pp397–420.Springer: New York, NY, USA [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Schiller JH, Spinola M, Minna JD. New molecularly targeted therapies for lung cancer. J Clin Invest. 2007;117:2740–2750. doi: 10.1172/JCI31809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B.2012. gplots: Various R programming tools for plotting data. URL, http://CRAN.R-project.org/package=gplots R package version 2.11.0.
- Whitfield ML, George LK, Grant GD, Perou CM. Common markers of proliferation. Nat Rev Cancer. 2006;6:99–106. doi: 10.1038/nrc1802. [DOI] [PubMed] [Google Scholar]
- Wilkerson MD, Yin X, Hoadley KA, Liu Y, Hayward MC, Cabanski CR, Muldrew K, Miller CR, Randell SH, Socinski MA, Parsons AM, Funkhouser WK, Lee CB, Roberts PJ, Thorne L, Bernard PS, Perou CM, Hayes DN. Lung squamous cell carcinoma mrna expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clin Cancer Res. 2010;16:4864–4875. doi: 10.1158/1078-0432.CCR-10-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistuba I, Bryant D, Behrens C, Milchgrub S, Virmani AK, Ashfaq R, Minna JD, Gazdar AF. Comparison of features of human lung cancer cell lines and their corresponding tumours. Clin Cancer Res. 1999;5:991–1000. [PubMed] [Google Scholar]
- Wu D, Lim E, Vaillant F, Asselin-Labat ML, Visvader JE, Smyth GK. Roast: rotation gene set tests for complex microarray experiments. Bioinformatics. 2010;26:2176–2182. doi: 10.1093/bioinformatics/btq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Smyth GK. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012;40:e133. doi: 10.1093/nar/gks461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model based background adjustment for oligonucleotide expression array. J Am Stat Assoc. 2004;99:909–917. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.