

Abstract
Background:
Antibody drug conjugates (ADCs) and immunotoxins (ITs) are promising anticancer immunotherapeutics. Despite their encouraging performance in clinical trials, both ADCs and ITs often suffer from disadvantages such as stoichiometrically undefined chemical linkage of the cytotoxic payload (ADCs) and the potential immunogenicity of toxins derived from bacteria and plants (ITs).
Methods:
Human microtubule-associated protein tau (MAP) was cloned in-frame with human EGF, expressed in E. coli and purified by standard chromatographic methods. The in vitro activity was confirmed by flow cytometry, cell viability assays and tubulin polymerisation assay. The in vivo efficacy was demonstrated using noninvasive far-red in vivo imaging.
Results:
The EGF-MAP selectively induced apoptosis in EGFR-overexpressing proliferating cancer cells through stabilisation of microtubules. Nonproliferating cells were not affected, demonstrating superior selectivity of EGF-MAP for cancer cells. The EGF-MAP was well tolerated at high doses in mice compared with the ETA'-based control. The in vivo efficacy of EGF-MAP was demonstrated in a tumour xenograft mouse model.
Conclusion:
Our data indicate the general mechanism of action for a new class of human immunotherapeutic reagents suitable for the treatment of cancer. This approach combines the binding specificity of targeting ligands with the selective cytotoxicity of MAP towards proliferating cells.
Keywords: immunotoxins, human cytolytic fusion proteins, ADCs, immunotherapy
One prominent goal in passive immunotherapy is the targeted delivery of highly cytostatic payloads to malignant cells. This can be achieved using bifunctional molecules, comprising a targeting component and a cytotoxic component. The targeting component can be a full-size monoclonal antibody (mAb), a fragment thereof or a protein ligand (e.g., a growth factor or cytokine) (Cawley et al, 1980; Williams et al, 1987; von Minckwitz et al, 2005). A variety of substances have been used as cytotoxic agents, including radioisotopes, small-molecule drugs and protein-based toxins derived from bacteria or plants. Depending on the linkage between the targeting and cytotoxic components, these molecules are described as antibody drug conjugates (ADCs) with chemical crosslinking between synthetic cytotoxic molecules and antibodies, or immunotoxins (ITs) if cytotoxic proteins are genetically fused to the targeting protein (Wu and Senter, 2005; Pastan et al, 2006; Carter and Senter, 2008). Both ADCs and ITs can overcome the disadvantages of naked mAbs, which often require a functional host immune system for their therapeutic effect (Weiner et al, 2010).
At least 22 ADCs are currently undergoing clinical trials. Two ADCs, Kadcyla (ado-trastuzumab emtansine) and brentuximab vedotin, have already been approved by the FDA for the treatment of HER2- and CD30-positive cancers, respectively (Lambert, 2005; Goncalves et al, 2012; Sievers and Senter, 2013). Interestingly, the cytotoxic domain of brentuximab vedotin is monomethyl auristatin E, a synthetic analogue of the tubulin polymerisation inhibitor dolastatin 10 (Tanaka et al, 2009). The success of this ADC underlines the enormous potential of cytostatic agents delivered by targeted immunotherapy. Classically, the cytotoxic compound is conjugated to the targeting mAb via nondirected chemical linkage, often resulting in an undefined stoichiometry of ADCs and heterogenic positioning of the cytotoxic payload in relation to the binding protein, which can directly affect its therapeutic efficacy (Ducry and Stump, 2010). Similarly, one disadvantage of chimeric ITs is their combination of a human or humanised binding component (e.g., a single-chain antibody fragment, scFv) and a toxin derived from bacteria or plants that can induce undesirable immune responses and dose limitations, for example, because of vascular leak syndrome (Li et al, 2005; Kreitman, 2006).
These shortcomings have been addressed by using pro-apoptotic human enzymes in the context of fully human cytolytic fusion proteins (CFPs). Human RNases 1, 2, 3 and 5 (angiogenin), which degrade RNA and induce apoptosis by inhibiting protein synthesis, have been used to replace non-human toxins (Mathew and Verma, 2009). The specific cytotoxicity of human angiogenin towards CD30-overexpressing Hodgkin's lymphoma-derived cell lines has been demonstrated by fusion with the CD30 ligand (Huhn et al, 2001). Another CD30-targeted CFP was shown to kill CD30-overexpressing tumour cells efficiently when combined with human death-associated protein kinase 2 (Tur et al, 2009). In 2008, a completely human granzyme B-based CFP directed against CD64 was found to be toxic towards an acute myeloid leukemia (AML)-related cell line and primary AML cells (Stahnke et al, 2008). Thus far, various human proteins including RNases, perforin, Bik, Bak, Bax, DNA fragmentation factor 40, FAS ligand and TNF-related apoptosis-inducing ligand have been used as toxic effector molecules (ten Cate et al, 2010). Although most of these have been tested for toxicity towards cancer cells, they could potentially be applied in other indication areas such as autoimmune and chronic inflammatory disorders.
Generally, only the binding domain of CFPs confers selectivity and specificity, whereas the cytotoxic component is chosen according to its ability to induce apoptosis based on different modes of action. However, most tumour markers are also expressed on physiologically normal cells, resulting in undesirable off-target effects. Cytostatic agents such as taxanes (e.g., paclitaxel) have superior selectivity. Taxanes are microtubule-stabilising agents and are currently used as chemotherapeutics in a wide range of indications including cancer (e.g., breast cancer, ovarian cancer, non-small-cell lung carcinoma) and haematological malignancies (Norris et al, 2000; Dimitroulis and Stathopoulos, 2005; Pajk et al, 2008). Taxanes conjugated to mAbs have also been successfully tested in tumour therapy. The mode of action is based on binding to and stabilising polymerised microtubule filaments, resulting in a change in the total microtubule mass (if high concentrations of the stabilising agent are present) or microtubule dynamics. Both changes lead to cell cycle arrest and the induction of apoptosis (Jordan et al, 1993; Zhou and Giannakakou, 2005; Jordan and Kamath, 2007).
Taxanes can be chemically conjugated to the targeting component using state-of-the-art chemistry. In contrast, microtubule-associated proteins, which are known to regulate microtubule growth and shortening (e.g., survivin, stathmin, dynactin-1 and MCAK), could be used as fusion proteins (Maney et al, 2001; Cassimeris, 2002; Giodini et al, 2002; Ligon et al, 2003). In physiologically healthy cells, these proteins orchestrate the fine-tuning of microtubule dynamics, thus allowing these polymers to fulfill diverse functions. In resting cells (interphase), microtubules are replaced slowly, with half-lives of minutes to hours, whereas the microtubule disassembly and polymerisation from spindle microtubules occurs up to 100-fold more rapidly during mitosis, and this increased rate is crucial for successful cell division (Saxton et al, 1984; Zhai et al, 1996). The dynamics of microtubule assembly and disassembly is under tight spatiotemporal control by microtubule-associated proteins, which are themselves regulated by phosphorylation (Billingsley and Kincaid, 1997). Therefore, we hypothesised that the presence of nonphosphorylated microtubule-associated proteins during mitosis would slow down the process and induce apoptosis.
Here we describe a new cytostatic effector protein, called human microtubule-associated protein tau (hereafter MAP), that is suitable for the generation of fully human CFPs. We used the human epidermal growth factor (EGF) as a binding component in an EGF-MAP fusion protein to test the efficacy of MAP. We removed the major serine phosphorylation sites from MAP (S156 and S204) to prevent negative regulation. The CFP was expressed successfully in Escherichia coli (E. coli) and enriched by immobilised metal ion affinity chromatography (IMAC) and size exclusion chromatography (SEC). The MAP-mediated and proliferation-dependent antitumour activity was demonstrated in vitro using EGF receptor (EGFR)-overexpressing cell lines including the pancreatic cancer cell line L3.6pl. The L3.6pl cells transfected with the far-red fluorescent protein Katushka-2 were then used in a mouse xenograft model in vivo, confirming that EGF-MAP can significantly inhibit tumour growth. Our data highlight the properties of a new class of human immunotherapeutic reagents for the treatment of cancer, combining the selectivity of the binding component and the cytotoxicity of MAP that is specific towards proliferating cancer cells.
Materials and methods
Cloning of EGF-MAP
The open reading frame (ORF) for human EGF (GeneID: 1950) was modified with 5′-SfiI and 3′-NotI restriction sites by PCR followed by ligation into a SfiI/NotI-linearised pMT vector. The ORF for MAP (Gene ID: 4137; NM_016834.4) was modified with 5′-NotI and 3′-BlpI restriction sites by PCR followed by ligation into a NotI/BlpI-linearised pMT vector already containing the EGF sequence (Figure 1A). Two point mutations were introduced to remove phosphorylation sites S156A and S204A and a 3′ nuclear localisation sequence (NLS) was introduced into the ORF for MAP by PCR. Successful cloning was confirmed by test digestion and sequencing.
Figure 1.

Generation and purification of EGF-MAP. (A) Vector map of pMT-EGF-MAP. The ORF contains the EGF ligand at the N-terminus and MAP at the C-terminus. The other elements are: F1 ori, origin of replication for M13 phage; nptI, kanamycin resistance gene; pBR322 origin for E. coli, lacI, repressor; T7 promoter, IPTG-inducible promoter; lac operator, for transcriptional regulation; pelB leader peptide, signal peptide for periplasmic secretion; His-10, poly-histidine sequence used for purification and detection of the recombinant protein; EK site, enterokinase cleavage site; S156A/S204A, mutations introduced to remove phosphorylation sites; repeats 1–4, microtubule-binding repeats 1–4; NLS, nuclear localisation sequence derived from the Simian Vacuolating Virus 40 large T antigen; T7 terminator, terminates transcription. (B) The SDS–PAGE and western blot analysis of EGF-MAP. The EGF-MAP was purified by IMAC and SEC. The enrichment was determined by SDS–PAGE followed by staining with Coomassie brilliant blue (lane 1). Protein identity was verified by western blotting (lane 2) using mouse-anti-penta-His (1 : 5000, Qiagen). The primary antibody was detected using an alkaline phosphatase-conjugated anti-mouse-IgG mAb (1 : 5000; Sigma, Germany) followed by staining with NBT/BCIP substrate (Life Technologies). (C) The EGF-MAP cell-binding analysis. Purified EGF-MAP was tested for its ability to bind EGFR+ L3.6pl cells (EGFR– HEK293 cells were used as a negative control). The 425(scFv)-ETA' has previously been shown to bind L3.6pl cells and was used as a positive control. Bound EGF-MAP and 425(scFv)-ETA' were detected with anti-penta-His-Alexa Fluor 488 antibody (Qiagen).
Expression and purification of recombinant EGF-MAP
Recombinant EGF-MAP was expressed in E. coli BL21 (DE3) cells using the protocol for periplasmic stress expression in the presence of compatible solutes as described previously (Barth et al, 2000). Briefly, bacteria were grown after transformation to an OD of 1.6 followed by stress induction with 500 mM D-sorbitol, 10 mM betaine monohydrate and 4% (w/w) NaCl. After incubation for 30 min at 26 °C with shaking (180 r.p.m.), protein expression was induced with 2 mM IPTG. Bacteria were harvested 18 h after induction by centrifugation (4000 × g, 10 min, 4 °C) and frozen at –80 °C overnight. The frozen pellet was resuspended in preparation buffer (75 mM Tris-HCl, 300 mM NaCl, 5 mM DTT, 10 mM EDTA, 10% (v/w) glycerol, pH 8.0) containing a complete protease inhibitor cocktail (Roche, Mannheim, Germany) at 4 °C and sonicated 5 times for 60 s at 200 W. Cell debris was removed by centrifugation (24 000 × g, 20 min, 4 °C) and EDTA was removed by dialysis against PBS (pH 7.4) at 4 °C overnight. The EGF-MAP was purified by IMAC on an Äkta Purifier System (GE Healthcare, Freiburg, Germany) using IMAC Sepharose 6 Fast Flow resin and a XK16-20 column (GE Healthcare). Preparative SEC was carried out using an Äkta Purifier System (GE Healthcare) with a XK16-70 column packed with Superdex 75 (GE Healthcare), and a mobile phase of PBS (pH 7.4) at a flow rate of 1.0 ml min−1.
SDS–PAGE and western blot analysis
Protein enrichment and identity were determined by sodium dodecylsulphate polyacrylamide gel electrophoresis (SDS–PAGE) followed by Coomassie brilliant blue staining and western blotting, respectively, as previously described (Hetzel et al, 2008). The protein concentration was determined by densitometry against bovine serum albumin standards using AIDA software (Raytest GmbH, Straubenhardt, Germany) after staining with Coomassie brilliant blue, and was verified using the Bradford assay (Bio-Rad, Munich, Germany). The EGF-MAP was detected by western blot using mouse-anti-penta-His (1 : 5000; Qiagen, Hilden, Germany) for 45–60 min at room temperature. The primary antibody was detected using an alkaline phosphatase-conjugated anti-mouse-IgG mAb (1 : 5000; Sigma, Steinheim, Germany). Final staining was carried out using NBT/BCIP substrate (Life Technologies, Darmstadt, Germany). The molecular masses of the proteins were determined by comparing with pre-stained broad-range protein markers (New England BioLabs, Schwalbach, Germany).
Cell culture
Human pancreatic cancer cell line L3.6pl (Bruns et al, 1999), human prostate cancer cell lines PC-3 (ATTC: CRL-1435), C4-2 (kindly provided by Dr Professor Elsässer-Beile, University Hospital, Freiburg, Germany) and human embryonic kidney cell line HEK 293T (ATCC, Manassas, VA, USA: CRL-11268) were grown in RPMI-1640 medium supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine, 50 μg ml−1 penicillin and 100 μg ml−1 streptomycin at 37 °C, 100% humidity and 5% CO2.
Transfection of L3.6pl cells with Katushka-2
Transfection was carried out as described previously (Pardo et al, 2012). Briefly, we seeded 1 × 105 L3.6pl cells in a 12-well plate and transfected them with DNA encoding the far-red fluorescent protein Katushka-2 (pTag-Katushka2-N; Evrogen, Heidelberg, Germany) using Roti-Fect (Roth, Karlsruhe, Germany) according to the manufacturer's instructions. Transfected cells were selected with G418 (InvivoGen, Toulouse, France) and sorted using the FACS Vantage Cell Sorter (Becton Dickinson, Heidelberg, Germany).
Binding analysis by flow cytometry
The cell-binding activity of purified EGF-MAP was analysed by flow cytometry. We incubated 4 × 105 cells with 1 μM EGF-MAP in PBS (pH 7.4) containing 2 mM EDTA and 0.5% (w/v) BSA for 30 min on ice followed by washing with PBS. Fluorescence staining was performed using an anti-penta-His-Alexa Fluor 488 antibody (1 : 100; Qiagen, Germany) for 30 min on ice in the dark. Finally, the cells were washed twice with PBS and analysed on a FACS Calibur flow cytometer (Becton Dickinson, Germany).
In vitro cellular cytotoxicity
The cytotoxic effect of EGF-MAP was assessed by measuring the conversion of XTT to a water-soluble orange formazan dye. We seeded 1 × 105 cells per well into a 96-well microtitre plate and incubated the cells with various dilutions of the recombinant protein for 72 h at 37 °C, 5% CO2 and 100% humidity. We used 425(scFv)-ETA', human recombinant EGF and the EGFR– cell line HEK293 as controls. We added 50 μl XTT/phenanzine methosulphate (100 : 1; Serva and Sigma, Steinheim, Germany) to each well and incubated the plates as above for 3–4 h before measuring the absorbance at 450 and 630 nm using an Epoch Microplate Spectrophotometer (Biotek, Bad Friedrichshall, Germany). The concentration required to achieve 50% reduction of protein synthesis (IC50) relative to untreated control cells was calculated using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). All experiments were carried out in triplicate.
Apoptosis assay
An AnnexinV/propidium iodide assay was used to determine the pro-apoptotic impact of EGF-MAP. We incubated 2.5 × 105 cells per ml with 100 nM EGF-MAP in a 12-well plate (Greiner, Frickenhausen, Germany) for 48 h at 37 °C, 5% CO2 and 100% humidity. We used 425(scFv)-ETA' (10 nM) and EGFR– HEK293 cells as controls. After incubation, the cells were washed twice with PBS (pH 7.4) and stained with AnnexinV-FITC (eBioscience, Frankfurt, Germany) in AnnexinV binding buffer (10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM CaCl2) for 30 min at room temperature. Finally, the cells were washed again as described above, resuspended in AnnexinV buffer containing 10 μg ml−1 propidium iodide and analysed by flow cytometry using a FACS Calibur (Becton Dickinson, Germany).
Tubulin polymerisation assay
The stabilising effect of EGF-MAP on tubulin polymerisation was tested using a Tubulin Polymerization Assay Kit (tebu-bio, Offenbach, Germany) according to the manufacturer's instructions. Paclitaxel and general tubulin buffer were used as controls. All measurements were carried out in duplicate. Vmax represents the slope of the linear phase in milli-extinction (mE) per min.
In vivo analysis
Animal experiments were officially approved by the local Animal Care and Use Review Committee. All animals received humane care in accordance with the requirements of the German Tierschutzgesetz, §8 Abs. 1 and in accordance with the guide for the care and use of laboratory animals published by the National Institutes of Health in 2011. Animal experiments were carried out as described previously (Pardo et al, 2012). Briefly, 1 × 106 L3.6pl cells expressing Katushka-2 were suspended in ∼20 μl PBS (pH 7.4) and subcutaneously injected into the right hind limb of 6–8-week-old female Balb/C nu/nu mice (Charles River, Sulzfeld, Germany). For imaging experiments, mice were placed on a purified, chlorophyll-free diet (AIN93G, SSNIFF GmbH, Soest, Germany) 11 days before the imaging experiments began. The animals were injected intravenously with EGF-MAP (4 mg kg−1) or PBS (pH 7.4) according to the treatment regimen shown in Figure 6. Readouts were taken on the same days as the treatment, using the Maestro CRi optical imaging system (Maestro CRi Inc., Waltham, MA, USA). Images were analysed using the Maestro spectral imaging software as previously described (Kampmeier et al, 2010). The Katushka-2 signal was detected using the yellow filter set (630–850 nm).
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 5 (GraphPad Software). Data were expressed as the mean±s.d. or s.e.m. as indicated. Statistical comparisons were made using a two-tailed unpaired Student's t-test. *P⩽0.05, **P⩽0.01, ***P⩽0.001.
Results
Generation of EGF-MAP and characterisation of binding activity
We considered a range of candidate effector molecules for CFPs based on the need to use a fully human sequence with a low molecular weight suitable for fusion proteins, and with the ability to induce apoptosis in target cells without nonspecific toxicity to healthy cells (e.g., by nonspecific surface binding). Human MAP met these criteria. The protein exists in eight isoforms, all derived from a single precursor RNA by alternative splicing. We selected isoform 3 because this was the smallest protein that retained all four highly conserved microtubule-binding repeats.
In contrast to the hyperphosphorylated tau protein that can form paired helical filaments and neuronal tangles associated with Alzheimer's disease (Morris et al, 2011), MAP is unable to aggregate. However, the MAP amino acid sequence includes numerous potential phosphorylation sites, many of which regulate its activity. We removed two phosphorylation sites, S156 and S204, by site-directed mutagenesis, which have been shown to critically regulate microtubule binding (Schneider et al, 1999). Both serine residues were replaced with alanine by PCR. Furthermore, we added a C-terminal NLS derived from the SV40 T-antigen by adding the sequence (5′-CCCAAAAAAAAAAGGAAAGTG-3′) to the 3′ end of the ORF to foster nuclear enrichment of the internalised protein. The complete ORF was then transferred to the pMT vector (Figure 1A) linearised with NotI/BlpI, placing it in frame with an upstream ORF representing human EGF.
The construct was introduced into E. coli BL21 (DE3) cells and EGF-MAP was expressed at yields of up to 1 mg of purified protein per litre of bacterial culture. After one IMAC purification step and SEC, the enrichment and identity of EGF-MAP were determined by SDS–PAGE followed by Coomassie brilliant blue staining and western blotting, respectively (Figure 1B). Although purity was low, subsequent functional analyses could be carried out always including appropriate negative controls. Specific binding activity to L3.6pl cells was confirmed by flow cytometry, which also revealed the absence of nonspecific binding to the EGFR– HEK293 cells (Figure 1C).
EGF-MAP shows dose- and proliferation-dependent cytotoxicity towards L3.6pl cells
The in vitro cytotoxicity of EGF-MAP was determined using an XTT cell viability assay. The EGF-MAP showed specific toxicity towards L3.6pl cells with an IC50 value of 1 μM, whereas the EGFR– cell line HEK293 was unaffected (Figure 2). Recombinant human EGF was used as control with no adverse effects on cell viability. In addition to our model cell line L3.6pl, EGF-MAP was cytotoxic to other cancer cell lines expressing different levels of EGFR (Table 1). As we used EGF-MAP as a model protein to demonstrate the cytotoxic potential of MAP as an effector, we were not interested in establishing an elaborated purification protocol for this fusion protein, which might lead to underestimated IC50 values.
Figure 2.

Dose-dependent cytotoxicity of EGF-MAP. The XTT cell viability assays were carried out using EGF-MAP or human EGF ligand against L3.6pl and HEK293 cells. Nonlinear regression and IC50 calculations were performed using GraphPad Prism 5 software (GraphPad Software). Values are presented as means±s.d.
Table 1. The IC50 (μ M) of EGF-MAP and 425(scFv)-ETA' to cell lines with different EGFR levels.
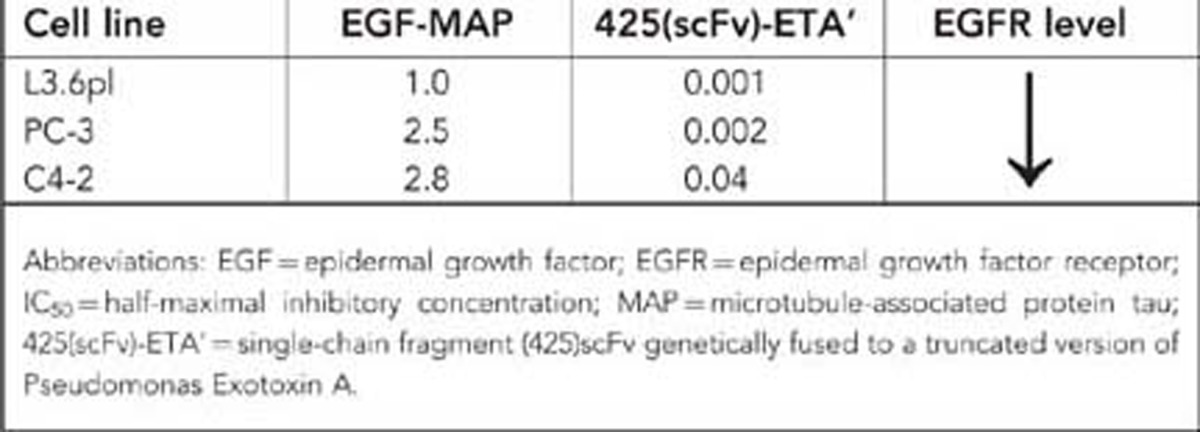
To test our hypothesis that MAP acts in a proliferation-dependent manner, we generated another fusion protein comprising MAP in combination with the EGFR-specific antibody fragment 425(scFv). The 425(scFv) is an EGFR-blocking antibody fragment. Antibodies that block EGFR have previously been shown to induce G1 arrest in EGFR-overexpressing cell lines, thus blocking proliferation (Kiyota et al, 2002). When we tested the 425(scFv)-MAP construct, we observed the binding to L3.6pl cells (data not shown) but no cytotoxicity (Figure 3A), confirming that the cytotoxicity of MAP is proliferation dependent. In contrast, the control immunotoxin 425(scFv)-ETA' was highly cytotoxic to L3.6pl cells (IC50=1 nM) even in their G1 arrested state (Figure 3A). To confirm this finding, we induced mitotic block in L3.6pl cells using a previously pre-titrated cytostatic but not apoptosis-inducing (Pasquier et al, 2004) concentration of paclitaxel (100 pM for 24 h) followed by the addition of 1 μM EGF-MAP. There was no cytotoxicity of EGF-MAP to nonproliferating L3.6pl cells, whereas apoptosis was strongly induced in L3.6pl cells that have not received paclitaxel before (Figure 3B).
Figure 3.

Cytotoxicity of EGF-MAP is proliferation dependent. (A) The XTT cell viability assays were carried out using 425(scFv)-MAP or 425(scFv)-ETA' against L3.6pl and HEK293 cells. Nonlinear regression and IC50 calculations were performed using GraphPad Prism 5 software (GraphPad Software). Values are presented as means±s.d. (B) Cell cycle arrest was achieved by preincubating L3.6pl cells with 100 pM paclitaxel for 24 h. The EGF-MAP (1 μM=IC50) failed to induce apoptosis in nonproliferating target cells. Values are presented as means±s.d. Statistical analysis was carried out using a two-tailed unpaired Student's t-test. ***P⩽0.001 compared with the corresponding PBS control.
EGF-MAP cytotoxicity involves the induction of apoptosis
The EGF-MAP mechanism of action was investigated by incubating L3.6pl cells with EGF-MAP followed by staining with AnnexinV-FITC and propidium iodide. As this assay was performed to demonstrate qualitatively the pro-apoptotic effect of the EGF-MAP, the concentration (100 nM for EGF-MAP and 10 nM for 425(scFv)-ETA') and the incubation time (72 h) were appropriately chosen to capture the cells within the early and late apoptotic stage. The population of early and late apoptotic cells (lower and upper right corner) increased significantly, demonstrating that the cytotoxicity of EGF-MAP towards L3.6pl cells reflects the induction of apoptosis (Figure 4A). There was comparatively little induction of apoptosis in HEK293 cells (Figure 4B).
Figure 4.

Apoptosis assays to determine the cytotoxicity mechanism of EGF-MAP. (A) Dot plots of L3.6pl and HEK293 cells after incubation with 100 nM EGF-MAP or 10 nM 425(scFv)-ETA' for 48 h. (B) Quantitative analysis of the dot blots shown in (A). Values are representative of three experiments and presented as means±s.d. Statistical analysis was carried out using a two-tailed unpaired Student's t-test. ***P⩽0.001 compared with the corresponding PBS control.
EGF-MAP stabilises microtubules in targeted cells
Apoptosis can be induced by modifying the polymerisation dynamics of microtubules, either by stabilising (e.g., paclitaxel) or destabilising them (e.g., nocodazole). The effector protein MAP was anticipated to interfere with mitosis by binding to and stabilising microtubules. To determine the mechanism of action, we carried out a tubulin polymerisation assay. A total of 1 μM (=IC50) EGF-MAP was able to stabilise microtubules effectively and promoted polymerisation, as reflected by the increased polymerisation velocity Vmax. As a control, 10 μM paclitaxel (concentration recommended by the manufacturer) was used (Figure 5).
Figure 5.

The EGF-MAP stabilises microtubules and promotes polymerisation. Tubulin polymerisation assay using 1 μM (=IC50) EGF-MAP, with general tubulin buffer and paclitaxel (10 μM) as controls. Vmax values were calculated using the slope of the linear phase (milli-extinction (mE)). Statistical analysis was carried out using a two-tailed unpaired Student's t-test. ***P⩽0.001 compared with the buffer control.
EGF-MAP is well tolerated and inhibits tumour growth in vivo
Before treatment experiments, EGF-MAP was tested in a dose-escalation study in tumour-free mice to determine the maximum tolerated dose. Three mice per group received 0.5, 1.0, 2.0 or 4.0 mg kg−1 of EGF-MAP intravenously on days 0, 1, 3 and 5. The overall health status and body weight of the mice was monitored daily. All mice survived and showed normal behaviour. No reduction of body weight was observed (Figure 6A).
Figure 6.

In vivo efficacy of EGF-MAP. (A) To determine the maximum tolerated dose of EGF-MAP in vivo, four different doses (0.5, 1.0, 2.0 and 4.0 mg kg−1) were administered to mice (n=3) on days 0, 1, 3 and 5. Values are presented as means±s.e.m. (B) Example of the detection of the Katushka-2 signal in vivo, with an arrow indicating the tumour. (C) Tumour areas of one representative mouse per group. (D) Comparison of tumour growth between untreated (PBS) and treated (EGF-MAP) groups. The mean tumour volumes of both groups were normalised to 100 on day 0. Values are presented as means±s.e.m. (n=5). Statistical analysis was carried out using a two-tailed unpaired Student's t-test. **P⩽0.01, ***P⩽0.001 compared with the respective PBS control.
To test the efficacy of EGF-MAP in vivo, we transfected L3.6pl cells with the far-red fluorescent protein Katushka-2, and injected 1 × 106 transfected cells subcutaneously into the right hind leg of 10 Balb/C nu/nu mice to induce tumours. We monitored tumour growth using the in vivo imaging system Maestro Cri, based on the premise that the total Katushka-2 signal correlates with the cell number. We tested the injected mice 11 days after inoculation with tumour cells, and all mice showed palpable tumours (Figure 6B). Mice were then randomly divided into two groups (n=5) that were allocated one treatment cycle with EGF-MAP (4 mg kg−1) or PBS (pH 7.4). Data are presented as means±s.e.m. Statistical analysis was carried out using a two-tailed unpaired Student's t-test (*P⩽0.05, **P⩽0.01, ***P⩽0.001). Our results clearly demonstrated that EGF-MAP caused a significant reduction in tumour growth compared with the PBS control group (Figure 6C and D).
Discussion
Progress towards the development of therapeutically effective ADCs, ITs and CFPs depends on the selection of an appropriate target and the corresponding binding component, on the efficacy of the cytotoxic payload and on the nature of the linkage between these two modalities. Most ADCs currently undergoing clinical evaluation contain small synthetic cytotoxic molecules, such as maytansinoids or auristatins (Sievers and Senter, 2013). The architecture of ITs is exclusively chimeric, comprising a protein ligand chemically or genetically linked to a cytotoxic protein (Kreitman et al, 2001, 2009). Both formats often suffer from inherent limitations that can either be encountered during synthesis (in the case of ADCs, where non-directed coupling produces heterogeneous products) or during testing (in the case of ITs, where non-human proteins such as ETA induce undesirable immune responses). To address these challenges, we have developed a new fully human CFP containing human MAP as the cytotoxic effector that kills cancer cells in a proliferation-dependent manner. The MAP was selected as a potential cytostatic effector against rapidly proliferating cancer cells based on its ability to regulate mitosis. It binds to microtubules directly through a common structural motif of 18 amino acids called microtubule-binding repeat (MBRs). These MBRs have a positive net charge that facilitates the interaction with negatively charged residues in tubulin (Jho et al, 2010). Binding to spindle microtubules during mitosis finally promotes microtubule assembly and stability (Weingarten et al, 1975). The intrinsic proliferative nature of cancer cells makes them a suitable target for this new effector protein. However, in addition to the selectivity conferred by using a cytotoxic effector that only affects proliferating cells, the inclusion of a targeting component ensures that the fusion protein is delivered only to cells expressing the target antigen. We tested EGF-MAP using EGFR-overexpressing L3.6pl cells as a model, as previously used to evaluate EGFR-targeted ITs such as 425(scFv)-ETA' (Bruell et al, 2005; Pardo et al, 2012). These in vitro tests confirmed the efficacious induction of apoptosis in L3.6pl cells. We then used in vivo optical imaging to determine the efficacy of EGF-MAP on the prevention of tumour growth in vivo (Kampmeier et al, 2010; Pardo et al, 2012). This approach allowed us to monitor the therapeutic effect of EGF-MAP in a single animal, thus reducing the number of total animals required per experiment. Tumour growth was significantly inhibited by EGF-MAP compared with the untreated control group.
Compared with the maximum tolerated dose of 425(scFv)-ETA' (1.0 mg kg−1) (Bruell et al, 2005), a dose of 4.0 mg kg−1 of EGF-MAP could be administered without adverse effects and a lethal dose was not reached. Cancer immunotherapy using protein-based therapeutics such as EGF-MAP will require repetitive administration, and thus the relatively high tolerability of CFPs make them superior to classical ITs.
Because ‘tau' is also the name of a protein associated with Alzheimer's diseases, it is important to emphasise that the MAP tau protein described herein is not able to form the pathologically relevant paired helical filaments and our proposed therapeutic strategy does not increase the risk of neurodegenerative disorders (Dickson, 2004; Feinstein and Wilson, 2005; Iovino et al, 2010; Morris et al, 2011). Because of removal of two critical phosphorylation sites, our MAP tau is not subject to hyperphosphorylation. Beyond that, the targeted nature of MAP-based CFPs prevents them from accumulating in the extracellular space of the central and peripheral nervous system.
The cytotoxic efficacy of human effector proteins is not yet comparable to that of bacterial ETA. The higher IC50 values we observed for MAP in our in vitro experiments partially reflects its lack of enzymatic activity (ETA is an ADP-ribosyl transferase that acts catalytically against its targets, whereas MAP binds stoichiometrically to microtubules). However, the cytotoxic activity of CFPs such as MAP could be improved by the insertion of an adapter sequence that could facilitate transfer of the effector molecule from the endosome to the cytosol (Hetzel et al, 2008). For example, it has been reported that only 3–4% of internalised ETA is ultimately translocated (Ogata et al, 1990). Improving the translocation efficiency could therefore significantly enhance the therapeutic potential of CFPs and make them even more suitable for clinical applications. Furthermore, we used the natural EGF ligand as the targeting component to provide a model suitable for testing of the new effector protein MAP. Potential differences in binding affinity between EGF and the anti-EGFR 425(scFv) might also contribute to disparities in efficacy. In regard to future development and improvements of EGFR-targeting CFPs, nonblocking anti-EGFR mAbs instead of EGF could also be used to improve the therapeutic efficacy of MAP.
Because of their human nature and the anticipated low immunogenicity, MAP-based CFPs could also be applied in indication areas beyond cancer. Non-life-threatening diseases, including many inflammatory and autoimmune disorders, still lack effective and targeted therapeutic approaches. The modular architecture of CFPs allows the rapid replacement of the binding component with new ligands, antibodies or antibody fragments, potentially allowing the targeting of proliferating inflammatory cells, such as macrophages and T cells (Hristodorov et al, 2012).
In conclusion, we have developed a new fully human CFP comprising the cytostatic effector MAP, allowing the induction of apoptosis specifically in proliferating target cells. Using EGF ligand as the binding component and L3.6pl cells as a model, we have demonstrated the cytotoxic potential of EGF-MAP in vitro and in vivo. Based on our findings, MAP-based immunotherapeutic reagents appear to be promising candidates for targeted immunotherapy in cancer and other immunological diseases.
Acknowledgments
We thank Reinhard Rosinke for the purification of EGF-MAP and Dr Richard M Twyman for critically reading the manuscript. This work was funded in part by the ForSaTum project, which is sponsored within the NRW-EU Ziel 2-Programm ‘Regionale Wettbewerbsfähigkeit und Beschäftigung 2007–2013' (ERFE).
Author Contributions
Conception and design: D Hristodorov, A Pardo, M Huhn and S Barth; acquisition of data: D Hristodorov, A-T Pham and R Mladenov; analysis and interpretation of data: D Hristodorov; writing, review and/or revision of the manuscript: D Hristodorov, R Fischer, S Barth and T Thepen; animal study supervision: A Pardo and T Thepen.
DH, RM, TT and SB are co-inventors on a corresponding patent assigned to Fraunhofer. The other authors declare no conflict of interest.
Footnotes
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
References
- Barth S, Huhn M, Matthey B, Klimka A, Galinski EA, Engert A. Compatible-solute-supported periplasmic expression of functional recombinant proteins under stress conditions. Appl Environ Microbiol. 2000;66 (4:1572–1579. doi: 10.1128/aem.66.4.1572-1579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323 (Pt 3:577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruell D, Bruns CJ, Yezhelyev M, Huhn M, Muller J, Ischenko I, Fischer R, Finnern R, Jauch KW, Barth S. Recombinant anti-EGFR immunotoxin 425(scFv)-ETA' demonstrates anti-tumor activity against disseminated human pancreatic cancer in nude mice. Int J Mol Med. 2005;15 (2:305–313. [PubMed] [Google Scholar]
- Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1 (1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter PJ, Senter PD. Antibody-drug conjugates for cancer therapy. Cancer J. 2008;14 (3:154–169. doi: 10.1097/PPO.0b013e318172d704. [DOI] [PubMed] [Google Scholar]
- Cassimeris L. The oncoprotein 18/stathmin family of microtubule destabilizers. Curr Opin Cell Biol. 2002;14 (1:18–24. doi: 10.1016/s0955-0674(01)00289-7. [DOI] [PubMed] [Google Scholar]
- Cawley DB, Herschman HR, Gilliland DG, Collier RJ. Epidermal growth factor-toxin A chain conjugates: EGF-ricin A is a potent toxin while EGF-diphtheria fragment A is nontoxic. Cell. 1980;22 (2 Pt 2:563–570. doi: 10.1016/0092-8674(80)90366-9. [DOI] [PubMed] [Google Scholar]
- Dickson DW. Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: cause or effect. J Clin Invest. 2004;114 (1:23–27. doi: 10.1172/JCI22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitroulis J, Stathopoulos GP. Evolution of non-small cell lung cancer chemotherapy (Review) Oncol Rep. 2005;13 (5:923–930. [PubMed] [Google Scholar]
- Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21 (1:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- Feinstein SC, Wilson L. Inability of tau to properly regulate neuronal microtubule dynamics: a loss-of-function mechanism by which tau might mediate neuronal cell death. Biochim Biophys Acta. 2005;1739 (2-3:268–279. doi: 10.1016/j.bbadis.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Giodini A, Kallio MJ, Wall NR, Gorbsky GJ, Tognin S, Marchisio PC, Symons M, Altieri DC. Regulation of microtubule stability and mitotic progression by survivin. Cancer Res. 2002;62 (9:2462–2467. [PubMed] [Google Scholar]
- Goncalves A, Tredan O, Villanueva C, Dumontet C. [Antibody-drug conjugates in oncology: from the concept to trastuzumab emtansine (T-DM1)] Bull Cancer. 2012;99 (12:1183–1191. doi: 10.1684/bdc.2012.1669. [DOI] [PubMed] [Google Scholar]
- Hetzel C, Bachran C, Fischer R, Fuchs H, Barth S, Stocker M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J Immunother. 2008;31 (4:370–376. doi: 10.1097/CJI.0b013e31816a2d23. [DOI] [PubMed] [Google Scholar]
- Hristodorov D, Mladenov R, Huhn M, Barth S, Thepen T. Macrophage-targeted therapy: CD64-based immunotoxins for treatment of chronic inflammatory diseases. Toxins (Basel) 2012;4 (9:676–694. doi: 10.3390/toxins4090676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhn M, Sasse S, Tur MK, Matthey B, Schinkothe T, Rybak SM, Barth S, Engert A. Human angiogenin fused to human CD30 ligand (Ang-CD30L) exhibits specific cytotoxicity against CD30-positive lymphoma. Cancer Res. 2001;61 (24:8737–8742. [PubMed] [Google Scholar]
- Iovino M, Patani R, Watts C, Chandran S, Spillantini MG. Human stem cell-derived neurons: a system to study human tau function and dysfunction. PLoS One. 2010;5 (11:e13947. doi: 10.1371/journal.pone.0013947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho YS, Zhulina EB, Kim MW, Pincus PA. Monte carlo simulations of tau proteins: effect of phosphorylation. Biophys J. 2010;99 (8:2387–2397. doi: 10.1016/j.bpj.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Kamath K. How do microtubule-targeted drugs work? An overview. Curr Cancer Drug Targets. 2007;7 (8:730–742. doi: 10.2174/156800907783220417. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA. 1993;90 (20:9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmeier F, Niesen J, Koers A, Ribbert M, Brecht A, Fischer R, Kiessling F, Barth S, Thepen T. Rapid optical imaging of EGF receptor expression with a single-chain antibody SNAP-tag fusion protein. Eur J Nucl Med Mol Imaging. 2010;37 (10:1926–1934. doi: 10.1007/s00259-010-1482-5. [DOI] [PubMed] [Google Scholar]
- Kiyota A, Shintani S, Mihara M, Nakahara Y, Ueyama Y, Matsumura T, Tachikawa T, Wong DT. Anti-epidermal growth factor receptor monoclonal antibody 225 upregulates p27(KIP1) and p15(INK4B) and induces G1 arrest in oral squamous carcinoma cell lines. Oncology. 2002;63 (1:92–98. doi: 10.1159/000065726. [DOI] [PubMed] [Google Scholar]
- Kreitman RJ. Immunotoxins for targeted cancer therapy. AAPS J. 2006;8 (3:E532–E551. doi: 10.1208/aapsj080363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, Fitzgerald DJ, Wilson WH, Pastan I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27 (18:2983–2990. doi: 10.1200/JCO.2008.20.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ, Pastan I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345 (4:241–247. doi: 10.1056/NEJM200107263450402. [DOI] [PubMed] [Google Scholar]
- Lambert JM. Drug-conjugated monoclonal antibodies for the treatment of cancer. Curr Opin Pharmacol. 2005;5 (5:543–549. doi: 10.1016/j.coph.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Li Z, Yu T, Zhao P, Ma J. Immunotoxins and cancer therapy. Cell Mol Immunol. 2005;2 (2:106–112. [PubMed] [Google Scholar]
- Ligon LA, Shelly SS, Tokito M, Holzbaur EL. The microtubule plus-end proteins EB1 and dynactin have differential effects on microtubule polymerization. Mol Biol Cell. 2003;14 (4:1405–1417. doi: 10.1091/mbc.E02-03-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maney T, Wagenbach M, Wordeman L. Molecular dissection of the microtubule depolymerizing activity of mitotic centromere-associated kinesin. J Biol Chem. 2001;276 (37:34753–34758. doi: 10.1074/jbc.M106626200. [DOI] [PubMed] [Google Scholar]
- Mathew M, Verma RS. Humanized immunotoxins: a new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009;100 (8:1359–1365. doi: 10.1111/j.1349-7006.2009.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70 (3:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris B, Pritchard KI, James K, Myles J, Bennett K, Marlin S, Skillings J, Findlay B, Vandenberg T, Goss P, Latreille J, Rudinskas L, Lofters W, Trudeau M, Osoba D, Rodgers A. Phase III comparative study of vinorelbine combined with doxorubicin versus doxorubicin alone in disseminated metastatic/recurrent breast cancer: National Cancer Institute of Canada Clinical Trials Group Study MA8. J Clin Oncol. 2000;18 (12:2385–2394. doi: 10.1200/JCO.2000.18.12.2385. [DOI] [PubMed] [Google Scholar]
- Ogata M, Chaudhary VK, Pastan I, FitzGerald DJ. Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J Biol Chem. 1990;265 (33:20678–20685. [PubMed] [Google Scholar]
- Pajk B, Cufer T, Canney P, Ellis P, Cameron D, Blot E, Vermorken J, Coleman R, Marreaud S, Bogaerts J, Basaran G, Piccart M. Anti-tumor activity of capecitabine and vinorelbine in patients with anthracycline- and taxane-pretreated metastatic breast cancer: findings from the EORTC 10001 randomized phase II trial. Breast. 2008;17 (2:180–185. doi: 10.1016/j.breast.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Pardo A, Stocker M, Kampmeier F, Melmer G, Fischer R, Thepen T, Barth S. In vivo imaging of immunotoxin treatment using Katushka-transfected A-431 cells in a murine xenograft tumour model. Cancer Immunol Immunother. 2012;61 (10:1617–1626. doi: 10.1007/s00262-012-1219-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier E, Carre M, Pourroy B, Camoin L, Rebai O, Briand C, Braguer D. Antiangiogenic activity of paclitaxel is associated with its cytostatic effect, mediated by the initiation but not completion of a mitochondrial apoptotic signaling pathway. Mol Cancer Ther. 2004;3 (10:1301–1310. [PubMed] [Google Scholar]
- Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6 (7:559–565. doi: 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- Saxton WM, Stemple DL, Leslie RJ, Salmon ED, Zavortink M, McIntosh JR. Tubulin dynamics in cultured mammalian cells. J Cell Biol. 1984;99 (6:2175–2186. doi: 10.1083/jcb.99.6.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38 (12:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- Stahnke B, Thepen T, Stocker M, Rosinke R, Jost E, Fischer R, Tur MK, Barth S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol Cancer Ther. 2008;7 (9:2924–2932. doi: 10.1158/1535-7163.MCT-08-0554. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Kano Y, Akutsu M, Tsunoda S, Izumi T, Yazawa Y, Miyawaki S, Mano H, Furukawa Y. The cytotoxic effects of gemtuzumab ozogamicin (mylotarg) in combination with conventional antileukemic agents by isobologram analysis in vitro. Anticancer Res. 2009;29 (11:4589–4596. [PubMed] [Google Scholar]
- ten Cate B, de Bruyn M, Wei Y, Bremer E, Helfrich W. Targeted elimination of leukemia stem cells; a new therapeutic approach in hemato-oncology. Curr Drug Targets. 2010;11 (1:95–110. doi: 10.2174/138945010790031063. [DOI] [PubMed] [Google Scholar]
- Tur MK, Neef I, Jost E, Galm O, Jager G, Stocker M, Ribbert M, Osieka R, Klinge U, Barth S. Targeted restoration of down-regulated DAPK2 tumor suppressor activity induces apoptosis in Hodgkin lymphoma cells. J Immunother. 2009;32 (5:431–441. doi: 10.1097/CJI.0b013e31819f1cb6. [DOI] [PubMed] [Google Scholar]
- von Minckwitz G, Harder S, Hovelmann S, Jager E, Al-Batran SE, Loibl S, Atmaca A, Cimpoiasu C, Neumann A, Abera A, Knuth A, Kaufmann M, Jager D, Maurer AB, Wels WS. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005;7 (5:R617–R626. doi: 10.1186/bcr1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10 (5:317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72 (5:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DP, Parker K, Bacha P, Bishai W, Borowski M, Genbauffe F, Strom TB, Murphy JR. Diphtheria toxin receptor binding domain substitution with interleukin-2: genetic construction and properties of a diphtheria toxin-related interleukin-2 fusion protein. Protein Eng. 1987;1 (6:493–498. doi: 10.1093/protein/1.6.493. [DOI] [PubMed] [Google Scholar]
- Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23 (9:1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Kronebusch PJ, Simon PM, Borisy GG. Microtubule dynamics at the G2/M transition: abrupt breakdown of cytoplasmic microtubules at nuclear envelope breakdown and implications for spindle morphogenesis. J Cell Biol. 1996;135 (1:201–214. doi: 10.1083/jcb.135.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Giannakakou P. Targeting microtubules for cancer chemotherapy. Curr Med Chem Anticancer Agents. 2005;5 (1:65–71. doi: 10.2174/1568011053352569. [DOI] [PubMed] [Google Scholar]