

Abstract
Motivation: The research area metabolomics achieved tremendous popularity and development in the last couple of years. Owing to its unique interdisciplinarity, it requires to combine knowledge from various scientific disciplines. Advances in the high-throughput technology and the consequently growing quality and quantity of data put new demands on applied analytical and computational methods. Exploration of finally generated and analyzed datasets furthermore relies on powerful tools for data mining and visualization.
Results: To cover and keep up with these requirements, we have created MeltDB 2.0, a next-generation web application addressing storage, sharing, standardization, integration and analysis of metabolomics experiments. New features improve both efficiency and effectivity of the entire processing pipeline of chromatographic raw data from pre-processing to the derivation of new biological knowledge. First, the generation of high-quality metabolic datasets has been vastly simplified. Second, the new statistics tool box allows to investigate these datasets according to a wide spectrum of scientific and explorative questions.
Availability: The system is publicly available at https://meltdb.cebitec.uni-bielefeld.de. A login is required but freely available.
Contact: nkessler@cebitec.uni-bielefeld.de
1 INTRODUCTION
Metabolomics research covers all aspects of the investigation of small molecule metabolite compositions resulting from cellular processes and constitutes an integrated part of systems biology (Bino et al., 2004). Like transcriptomics and proteomics, metabolomics is capable of measuring extrinsically initiated changes in organisms. The metabolome, the entity of all small molecules in a cell, organism or tissue, is considered to be the closest to the phenotype of all ‘-omes’ (Fiehn, 2002).
Compared with other molecular levels or -omics methods, metabolomics is challenging in its high degree of interdisciplinarity, interlinking experts from research fields as diverse as engineering, physics, chemistry and biology and from cheminformatics over bioinformatics to statistics, data mining and finally visualization.
Both sample acquisition and subsequent analysis are automated in high-throughput instruments, which has continuously posed challenges on the systematic storage and computational processing of the gathered experimental datasets, starting in the early 2000s. The increasing number and quality of measurements not only raised the generated data volume but also allowed to address more complex biological questions within conducted experiments. To comprehensively address these demands, bioinformatics internet applications were developed. MeltDB, ‘a software platform for the analysis and integration of data from metabolomics experiments’, has been published by Neuweger et al. (2008). Xia et al. (2009) released MetaboAnalyst, ‘a comprehensive tool suite for metabolomic data analysis’. Carroll et al. (2010) published the MetabolomeExpress web server as ‘a public place to process, interpret and share GC/MS metabolomics datasets’.
Since around 2008, we have observed that the requirements to comprehensive metabolomics software platforms have changed: The general growth of the field of metabolomics and the increasing number of collaborations diversified the user community of researchers and their individual scientific goals. It is obvious that the success of a metabolomics study depends on an efficient and effective collaboration of this interdisciplinary research community. Thus, not only the availability and sharing of the data is important but also special functions have to be significantly extended with specific features to consider all researcher’s demands and perspectives. In addition, the ever-increasing throughput and the constant lack of time makes it immensely important that automated pre-processing methods are reliable and that analyses and manual intervention are fast and easy. Since Metabolomics approaches are applied to more and more scientific objectives, a powerful set of statistical methods is mandatory, ranging from hypothesis-driven statistical tests to less specified and untargeted data-mining methods, such as clustering and dimension reduction. Finally, the wealth of generated data poses a necessity for exploratory data analysis tools and information visualization.
To tackle these new challenges systematically, a next generation of bioinformatics tools needed to be developed, covering all of the aforementioned aspects of metabolome data analysis, ranging from processing raw data (RD) to finishing and finally the derivation of biological knowledge. During the stages of that process, one can identify four successive data categories that represent different levels of data classification and annotation as well as different levels of abstraction. First, RD, stored and organized in meaningful groups, build the basis. Then, pre-processed data (PD) is computed, where peaks and their quantities have been detected. It follows integrated data (ID), where peaks that putatively originate from the same compound are consistently annotated over chromatograms of an experiment and thus become comparable. Last, derivative data (DD) is achieved by statistical analyses of metabolite quantities in an experiment and then visualized to allow effective exploration and to draw conclusions.
In this manuscript, we present MeltDB 2.0, which offers novel tools to challenge the rising wealth of data quality and quantity and support the analysis of all four categories RD, PD, ID and DD and includes a multitude of updates. New and improved preprocessing methods underpin the reliability of automatically created annotations. At the same time, straightforward tools for manual peak annotation simplify the curation even of large experiments. To help answering questions of different scientific objectives, the set of statistical analyses and data-mining tools has been strongly enriched. To finally nail down the quintessence of an experiments outcome, data exploration is supported by new interactive and telling information visualizations.
2 IMPLEMENTATION AND METHODS
The first version of the MeltDB software platform, a three-tiered web application and database server published in 2008 (Neuweger et al., 2008), provides means for the standardization, systematic storage and analysis of gas chromatography–mass spectrometry (GC-MS) metabolomics experiments. Within a powerful project and user management, raw chromatograms of various file formats can be uploaded and organized into chromatogram groups (e.g. replicates, factor levels) and experiments. A flexible processing pipeline allows to find, quantify and identify peaks in the raw chromatograms. Subsequently, a set of statistical tools and visualizations can be applied to analyze the gathered data tables. This fast growing, free online platform today hosts >25 distinct projects conducted by >150 registered users from around the world. More than 17 000 chromatograms have been uploaded and analyzed yet.
In the following, all major improvements to the entire process from RD to DD will be described in more details. Figure 1 summarizes the four stages of data processing and associates visualizations and data mining methods that can be performed in MeltDB 2.0 to each stage.
Fig. 1.

The overview shows the information processing in MeltDB 2.0 as well as visualizations and tools that are applicable to each level of data: RD, PD, ID and DD. Although different chromatogram viewers are available immediately after RD upload, heatmaps and data matrices can only be computed as soon as data have been integrated, i.e. there are peaks that are consistently named across chromatograms. To finally derive knowledge from the data, MeltDB 2.0 offers a versatile set of statistics and data-mining tools
2.1 From RD to PD: improved pre-processing
In metabolomics data analysis, pre-processing is a critical step, as ID and DD build on PD. To ensure a reliable data basis for statistical data exploration, MeltDB 2.0 is equipped with several new and updated algorithms for the early steps of experiment data analysis.
The growing list of pre-processing methods now includes support for the centWave algorithm by Tautenhahn et al. (2008) for chromatographic peak detection, which features a high sensitivity, and updates of the XCMS package (Smith et al., 2006) for chromatogram alignment and profiling analyses. In addition, the ChromA (Hoffmann and Stoye, 2009) software is added to the list of supported chromatogram alignment tools. ChromA computes pairwise alignments of chromatograms without a priori knowledge, but it is capable of optionally using previously matched or identified peaks as anchor points, which speeds up the process.
The calculation of retention time indices in GC-MS measurements is improved and can now also be performed manually using the web interface. Peaks of added substances can be assigned with retention indices and will be used as anchors for interpolating other peaks retention indices (Ettre, 1994), which support subsequent peak identification (Kopka et al., 2005). The detection of alkanes as retention markers can be automated.
Furthermore, peak identification itself is facilitated with a powerful feature: MeltDB 2.0 offers a new Reference list tool to save peaks of measured reference substances as Reference in the MeltDB database. The stored data comprises retention indices, quantification masses and mass spectra of reference compounds. This helps to generate project specific databases that complement the Golm Metabolite Database (http://gmd.mpimp-golm.mpg.de/) (Kopka et al., 2005) or the National Institute of Standards and Technology standard reference database 1A (http://www.nist.gov/srd/nist1a.cfm). The tool allows to aggregate References and to use their underlying mass spectra for efficient peak identification and comparison.
2.2 From PD to ID: profiling methods
To complete the first step towards ID, peaks in different chromatograms that derive from the same small molecule have to be named consistently and need to be associated to each other. Thus, a new support for GC-MS-based metabolite profiling experiments has been implemented. The focus for the profiling approach in MeltDB 2.0 is to combine the results from chemometrics approaches with further identification.
The generic MeltDB approach can be applied on netCDF, mzXML (Pedrioli et al., 2004) and mzDATA (Orchard et al., 2007) measurements from any supported analytical system. The novel tool registers peaks for non-targeted metabolite profiling based on multiple criteria. These are similarity of mass spectra, retention time difference and the existence of common extracted-ion chromatogram (EIC) peaks above a given signal-to-noise threshold. It allows to annotate completely unknown peaks that are consistently detectable in several measurements of a metabolomics experiment. Thus, these potentially interesting peaks can be subjected to further statistical analyses in MeltDB and become accessible for profiling experiments, where the aim is in general to find differences in the metabolic composition of two or more sample groups.
Parameterizations of all pre-processing tools can be customized freely from within the web interface. Parameterizations are persisted project wide so that other users from the same project—who typically use similar instrumental setups—can reuse them. As pre-processing methods are usually too long running tasks as to complete during a web request or to even allow interactivity, these jobs are forwarded to our compute cluster and run decoupled from the web server.
As soon as pre-processing is completed at least to the point of profiling, data tables can be exported as XLS or TSV files. This allows to subsequently use the MeltDB 2.0 processing results in other external programs of choice.
2.3 Analyzing PD: efficient mass decomposition
To elucidate single mass signals in a spectrum, e.g. to identify an unknown compound or to explain a certain fragmentation, potential sum formulas explaining this signal may be insightful. Thus, a combination of the efficient mass decomposition algorithm described by Böcker et al. (2006) with the filter criteria described by Kind and Fiehn (2007) has been implemented.
To improve the runtime of the approach and reduce the number of false positives, the maximal numbers of heteroatoms in the molecular formulae of metabolites in the mass ranges of 100 Da from 0 to 2000 Da have been extracted from public metabolite databases. These numbers act as upper limits for the molecular formula generation and drastically reduce the number of formulae that are evaluated by the algorithm.
To evaluate the runtime improvement of the newly implemented algorithm compared with the filter-free version, 200 compounds from the Kyoto Encyclopedia of Genes and Genomes (KEGG) compound database with specified sum formulas and known molecular masses were chosen in the mass interval from 0 to 1000 Da. For each of these compounds, mass decomposition was performed using the filter-free and the improved algorithm with a maximal mass deviation of 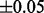 Da. Runtime information was obtained using a 64 bit 2600 MHz AMD system running Solaris OS 5.10. Figure 2 shows the runtime in relation to the molecular mass of the compound. It can be observed that the filtered version is up to seven times faster, and the improvement is observable especially for large molecules and masses.
Da. Runtime information was obtained using a 64 bit 2600 MHz AMD system running Solaris OS 5.10. Figure 2 shows the runtime in relation to the molecular mass of the compound. It can be observed that the filtered version is up to seven times faster, and the improvement is observable especially for large molecules and masses.
Fig. 2.

The improved algorithm was compared with the original mass decomposition approach. For compounds of molecular mass close to 1000 Da, 7-fold runtime improvements can be observed. As presented in the graph, the relation of the runtimes of both methods is not linear. Especially for large compounds, the improved variant becomes most beneficial
In addition to the runtime improvement, the filtering of the potential candidate formulae is applied afterwards to remove infeasible formulae from the generated candidate list. Simple filters can generally be computed in constant time (Degree of Unsaturation, Lewis check, Senior check, Heteroatom rule) (Kind and Fiehn, 2007), but especially the computation of theoretical isotope patterns needs at least 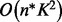 time for each sum formula with n being the size of the atom alphabet and K representing the length of the computed distribution (Böcker et al., 2006). The matching and scoring of the measured and simulated isotope patterns adds O(K) runtime. Every infeasible sum formula that is filtered out implicitly by the improved algorithm does not need to be post processed in the filtering stage, which furthermore improves the overall runtime of the method.
time for each sum formula with n being the size of the atom alphabet and K representing the length of the computed distribution (Böcker et al., 2006). The matching and scoring of the measured and simulated isotope patterns adds O(K) runtime. Every infeasible sum formula that is filtered out implicitly by the improved algorithm does not need to be post processed in the filtering stage, which furthermore improves the overall runtime of the method.
The improved and extended implementation is offered through the MeltDB 2.0 web interface, and it can be applied on every detected chromatographic peak. Users can specify the expected mass error of the instrument and correct the effect of potential adduct ions, which is especially important in LC-MS measurements. Several filters can be activated to reduce the number of computed sum formulas. As the implementation does automatically extract and sort, the dominant ions found in the mass spectrum, the access to the mass decomposition is greatly simplified for the researcher.
After the efficient generation and filtering step, the sum formulae are compared with the KEGG compound database. Matching sum formulae are highlighted, and both compound name and synonyms are presented to the user.
2.4 From RD to DD: new user interfaces
Visual inspection and the possibility for manual curation is important at all data abstraction levels, i.e. from RD to DD. Intuitive and responsive data visualizations are required as well as intelligent tools that allow to solve common tasks, such as the inspection of processing results or the navigation from an experiments overview to the spectrum of a single peak, in a few steps.
Introducing Asynchronous Javascripting and XML to MeltDB 2.0 using the jQuery library (http://jquery.com) allows a new quality of interactive and dynamic data display (The user can interact with visualizations to cause small changes to the data representation. These changes are performed in place without requiring a reload of the visualization) in terms of speed, responsiveness and user guidance. This was utilized to improve the Experiment Total Ion Current view [cmp. (Fig. 3b)] with peak specific tooltips containing information about the associated compounds, latest annotations, and quantities (Fig. 3b.1). On demand, the complete peak object can be loaded and displayed inside the experiment TIC view, giving i.a. access to its spectrum, the complete list of annotations and observations.
Fig. 3.

This figure presents representative screenshots from the MeltDB 2.0 interface, which are part of an efficient workflow for manual data curation and quality improvement. (a) The Show Absent/Present Compounds view lists which compounds were identified in which chromatogram groups in how many chromatograms (pie charts). Checked fields tell that the respective compound was identified in all chromatograms of that group. The darker column in the right represents the entire experiment. (b) The Experiment Total Ion Chromatogram view depicts the total intensities over time for all chromatograms of an experiment. Detected peaks are marked with colored dots. Blue: Detected peak; Green: peak that was consistently detected throughout chromatograms; Red: Peak that was identified using any database. Hovering a peak highlights other peaks (link-and-brush) with the same annotation and description and shows a tooltip with detailed information (b.1). From this view, semi-automated tools for annotation (c) can be accessed. The Annotate tool (screenshot shown) allows to manually define a description and select a KEGG compound. Similar peaks from other chromatograms are suggested and can be annotated in a batch. Alternatively, the MeltDB-own Warped Peak Detection can be applied to find and quantify peaks, which are similar to ones that are already annotated in other chromatograms using the tool ReQuantify Peaks for Exisiting Annotations. Sample names in the sreenshots were modified
One additional major improvement that benefits from the employment of Asynchronous Javascripting and XML is the dynamic manual annotation dialog [cmp. (Fig. 3c)]. The interactive annotation functionality for whole experiments has been improved so that aligned peaks with high mass spectral similarity can be annotated in parallel. The streamlined web interface helps to annotate peaks across chromatograms in a consistent manner, when researchers annotate manually, correct errors of automated annotation tools or correct data that have been imported beforehand. This consistency is ensured by an autocompletion of compound names according to the KEGG database and is important for statistical analyses and comparison of results among different experiments.
2.5 From ID to DD: extended statistics and data mining
In metabolomics, data analysis and mining can be driven by various intentions and will strive for DD with different purposes. One way to group these aims is to relate them to one of the following questions: (i) What are the significant features of a sample group separating it from other sample groups? (ii) Do groups of samples form clusters according to their features and quantities? (iii) Can a sample be assigned to a class based on its spectral features? For each of these questions, a variety of statistical analysis methods exists.
A binding to the R software (R Development Core Team, 2011) makes numerous statistical tools available from within the MeltDB software (Neuweger et al., 2008). Its database objects are converted to R objects in a standardized manner. Information about chromatogram associations to chromatogram groups is pertained in the data representation. This ID conversion avoids the cumbersome process of converting data tables from proprietary software to a format a statistics software package can interpret and analyze. By default, data are gathered from the database on the fly, but snapshots can be stored to speed up statistical analyses vastly, especially for large experiments.
Statistical analyses and data mining tools in MeltDB are accessed through a standardized parameterization and data selection form. Where appropriate, this basic form is extended with specific options and parameters. The basic form consists of a list of all chromatograms of the experiment from which the user may select. Additionally, features can be selected to be considered in the analysis. Features can either be identified compounds or unidentified features that have been detected consistently among most chromatograms. A feature or compound can be chosen as reference to normalize to. When available, ribitol is preselected. One can select whether peak intensities or peak areas will be used for quantitation. Quantitations can be scaled lineary or logarithmically. Missing values can be handled in different ways.
To determine the significance and variances of features (see question a), the t-test of the Perl CPAN package Statistics::TTest (Juan, 2003) and Statistics::KruskalWallis (Lee, 2003) is offered as well as analysis of variance using the aov method of the R statistical software (R Development Core Team, 2011), which fits a linear model. For all of these, Bonferroni, Holm and Benjamini & Hochberg corrections are calculated (Benjamini and Hochberg, 1995; Holm, 1979). For each feature (metabolite) presented in the analysis of variance and Kruskal–Wallis test results, a boxplot view and the extracted ion chromatograms of all samples can directly be accessed. In another view, the m-values (log-2 signal ratios) of features of all chromatogram groups of an experiment in reference to the same features in a user-selected chromatogram group are displayed tabularly. Volcano plots can be created plotting either the a-values (average log-2 signal values) or t-test values (as negative decadic logarithm of the P-value) against m-values. Variable importance estimation via the random forest algorithm from the caret R package (Kuhn et al., 2011) can be applied to find differing features in groups. The metabolite set enrichment analysis published by Persicke et al. (2011) is another powerful tool in MeltDB for the identification of differentially regulated metabolic pathways.
Samples may aggregate to clusters according to their features quantities (see question b), regardless of the groups they nominally belong to. To visualize these clusters, MeltDB provides the dimensionality reduction methods principal component analysis (PCA, prcomp method in R, cmp. Fig. 4b), independent component analysis (ICA, fastICA package for R) and partial least squares discriminant analysis (caret package for R) (Kuhn et al., 2011; Marchini et al., 2010; R Development Core Team, 2011). Hierarchical clustering allows to display dendrograms of chromatograms and is made available using the hclust method in R, which can be applied with different linkage methods (cmp. Fig. 4c). The heatmap method of R is used to show false color maps of feature signals in chromatograms, sorting columns and rows according to the before-mentioned hierarchical clustering of feature signals and chromatograms, respectively. Here, data can be normalized for either chromatograms or features.
Fig. 4.
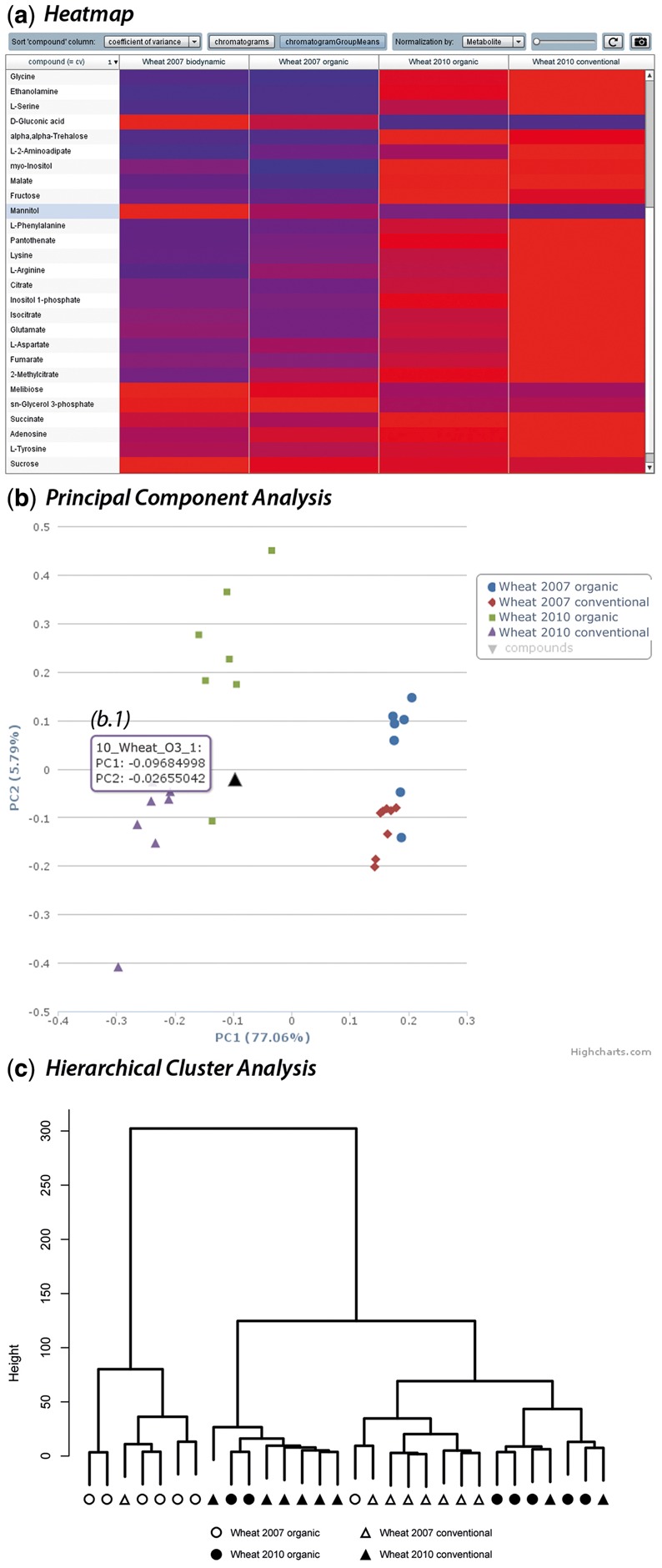
A small collection of visualization and statistical tools in MeltDB 2.0. (a) The Flash® heatmap visualization tabularizes color-encoded relative abundances of metabolites in either chromatograms or chromatogram groups (shown). Abundances may be normalized on the entire experiment or per metabolite (shown). Rows and columns can be sorted freely or automatically by alphabetical order or according to the coefficient of variation. A gain factor may be set to reveal differences between relatively small abundances. (b) The principal component analysis is one of the most applied work horses in MeltDB 2.0. The visualization is implemented using Highcharts components. Single chromatogram groups can be shown and hidden. A zoom functionality is available, which is extremely useful in large experiments. Each data point chromatogram name, and coordinates can be revealed via tooltip (b.1). (c) Another work horse of data mining is the hierarchical cluster analysis. The static visualization was generated in R. Sample names in the screenshots were modified
Whenever a dataset is subdivided in k groups (see question c), as samples were for instance taken from k sites or treated with k different protocols, another suitable data mining strategy is to apply supervised machine-learning methods to learn to approximate a relationship between metabolic profiles to the k categories (Hastie et al., 2009). Such a classification can be helpful in the design of automated screening and identification processes or give insight into hidden links in small molecule patterns, which are characteristic for a group k’. This propelled to extend MeltDB with the powerful R package caret (Kuhn et al., 2011) of which the variable importance estimation has been aforementioned. Now, classification algorithms (support vector machine, random forest) can be trained with chromatogram groups representing c different classes 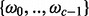 and then be applied to other chromatograms of samples that have not yet been assigned to any class
and then be applied to other chromatograms of samples that have not yet been assigned to any class 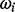 . For evaluation purposes, the user may opt to partition chromatograms into training and testing groups randomly. Additionally, MeltDB uses caret to compute and evaluate the classification performances of the algorithms random forest, k nearest neighbors, support vector machine, neural networks and partial least squares, to estimate which classification algorithm performs best on a problem.
. For evaluation purposes, the user may opt to partition chromatograms into training and testing groups randomly. Additionally, MeltDB uses caret to compute and evaluate the classification performances of the algorithms random forest, k nearest neighbors, support vector machine, neural networks and partial least squares, to estimate which classification algorithm performs best on a problem.
Generally, the computed results can be downloaded as TSV, XLS, PNG or PDF files in addition to the representation in the web browser.
2.6 Diving into DD: new interactive visualizations
To ultimately grasp the gist of gathered and calculated DD information, visualizations will be powerful tools, if they are intuitive, fast, responsive, easily customizable and—most important—well represent the underlying data.
The R software is not only useful for statistical analysis computation but also for the results visualization. Parameterization of the analyzes and customization of the visualizations is realized with MeltDBs tool forms, which can be easily extended on request or requirement. Nevertheless, these visualizations are static, and even small changes require server side computing and a page reload. This makes visual data exploration a time-consuming process. Therefore, new visualizations have been developed for MeltDB 2.0, which are based on the R data output and thus are consistent with their static ‘sister’ visualizations, but prepare the results in a novel dynamic and interactive fashion.
There are several web technologies for the development of rich internet applications (RIAs) available. Oracle®’s JavaTM Webstart applications, using the Java Network Launch Protocol, belong to the first RIAs created for the web (Farrell and Nezlek, 2007). The Adobe® Flash® technology and the Flash® Builder® made platform-independent RIA development even easier and provided great functionality for user interface design, backed by a large vivid community. Most recent changes in the industry, namely, the development of HTML 5, made Javascript-based RIA development applicable and seem to shorten the unique qualities of Flash. In the development of interactive content for MeltDB, these concepts have been followed.
A Java Webstart application is introduced, displaying the first three components of PCA and ICA results in a rotatable 3D grid. Samples are color encoded according to their chromatogram groups and metabolites contributing to the three components are represented in a bi-plot in the same grid. The 3D viewer is accessible through the web interface and allows to effectively explore dimensionality reduction results of hundreds of samples.
Basic visualizations such as scatter plots and bar charts are now realized as Javascript-based interactive views that allow to filter, zoom and demand additional details, following the information visualization mantra of Shneiderman and Plaisant (2004). For that, the Javascript libraries jQuery(http://jquery.com) and Highcharts (http://highcharts.com) were used. An interactive table of quantities was realized based on the AdobeTM FlashTM platform. Metabolite signal intensities or areas are projected to a color scale and displayed for each chromatogram or as the mean of a chromatogram group. The FlashTM application is available through the MeltDB web interface and receives the data table as an XML document provided by the MeltDB API. Parameterization that is necessary for the compilation of the data table can be assigned via the standard MeltDB tool form.
To compare raw 2D chromatograms visually, a new tool called ColorizeMS was created. Users can chose three chromatograms to assign each to one channel red, green or blue of an overlay image. Although common signals will appear in white, signals that are missing in one or two chromatograms can easily be spotted due to their coloration.
3 APPLICATION EXAMPLE AND RESULTS
This application example makes use of a dataset from another study (in preparation) in which we investigated whether it is possible to distinguish between wheat samples of different years, farming schemes and cultivars. The data shown in the following result from the GC-MS measurements of a wheat cultivar that was grown in the years 2007 and 2010 and under organic (O) and conventional (C) treatment. Consequently, samples can be divided into the four groups 2007-O, 2007-C, 2010-O and 2010-C. Each group consists of four biological replicates, which were each analyzed as two technical replicates. This results in a total of eight chromatograms per group. Chromatograms of low quality have been discarded.
The MeltDB 2.0 processing pipelines and its versatile tool box for statistical analyses was applied to reveal potential differences in the metabolic compositions of wheat from two distinct years and treatments. These insights may lead to the determination of bio markers that allow to distinguish between the respective sample groups. However, the focus of this application example is on the software and its capabilities, not on the biological interpretation of the generated results.
The MeltDB setup started by uploading the chromatograms (RD) in the netCDF file format. These were then organized into an experiment of four chromatogram groups. Peak detection was performed using the MeltDB Warped Peak Detection method. Alkanes were detected by the RISimple method, and peaks were thus provided with interpolated retention indices, completing the steps toward PD. Subsequently, Multiple Profiling was performed to consistently name peaks with similar spectra and retention indices throughout chromatograms of the experiment. Finally, to obtain ID, peaks were matched against the user-curated Reference List database to annotate identified peaks with the respective compound names.
The tool Show Absent/Present Compounds reveals a tabular presentation of pie charts, which shows in how many chromatograms of a particular chromatogram group or the entire experiment a particular compound was identified (cmp. Fig. 3a). Compounds that were identified automatically in the majority of chromatograms are likely to be found by manual inspection in the remaining measurements. This is easy owing to the mean retention time that is provided for each compound to find the respective peaks in the Experiment Total Ion Chromatogram view (cmp. Fig. 3b). Here, peaks with the same annotation and description are visually connected by link-and-brush (Highlighting an annotated peak will cause all other peaks with the same annotation and description to highlight too. This visually links signals that derive from the same original molecule throughout measurements). Peaks that were not annotated automatically (or annotated incorrectly) are easily spotted and may be annotated semi-automatically now. This is possible using the Annotate tool that is offered for each peak and allows to fill an annotation form manually, but it also allows to select peaks with similar spectra and similar retention time from other chromatograms to be annotated in the same way (cmp. Fig. 3c). Another tool, ReQuantify Peaks for Existing Annotations, provides means to locally run the Warped Peak Detection with relaxed thresholds and using existing annotations of a certain compound as samples to be matched. Using these tools, even large experiments of several hundred chromatograms can be annotated with high coverage in a reasonable time frame.
In the following, tools for visualization and statistical analysis are demonstrated that finally help to gather DD. In MeltDB 2.0, these tools may generally be adjusted with an upstream form for selection of chromatograms and compounds to be used. Furthermore, the user can set the specific parameters of each tool as well as the common settings, such as how to deal with missing zeros in the data, whether to use peak intensities or peak areas or whether to scale them logarithmically.
To get a first overview of the measured compound abundances among chromatograms of the experiment, the Heatmap visualization is a helpful choice (cmp. Fig. 4a). For a quick insight or visual control, all chromatograms and all compounds should be selected. Abundances are taken from peak areas, without additional scaling. In this case, missing values were replaced with zeros to make them easily spottable in the heatmap, as zero values are displayed as black boxes. The Heatmap tabularly represents all compound abundances in all chromatograms or their mean abundances in chromatogram groups according to a blue-to-red (low to high abundance) color scale. Rows (compounds) and columns (chromatograms or chromatogram groups) can be rearranged freely. Rows may additionally be sorted alphabetically, by mean abundance or by coefficient of variance. The color scale is either normalized on the whole experiment or on each single compound. The color scale may be shifted by a factor using a slider. The Heatmap as currently set can be exported as a PNG file.
When analyzing data of multiple—and as in this case even overlapping classes—it is generally interesting whether and which clusters are formed. Typical workhorses to explore this are Principal Component Analysis (pca) and Hierarchical Cluster Analysis (hca). Figure 4b shows a screenshot of the interactive scatter plot of the pca, which was calculated on all chromatograms, and all compounds that were detected in all chromatograms (option ‘strict’ for missing values). Measured peak areas were taken for quantification. Data points with the same color belong to the same chromatogram group. As the pca shows, samples are clearly separated by years along the first principal component (x-axis), which explains 75.24% of the variance in the data. Less clear but still visible is a separation according to the cultivation type, mainly oriented along the second principal component (y-axis), which explains 6.28% of the data. A mouse over on a data point reveals its chromatogram name and coordinates in the plot (cmp. Fig. 4b.1). Similarly, the hca as shown in Figure 4c mainly clusters data according to their years, whereas samples of the same cultivation type group cluster hardly at all. The hca was performed as a Ward clustering with the same basic settings as the pca.
4 DISCUSSION AND CONCLUSION
MeltDB 2.0 was developed to comprehensively provide means to complete the entire process from RD to DD within a software platform that supports researchers of diverse scientific backgrounds and fosters collaborations in complex metabolomics research projects. It was a further goal to make the final exploration of produced results and statistical outcomes effective and efficient. This has been achieved by improving the MeltDB tool set throughout all four stages RD, PD, ID and DD. These recent developments leveraged MeltDB to an interactive RIA that allows to generate high-quality datasets and to dive deep into their analyses.
As MeltDB was first published in 2008, a few other tools have been released that take a similar line. The MetaboAnalyst (2.0) web server offers a feature set similar to MeltDB, also supplying means to cover the pipeline from RD to DD. MetaboAnalyst is merely made for a one-time web service-like usage though, whereas MeltDB offers a project and user management that supports collaborative work, allows to manually refine and annotate processing results and stores data for documentation purposes and to support larger and/or long-term projects.
The MetabolomeExpress web server is dedicated to making reviewed datasets publicly available. For that, it offers a fixed pipeline and a set of statistical tools that comprises a clearly smaller set of features, compared with MeltDB or MetaboAnalyst.
The hierarchical data model of MeltDB 2.0 serves single-factorial experiment designs best. It is still possible to address even complex multi-factorial designs, but then a careful organization (sometimes multiple organizations) of chromatogram groups is necessary. The application example shown is a multi-factorial experiment differentiating wheat samples of different years and farming schemes. It is part of a study that additionally considers different cultivars as a third factor, which was also investigated in MeltDB 2.0.
Despite the advantages and opportunities of web platforms, this technology also has its drawbacks. The most critical aspect probably is the lack of tools, which lets users browse the original RD and its raw signals in a smooth interactive way as known from desktop applications. This can be of particular importance especially for de novo identification of molecules. Thus, metabolomics web platforms, including MeltDB 2.0, are mostly useful for experiments with large numbers of chromatograms, which ask for the statistical comparison of sample groups.
Web platforms are also still limited in the consequent analysis of LC-MS data. Here, spectral deconvolution is of critical importance to reveal the interrelationships of mass signals, which may lead to the identification of the original molecules. Although software like the R tool CAMERA (Kuhl et al., 2012) exists to address this, it has not yet been integrated into a larger metabolomics software platform. We are currently working to overcome that limitation.
Further developments also must include support for multi-stage (MSn) data, another inevitable tool for proper de novo identification.
From a more global and systemic point of view into the future, the potential of integrated analysis with other omics data needs to be explored more intensively. As an example, the MeltDB 2.0 API allows the ProMeTra (Neuweger et al., 2009) software to map relative metabolite abundances to pathway maps, together with either proteome or transcriptome data. Nevertheless, to our knowledge, there is no software available for truly statistical multiomics approaches.
In total, MeltDB has undergone substantial improvements in its capacity as a ‘one-stop-shop’ providing a wide spectrum of necessary tools to answer biological and statistical questions, beginning from chromatographics RD files. The addition of supervised machine-learning tools now allows to directly apply gathered knowledge for classification purposes. Embedded in its powerful permission management system, MeltDB 2.0 delivers a powerful bioinformatics package for detailed systemic metabolomics research projects.
ACKNOWLEDGEMENTS
The authors A. Goesmann and Tim W. Nattkemper both want to be considered as senior authors for this manuscript. The authors thank Leonhard J Stutz for his support as a research assistant. The authors appreciate the helpful comments by the anonymous reviewers of this manuscript very much. N.K. is supported by a fellowship from the CLIB Graduate Cluster Industrial Biotechnology. The authors also thank the BRF team for expert technical support.
Funding: German Federal Ministry of Food, Agriculture and Consumer Protection under the Federal Scheme of Organic Farming and other forms of sustainable agriculture [Project 08OE023].
Conflict of Interest: none declared.
REFERENCES
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B. 1995;57:289–300. [Google Scholar]
- Bino RJ, et al. Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 2004;9:418–425. doi: 10.1016/j.tplants.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Böcker S, et al. Decomposing metabolomic isotope patterns. Algorithms Bioinform. 2006;4175:12–23. [Google Scholar]
- Carroll AJ, et al. The MetabolomeExpress Project: enabling web-based processing, analysis and transparent dissemination of GC/MS metabolomics datasets. BMC Bioinformatics. 2010;11:376. doi: 10.1186/1471-2105-11-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettre L. New, unified nomenclature for chromatography. Chromatographia. 1994;38:521–526. [Google Scholar]
- Farrell J, Nezlek G. Information Technology Interfaces, 2007. ITI 2007. 29th International Conference. Cavtat, Croatia: 2007. Rich internet applications the next stage of application development; pp. 413–418. [Google Scholar]
- Fiehn O. Metabolomics–the link between genotypes and phenotypes. Plant Mol. Biol. 2002;48:155–171. [PubMed] [Google Scholar]
- Hastie T, et al. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. 2nd edn. Springer, New York Inc; 2009. [Google Scholar]
- Hoffmann N, Stoye J. ChromA: signal-based retention time alignment for chromatography-mass spectrometry data. Bioinformatics (Oxford, England) 2009;25:2080–2081. doi: 10.1093/bioinformatics/btp343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand. J.Stat. 1979;6:65–70. [Google Scholar]
- Juan YF. Statistics::TTest. 2003. Comprehensive Perl Archive Network (CPAN) module version 1.1. [Google Scholar]
- Kind T, Fiehn O. Seven golden rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 2007;8:105. doi: 10.1186/1471-2105-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopka J, et al. GMD@CSB.DB: the Golm Metabolome Database. Bioinformatics (Oxford, England) 2005;21:1635–1638. doi: 10.1093/bioinformatics/bti236. [DOI] [PubMed] [Google Scholar]
- Kuhl C, et al. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012;84:283–289. doi: 10.1021/ac202450g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn,M. Contributions from Jed Wing, Steve Weston, Andre Williams, Chris Keefer and Allan Engelhardt (2012). caret: Classification and Regression Training. R package version 5.05-004. http://CRAN.R-project.org/package=caret (6 August 2013, date last accessed)
- Lee M. Statistics::KruskalWallis. 2003. Comprehensive Perl Archive Network (CPAN) module version 0.01, http://search.cpan.org/∼mglee/Statistics-KruskalWallis-0.01/KruskalWallis.pm (6 August 2013, date last accessed) [Google Scholar]
- Marchini JL, et al. fastICA: FastICA Algorithms to perform ICA and Projection Pursuit. 2010. R package version 1.1-16. http://CRAN.R-project.org/package=fastICA (6 August 2013, date last accessed) [Google Scholar]
- Neuweger H, et al. MeltDB: a software platform for the analysis and integration of metabolomics experiment data. Bioinformatics (Oxford, England) 2008;24:2726–2732. doi: 10.1093/bioinformatics/btn452. [DOI] [PubMed] [Google Scholar]
- Neuweger H, et al. Visualizing post genomics data-sets on customized pathway maps by ProMeTra-aeration-dependent gene expression and metabolism of Corynebacterium glutamicum as an example. BMC Syst. Biol. 2009;3:82. doi: 10.1186/1752-0509-3-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, et al. Five years of progress in the Standardization of Proteomics Data 4th Annual Spring Workshop of the HUPO-Proteomics Standards Initiative April 23–25, 2007 Ecole Nationale Supérieure (ENS), Lyon, France. Proteomics. 2007;7:3436–3440. doi: 10.1002/pmic.200700658. [DOI] [PubMed] [Google Scholar]
- Pedrioli PG, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- Persicke M, et al. MSEA: metabolite set enrichment analysis in the MeltDB metabolomics software platform: metabolic profiling of Corynebacterium glutamicum as an example. Metabolomics. 2011;8:310–322. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2011. [Google Scholar]
- Shneiderman B, Plaisant C. Designing the User Interface: Strategies for Effective Human-Computer Interaction. 4th edn. Boston: Pearson Addison Wesley; 2004. [Google Scholar]
- Smith CA, et al. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- Tautenhahn R, et al. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics. 2008;9:504. doi: 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, et al. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37:W652–W660. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]