

Abstract
Motivation: Pyrosequencing technology provides an important new approach to more extensively characterize diverse sequence populations and detect low frequency variants. However, the promise of this technology has been difficult to realize, as careful correction of sequencing errors is crucial to distinguish rare variants (∼1%) in an infected host with high sensitivity and specificity.
Results: We developed a new approach, referred to as Indel and Carryforward Correction (ICC), to cluster sequences without substitutions and locally correct only indel and carryforward sequencing errors within clusters to ensure that no rare variants are lost. ICC performs sequence clustering in the order of (i) homopolymer indel patterns only, (ii) indel patterns only and (iii) carryforward errors only, without the requirement of a distance cutoff value. Overall, ICC removed 93–95% of sequencing errors found in control datasets. On pyrosequencing data from a PCR fragment derived from 15 HIV-1 plasmid clones mixed at various frequencies as low as 0.1%, ICC achieved the highest sensitivity and similar specificity compared with other commonly used error correction and variant calling algorithms.
Availability and implementation: Source code is freely available for download at http://indra.mullins.microbiol.washington.edu/ICC. It is implemented in Perl and supported on Linux, Mac OS X and MS Windows.
Contact: jmullins@uw.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Massively parallel sequencing (MPS) technologies, such as 454 pyrosequencing (Margulies et al., 2005), are becoming common to rapidly and cost-effectively detect and quantitate rare sequence variants. Pyrosequencing generates up to millions of reads that can include rare variants to detect low frequency drug resistance and immune escape variants in viral (Human immunodeficiency virus [HIV] and Simian immunodeficiency virus [SIV]) populations (Bimber et al., 2009, 2010; Burwitz et al., 2011; Fischer et al., 2010; Hedskog et al., 2010; Henn et al., 2012; Love et al., 2010; O'Connor et al., 2012; Poon et al., 2010; Simen et al., 2009; Tsibris et al., 2009; Wang et al., 2007). However, the PCR required before pyrosequencing of HIV/SIV populations introduces misincorporation errors, and the pyrosequencing process introduces a significant number of indels and carryforward errors (Margulies et al., 2005). To accurately estimate population diversity by MPS, it is crucial to distinguish biological variants from process errors with high sensitivity and specificity. Previous studies of 454 pyrosequencing data have managed to reduce sequence-processing errors by improving PCR and sequencing platforms (Gilles et al., 2011; Huse et al., 2007; Shao et al., 2013; Vandenbroucke et al., 2011; Wang et al., 2007). Also, several error correction and variant calling algorithms have been published (Archer et al., 2010; Bragg et al., 2012; Eriksson et al., 2008; Huse et al., 2010; Macalalad et al., 2012; Prosperi and Salemi, 2012; Quince et al., 2009, 2011; Ramirez-Gonzalez et al., 2013; Reeder and Knight, 2010; Salmela and Schröder, 2011; Wang et al., 2007; Zagordi et al., 2010a, b, 2011). Salmela and Schröder (Salmela and Schröder, 2011) used multiple alignments of reads as well as quality scores to distinguish correct base calls from erroneous ones, and their method is easily adjustable to reads derived from different MPS platforms. Prosperi and Salemi (Prosperi and Salemi, 2012) developed a program for viral population reconstruction with a built-in Poisson error correction method and post-reconstruction probabilistic clustering. Macalalad et al. (2012) introduced V-Phaser, a single nucleotide variant calling tool that uses phase and quality filtering with a probability model that incorporates and recalibrates individual base quality scores to increase both sensitivity and specificity. Sequence clustering is also a common way to reduce sequencing errors. One approach is to cluster sequences using genetic distances (Bragg et al., 2012; Huse et al., 2010). Another approach is based on flowgrams rather than sequences, which allows pyrosequencing errors to be modeled naturally, as performed by Quince et al.. (2009, 2011) and Reeder & Knight (2010). Both approaches require a distance cutoff value that combines substitutions, insertions and deletions in a single distance measure. The cluster centers are haplotypes, and the cluster sizes are interpreted as the haplotype frequency in the population. Error correction in a cluster is performed by collapsing variation within the cluster. Therefore, there is a risk for loss of real variants in a population if an inappropriate cutoff value is set, especially in a population of low genetic diversity.
To address the challenge of detecting genetic variants, especially those occurring at low frequencies, we developed a new approach that clusters sequences without substitutions and locally corrects only indel and carryforward errors within clusters to ensure that no rare variants are lost. Indel and Carryforward Correction (ICC) provides a complete suite for users to analyze pyrosequencing data, including read quality filtering and alignment, indel and carryforward error correction, variant calling and calculation of single nucleotide variant and haplotype frequencies. To determine the efficiency of ICC in correcting errors, we calculated and compared error rates before and after ICC correction on datasets derived from previously sequenced plasmid DNAs. Using pyrosequencing data of PCR fragments derived from a mixture of 15 HIV-1 plasmid clones at various frequencies, we used ICC to estimate sensitivity and specificity and compared these results with several other commonly used error correction and variant calling algorithms.
2 SAMPLE PREPARATION AND PYROSEQUENCING
2.1 Control datasets
pNL4-3, a plasmid containing a full-length HIV-1 genome, served as a control for pyrosequencing error calculations and comparisons. Two sets of first round PCR products were generated with primers that targeted portions of the gag-pol genes to produce a 2.6 kb amplicon and the env gp120 coding sequence for a 2.1 kb amplicon. Nested second round PCR reactions were multiplex PCRs that generated four amplicons (gag3, pol1, env3 and env5) from the mixture of two first round products that ranged from 391 to 597 bp in size with primers containing the 454 sequencing adapter and Multiplex Identifier (MID) adapter (Supplementary Table S1). PCR reactions are described in supplementary data.
2.2 HIV-1 plasmid mixture
Fifteen HIV-1 plasmid clones (Rousseau et al., 2006) were mixed to reach the final proportions (Supplementary Table S2), with individual DNA concentrations determined using a Nanodrop instrument (Thermo Scientific, USA). The plasmid mixture was further quantified by limiting dilution endpoint PCR (Rodrigo et al., 1997) with the program Quality (http://indra.mullins.microbiol.washington.edu/quality/). An estimated maximum of 1000 (691 ± 310) plasmid molecules were used as templates for PCR amplification and subsequent pyrosequencing. Primers and nested PCR conditions were the same as for pNL4-3 mentioned previously (and see Supplementary Data).
2.3 HIV-1 clinical sample
RNA was extracted from five plasma specimens from one HIV-1 infected individual from the Seattle Primary Infection Cohort (Schacker et al., 1996; Stekler et al., 2012) using the QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. A total of 560 µl of plasma was extracted in each case and eluted in 80 µl of elution buffer. cDNA was synthesized using Takara BluePrint First Strand Synthesis Kit (Clontech 6115A) according to the manufacturer’s protocol. cDNA was synthesized with gene specific primers, R3337-1 (5′-TTTCCYACTAAYTTYTGTATRTCATTGAC-3′) for gag-pol and R9048 (5′-AGCTSCCTTGTAAGTCATTGGTCTTARA-3′) for gp120, at final concentrations of 400 nM. Fragments of gag-pol (2.6 kb) and env (2.1 kb) were amplified in first round reactions separately, then mixed together and used as template for multiplex second round amplification of a 505 bp gag and a 597 bp env fragment (gag3 and env5) (Supplementary Table S1). PCR amplifications are described in supplementary data.
PCR products were visualized using a Qiaxcel (Qiagen, USA), purified using Agencourt AMPure beads (Beckman Coulter, USA) and then pyrosequenced on the 454 Life Sciences GS-FLX Titanium platform according to the manufacturer’s protocols.
3 ALGORITHM
3.1 Multiple sequence alignment
Sequences in a user-defined window were aligned using the Needleman–Wunsch algorithm (Needleman and Wunsch, 1970). First, sequences were collapsed so that only unique sequences were presented, and unique sequences were ranked by their abundance. The two most abundant unique sequences S1 and S2 were pairwise aligned using dynamic programming (Gusfield, 1997):
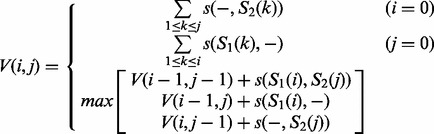 |
(1) |
V(i,j) is defined as the value of the optimal alignment of prefixes S1[1..i] and S2[1..j]. s(x,y) denotes the score obtained by aligning character x against character y. Second, the profile of the alignment was computed, taking into account the abundances of the aligned unique sequences. The third most abundant unique sequence was then aligned to the profile, which produced a new multiple sequence alignment including the first three most abundant unique sequences:
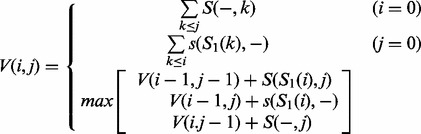 |
(2) |
V(i,j) denotes the value of the optimal alignment of prefix S1[1..i] with the first j columns of the profile. For a character y and column j, let p(y,j) be the frequency that character y appears in column j of the profile; S(x,j) denotes 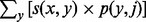 , the score for aligning x with column j. The process of calculating the profile of the newly produced multiple sequence alignment and aligning the next most abundant unique sequence to the updated profile was repeated until the last unique sequence was aligned. The default scoring parameters for alignment were as follows: match, 10; mismatch, -9; gap penalty, -15.
, the score for aligning x with column j. The process of calculating the profile of the newly produced multiple sequence alignment and aligning the next most abundant unique sequence to the updated profile was repeated until the last unique sequence was aligned. The default scoring parameters for alignment were as follows: match, 10; mismatch, -9; gap penalty, -15.
3.2 Computing sequence similarity and edit transcript
The similarity of two sequences, and associated optimal alignment and edit transcript, can be computed by dynamic programming (Gusfield, 1997). An edit transcript is a string over the alphabet I (insertion), D (deletion), R (replacement or substitution), M (match) that describes a transformation of one string to another. To compute the similarity of two sequences, a pairwise scoring matrix can be calculated by Equation (1). Therefore, the optimal edit transcript can be computed to describe the transformation between two sequences and distinguish sequence differences by insertion, deletion and substitution. Figure 1 shows an example of a pairwise alignment of two sequences S1 and S2, as well as the edit transcript. The differences between two sequences can be readily identified from their edit transcript. Moreover, the transcripts can be used along with the aligned sequences to distinguish indels in homopolymer and non-homopolymer regions, where a homopolymer is defined as two or more adjacent nucleotides with the same state, and to identify the pattern of carryforward errors.
Fig. 1.
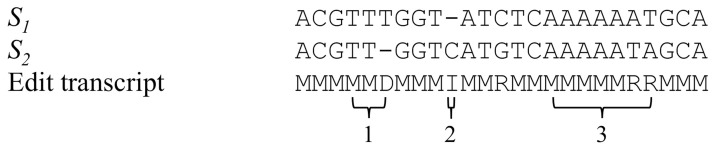
Pairwise alignment and edit transcript showing sequencing error patterns. ‘1’ homopolymer indel, ‘2’ non-homopolymer indel, ‘3’ carryforward error
4 IMPLEMENTATION
4.1 Error correction by ICC
ICC was written in the Perl scripting language and has been tested on Linux, Mac OS X and MS Windows systems. Starting with raw pyrosequencing reads and their quality scores, the software pipeline performs the following steps. The workflow of the implementation is shown in Figure 2.
Read quality filtering: Raw pyrosequencing reads are filtered based on ambiguous bases, length and average quality. The default parameters remove reads that are shorter than 100 bp, contain ambiguous bases or have average quality scores <25.
BLAST and retrieval by sequence window: Reads passing (i) are mapped to a reference sequence using the BLASTN algorithm (Altschul et al., 1990) with parameters for alignment as follows—match reward, 1; mismatch penalty, -1; gap existence, 1; gap extension, 2. User-defined window and stride size parameters retrieve windows of sequences across the reference sequence from the BLASTN output.
Non-substitution clustering and error correction: In each window of sequences, the similarity between each pair of sequences is computed using dynamic programming, along with an optimal edit transcript, which is then used for sequence clustering. Sequence errors are corrected through three sequential steps of non-substitution sequence clustering specifically designed for correction of homopolymer indels, indels and carryforward errors. A greedy scheme is used to cluster reads from the most to the least abundant. First, homopolymer indel errors are corrected by clustering sequences only differing by homopolymer indels. All sequences are condensed into unique sequences. Unique sequences with their abundance are used to perform sequence clustering. The most abundant sequence is used to cluster other sequences. Pairwise alignments of the most abundant sequence to all other sequences are computed along with the edit transcripts. The sequences with edit transcripts not containing substitutions and only showing the pattern of homopolymer indels relative to the most abundant sequence are clustered together with the most abundant sequence. For the next round, the most abundant sequence among the remaining sequences is chosen as a cluster seed, and the whole procedure iterates until the cluster seed reaches the first single sequence. Errors are corrected by collapsing variation within a cluster using the most abundant/consensus sequence for each cluster. The cluster size is now the abundance of the corrected sequence. Next, the sequences after homopolymer indel correction are further corrected for indel errors using the same strategy as homopolymer indel correction, except that it clusters sequences only differing in indels, i.e. sequences with edit transcripts not containing substitutions and only showing the pattern of indels to the most abundant sequence are clustered together with the most abundant sequence. Finally, after correcting homopolymer and non-homopolymer indels, carryforward errors are corrected by clustering sequences only showing carryforward patterns.
Variant calling and profiling: A Poisson probability model approximates the expected distribution of mismatch error rates to distinguish sequencing errors from authentic minor variants (Wang et al., 2007). The nucleotide variant frequencies at each site across the reference sequence are calculated. Local haplotypes in each window are constructed by the most abundant/consensus sequence in each cluster. The cluster size is interpreted as the haplotype frequency in the population.
Fig. 2.

Schematic of ICC workflow
4.2 Error rate calculation
Pyrosequencing reads that served as the control for error rate calculations were processed by read quality filtering [step (i) mentioned previously]. The remaining qualified reads were aligned to the pNL4-3 reference sequence using BLASTN with the alignment parameters as follows—match reward, 1; mismatch penalty, -1; gap existence, 1; gap extension, 2. Each type of error rate (number of errors/total number of mapped reference bases) was calculated by parsing the BLASTN output file, categorized by insertion, deletion and mismatch.
5 RESULTS AND DISCUSSION
5.1 Efficiency of sequencing error correction
To characterize the frequency of pyrosequencing errors and determine the efficiency of the ICC method in correcting errors, we sequenced a control dataset of six PCR-derived amplicons from the HIV-1 gag, pol and env genes within the pNL4-3 plasmid. We compared these reads with the Sanger-derived sequence of the clone. Table 1 shows the comparison of sequencing error frequencies of the six amplicons before and after error correction by ICC. These errors include both those introduced by PCR and those introduced during pyrosequencing. Overall error frequencies were reduced by 93–95%, whereas the frequencies of insertion and deletion errors were reduced by 98–99%, and mismatch errors were reduced by 48–71%, the latter due to correction of carryforward errors because no substitution mutations were corrected (see Section 5.2 later in the text).
Table 1.
Comparison of error rates before and after ICC
| Before correction |
After correction |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Amplicon | Size (bp) | Reads passing quality filter | Insertion (%) | Deletion (%) | Mismatch (%) | Overall (%) | Insertion (%) | Deletion (%) | Mismatch (%) | Overall (%) |
| Run3 gag3 | 383 | 16 987 | 0.2017 | 0.1242 | 0.0412 | 0.3671 | 0.0041 | 0.0011 | 0.0146 | 0.0198 |
| Run3 pol1 | 317 | 22 456 | 0.29 | 0.0358 | 0.0305 | 0.3563 | 0.0027 | 0.0005 | 0.0097 | 0.0129 |
| Run3 env3 | 269 | 21 271 | 0.2131 | 0.0299 | 0.0297 | 0.2727 | 0.0011 | 0.0001 | 0.0135 | 0.0147 |
| Run3 env5 | 443 | 20 593 | 0.1915 | 0.1585 | 0.0432 | 0.3932 | 0.0032 | 0.003 | 0.0125 | 0.0187 |
| Run4 gag3 | 383 | 8247 | 0.1602 | 0.055 | 0.0282 | 0.2434 | 0.0017 | 0.0008 | 0.0144 | 0.0169 |
| Run4 pol1 | 317 | 12 001 | 0.1348 | 0.0729 | 0.0254 | 0.2331 | 0.0024 | 0.0009 | 0.0131 | 0.0164 |
We also applied ICC to data from an individual (PIC64236) with HIV-1 infection. We pyrosequenced PCR-derived amplicons from the viral gag and env genes found in the patient at five different time points, and we analyzed variants by their frequency. Table 2 shows the analysis of one of the amplicons (gag3, 383 bp with primers trimmed). ICC reduced the number of variable sites by an average of 84%, and the overall trend was as expected for early HIV infection, with a slightly higher level of diversity early and then increasing diversity through time (Herbeck et al., 2011).
Table 2.
Called variants in HIV-1 clinical data by ICC
| Days post infection | Reads | Variable sites |
Reduction in variable sites (%) | |
|---|---|---|---|---|
| Raw data | After ICC | |||
| 29 | 5508 | 197 | 31 | 84.26 |
| 49 | 8281 | 223 | 12 | 94.62 |
| 146 | 9959 | 258 | 27 | 89.53 |
| 257 | 11 028 | 282 | 68 | 75.89 |
| 428 | 9863 | 282 | 74 | 73.76 |
Note: Pyrosequencing data were derived from a PCR amplicon from the HIV-1 gag gene from one infected individual at five time points over the first 14 months of infection. Variable sites correspond to aligned nucleotide positions with at least two different nucleotides.
5.2 Correction of carryforward errors
The 454 pyrosequencing is known to be particularly prone to errors in homopolymeric regions due to carryforward and incomplete extension errors (Margulies et al., 2005). Incomplete extension refers to a homopolymer that is not completed due to insufficient local nucleotide concentrations within a flow. Carryforward errors occur when reagent flushing between the flows is insufficient, and leftover nucleotides are introduced near but not adjacent to homopolymers. With commonly used parameters for alignment, including the settings we used, one carryforward error was usually interpreted as two mismatches when aligned to the reference sequence. Because most carryforward errors were found immediately 3′ to homopolymers, we investigated carryforward errors as a function of homopolymer length using the same control dataset of six amplicons. Carryforward errors increase as the length of the homopolymer increases, with a large increase noted with homopolymers of 6 nt in length (mean rate increased from 0.03 to 1.3% as homopolymer length increased from 3 to 6) (Fig. 3). According to these results, we conservatively corrected carryforward errors when found in up to 5% of reads. However, users can set different cutoff values. Because ICC does not correct mismatches, the reduction in mismatch errors noted above is attributable solely to the correction of carryforward errors.
Fig. 3.

Carryforward error rates as a function of different homopolymer lengths. Data were generated from six PCR-derived amplicons from the pNL4-3 plasmid clone in two separate pyrosequencing runs. Carryforward error rates were calculated by dividing the number of reads that contained carryforward errors by the total number of reads covering the homopolymer region
5.3 Comparison with other error correction and variant calling algorithms
We used several other error correction and variant calling programs to process our control datasets: V-Phaser (Macalalad et al., 2012), QuRe (Prosperi and Salemi, 2012), Coral (Salmela and Schröder, 2011), Acacia (Bragg et al., 2012) and the PyroNoise component of AmpliconNoise v1.25 (Quince et al., 2011). All compared algorithms were run using their default parameters. To compare sensitivity and specificity across the various algorithms, we mixed 15 HIV-1 plasmid clones of known sequence at frequencies ranging from 0.1 to 80% (Table 1). A 534 bp amplicon in gag was PCR amplified and subjected to pyrosequencing. After pyrosequencing, we detected 12 of the mixture of 15 plasmid clones with one at 80% frequency and the remaining 20% divided among the other 11 minor variants. Three minor variants mixed in at 0.1% frequencies were not detected, as expected, as we sequenced only 691 ± 310 templates. Because the Coral, Acacia and PyroNoise algorithms do not have methods to call variants, to fairly compare sensitivity and specificity among different programs, we set a frequency cutoff at 0.1% according to our experimental setup of ∼1000 input templates, i.e. only variants at frequencies ≥0.1% were considered when calculating sensitivity and specificity (Table 3). The specificity of each method was similar. However, ICC outperformed the other programs in sensitivity with each of the 11 minor variants detected.
Table 3.
Sensitivity and specificity for variant detection from different algorithms
| Program | Minor variants detected | Sensitivity | Specificity |
|---|---|---|---|
| PyroNoise | 5 | 0.4545 | 0.9957 |
| Acacia | 8 | 0.7273 | 0.9808 |
| Coral | 9 | 0.8182 | 0.9893 |
| QuRe | 8 | 0.7273 | 0.9976 |
| V-Phaser | 9 | 0.8182 | 0.9872 |
| ICC | 11 | 1 | 0.9827 |
Note: Sensitivity is reported as the fraction of the known variants found in the raw data by each correction algorithm. Specificity is reported as the fraction of sites not containing the known variants observed in the raw data. Variant calling required a frequency of ≥0.001 with total input copy number of 691 ± 310.
We also calculated the raw and corrected nucleotide frequencies from pyrosequencing data using PyroNoise, Acacia, Coral, QuRe, V-Phaser and ICC. Except for PyroNoise, we found good correlations between raw and corrected nucleotide frequencies of the expected variants (Fig. 4). PyroNoise and QuRe showed a relatively high specificity, indicating that they performed well in reducing sequencing noise, but PyroNoise had the lowest sensitivity among the six methods (Table 3). Furthermore, some variants detected by PyroNoise had much lower frequencies compared with raw and expected frequencies (Fig. 4A). Using QuRe on the same dataset, we obtained results over only a portion of the amplicon (423 bp of the full length of 479 bp), as this program trimmed the amplicon at both ends, where high-sequencing noise is usually detected. QuRe also used more relaxed settings by default in calling variants by Poisson distribution than ICC did. Thus, QuRe reached the highest specificity by removing more errors; on the other hand, it also eliminated more real rare variants, resulting in a lower sensitivity (Table 3). These results show that ICC is able to reduce sequencing noise to a large extent while retaining the correct frequencies of real variants.
Fig. 4.

Correlation between raw and corrected frequencies of real variants by different algorithms. (A) PyroNoise, (B) Acacia, (C) Coral, (D) QuRe, (E) V-Phaser and (F) ICC. Pyrosequencing data were derived from a 479 bp PCR-derived amplicon of a mixture of different copies of 15 HIV-1 clones of known sequence with total input copy number of 691 ± 310
PyroNoise uses distances defined by flowgrams to assist in removing pyrosequencing errors. In our studies, PyroNoise eliminated some high-frequency variants, whereas some mutations were retained no matter how low their frequency. We found that mutations resulting in a flow cycle change were never corrected, whereas others that maintained flow cycles were subject to correction. Figure 5 shows an example of these two different types of mutations. When we compared the distance between (I) and (II) with the distance between (I) and (III) at the sequence level, there was no difference, with both having one mismatch. But at the flowgram level, the distance between (I) and (II) was different from that between (I) and (III). With a mutation from (I) to (II), the flows were maintained and still aligned. But when a mutation from (I) to (III) occurs, the flows were changed by an insertion of one flow cycle (highlighted in bold type); therefore, the flowgrams were no longer aligned. We therefore defined an In-Flow-Cycle (IFC) mutation as one that does not change the number of flows, and an Out-Flow-Cycle (OFC) mutation as one that changes the number of flows. Although there is no difference in distance between IFC and OFC mutations at the sequence level, there is a difference at the flowgram level. Because of their misalignment, each cycle after an OFC mutation increases the distance between flowgrams. To verify this finding, we simulated flowgrams having different frequencies of minor variants with various numbers of IFC or OFC mutations. The results of PyroNoise correction using the program’s default parameter settings are shown in Table 4. Variants with frequency up to 19% were eliminated if there was one IFC mutation between major and minor variants. The frequency threshold of minor variants to be eliminated decreased as the number of IFC mutations increased. However, minor variants were not eliminated if there was a single OFC mutation distinguishing major and minor variants (Table 4). The results from sequencing the mixture of 15 HIV-1 plasmid clones also confirmed these findings (Fig. 4A). Thus, it is crucial to choose proper parameters to not eliminate rare and sometimes abundant variants when using PyroNoise to correct sequencing errors.
Fig. 5.

The effect of different mutations on flowgrams. Flowgram data corresponding to the sequences on the left, with nucleotides flowed in the order of TACG. The ‘:’ indicates a new flow cycle series of the four nucleotides. Mutated nucleotides are highlighted in bold type. Inserted flow cycle is also highlighted in bold type
Table 4.
Simulation of maximum frequency of minor variant to be eliminated by PyroNoise
| IFC mutations | OFC mutations | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 |
| 19% | 2% | 0.30% | 0.04% | 0 | 0 |
Note: Flowgrams were simulated based on an amplicon of 339 bp. IFC mutations represent mutations that do not change the number of flow cycles. OFC mutations will change the number of flow cycles.
5.4 Determination of minor variants
ICC does not correct mismatches, and as a result, it maximizes the number of real rare variants retained. To distinguish mismatch errors from authentic minor variants, we applied a statistical analysis that assumes a Poisson distribution in the frequency of these errors (Wang et al., 2007). Variants whose frequency of occurrence yielded a P < 0.001 according to the Poisson model were considered highly unlikely to be sequencing errors. Pyrosequencing error rate is largely dependent on the sequencing platform, PCR amplification and sequence context (Gilles et al., 2011; Huse et al., 2007; Shao et al., 2013; Vandenbroucke et al., 2011; Wang et al., 2007). Wang et al. (2007) measured mismatch error rates using a Roche 454 GS20 sequencing platform and found that mismatches were six times more frequent in homopolymeric regions (0.0044) than in non-homopolymeric regions (0.0007). Therefore, they used two Poisson distributions of errors to distinguish sequence errors from authentic minor variants in homopolymeric and non-homopolymeric regions, respectively. Considering that carryforward error is a major source of 454 pyrosequencing errors and one carryforward error is usually interpreted as two mismatches when aligned to reference sequence, we asked whether the difference in mismatch rate between the two contexts could be caused by carryforward errors in homopolymeric regions. To test this, we examined six amplicons on two separate pyrosequencing runs using the 454 GS-FLX Titanium platform. We calculated different types of errors and categorized them according to their sequence context (inside or outside of homopolymer regions) by distinguishing carryforward errors from substitutions in alignments (Table 5). The average mismatch error rate of the six amplicons, excluding carryforward errors, was 0.00013. The rates of mismatch errors were equivalent in non-homopolymeric regions (0.00013) and homopolymeric regions (0.00012) when carryforward errors were correctly aligned to the reference. After ICC correction of indels and carryforward errors, we therefore required only one distribution of mismatch error rates, which approximated a Poisson distribution with µ = 0.00013. We then used this empirically observed distribution to distinguish sequence errors from authentic minor variants. Within ICC, users can provide specific values according to different sequencing platforms and PCR conditions.
Table 5.
Error frequency by type and sequence context
| Amplicon | Type |
Context |
||||
|---|---|---|---|---|---|---|
| Insertion (%) | Deletion (%) | Mismatch (%) | Overall (%) | Non-homopolymeric mismatch (%) | Homopolymeric mismatch (%) | |
| Run3 gag3 | 0.2861 | 0.1535 | 0.0121 | 0.4517 | 0.0097 | 0.02 |
| Run3 pol1 | 0.3496 | 0.0678 | 0.0114 | 0.4288 | 0.0115 | 0.011 |
| Run3 env3 | 0.261 | 0.04 | 0.0125 | 0.3135 | 0.0138 | 0.0039 |
| Run3 env5 | 0.3254 | 0.19 | 0.014 | 0.5294 | 0.0134 | 0.0171 |
| Run 4 gag3 | 0.2134 | 0.082 | 0.0141 | 0.3095 | 0.0151 | 0.011 |
| Run4 pol1 | 0.1729 | 0.1042 | 0.0127 | 0.2898 | 0.0135 | 0.0097 |
| Average | 0.2681 | 0.1063 | 0.0128 | 0.3871 | 0.0128 | 0.0121 |
In conclusion, ICC provides a complete software pipeline for users to analyze pyrosequencing data for both library and amplicon applications. It is specifically designed to correct indel and carryforward and incomplete extension errors in 454 pyrosequencing data and avoid the elimination of real variants during error correction by a novel approach of non-substitution clustering without need of a distance cutoff value. ICC can be applied in other next-generation sequencing platforms such as Ion Torrent and Illumina. Ion Torrent has similar technology and the same source of errors as 454 pyrosequencing. ICC is able to analyze Ion Torrent data with the same efficiency as 454 pyrosequencing data. It can also be run on Illumina data, although there is low indel error in Illumina platform. By applying a Poisson probability model, ICC is able to distinguish sequencing errors from authentic minor variants and remove sequencing noise to a large extent. With the high sensitivity and specificity achieved by ICC, it should expedite analysis of variable sequence populations.
Funding: This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health [PO1AI057005, UM1AI068618 and R37AI47734 to J.I.M.], including the University of Washington Centers for AIDS Research Computational Biology Core [P30AI027757].
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Altschul SF, et al. Basic local alignment search tool J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Archer J, et al. The evolutionary analysis of emerging low frequency HIV-1 CXCR4 using variants through time—an ultra-deep approach. PLoS Comput. Biol. 2010;6:e1001022. doi: 10.1371/journal.pcbi.1001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bimber BN, et al. Ultradeep pyrosequencing detects complex patterns of CD8+ T-lymphocyte escape in simian immunodeficiency virus-infected macaques. J. Virol. 2009;83:8247–8253. doi: 10.1128/JVI.00897-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bimber BN, et al. Whole-genome characterization of human and simian immunodeficiency virus intrahost diversity by ultradeep pyrosequencing. J. Virol. 2010;84:12087–12092. doi: 10.1128/JVI.01378-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg L, et al. Fast, accurate error-correction of amplicon pyrosequences using Acacia. Nat. Methods. 2012;9:425–426. doi: 10.1038/nmeth.1990. [DOI] [PubMed] [Google Scholar]
- Burwitz BJ, et al. Pyrosequencing reveals restricted patterns of CD8+ T cell escape-associated compensatory mutations in simian immunodeficiency virus. J. Virol. 2011;85:13088–13096. doi: 10.1128/JVI.05650-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson N, et al. Viral population estimation using pyrosequencing. PLoS Comput. Biol. 2008;4:e1000074. doi: 10.1371/journal.pcbi.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One. 2010;5:e12303. doi: 10.1371/journal.pone.0012303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles A, et al. Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC genomics. 2011;12:245. doi: 10.1186/1471-2164-12-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusfield D. Algorithms on Strings, Trees, and Sequences: Computer Science and Computational Biology. Cambridge CB2 1RP: Cambridge University Press; 1997. [Google Scholar]
- Hedskog C, et al. Dynamics of HIV-1 quasispecies during antiviral treatment dissected using ultra-deep pyrosequencing. PLoS One. 2010;5:e11345. doi: 10.1371/journal.pone.0011345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn MR, et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012;8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbeck JT, et al. Demographic processes affect HIV-1 evolution in primary infection before the onset of selective processes. J. Virol. 2011;85:7523–7534. doi: 10.1128/JVI.02697-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, et al. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, et al. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love TM, et al. Mathematical modeling of ultradeep sequencing data reveals that acute CD8+ T-lymphocyte responses exert strong selective pressure in simian immunodeficiency virus-infected macaques but still fail to clear founder epitope sequences. J. Virol. 2010;84:5802–5814. doi: 10.1128/JVI.00117-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macalalad AR, et al. Highly sensitive and specific detection of rare variants in mixed viral populations from massively parallel sequence data. PLoS Comput. Biol. 2012;8:e1002417. doi: 10.1371/journal.pcbi.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- O'Connor SL, et al. Conditional CD8+ T cell escape during acute simian immunodeficiency virus infection. J. Virol. 2012;86:605–609. doi: 10.1128/JVI.05511-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon AF, et al. Phylogenetic analysis of population-based and deep sequencing data to identify coevolving sites in the nef gene of HIV-1. Mol. Biol. Evol. 2010;27:819–832. doi: 10.1093/molbev/msp289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosperi MC, Salemi M. QuRe: software for viral quasispecies reconstruction from next-generation sequencing data. Bioinformatics. 2012;28:132–133. doi: 10.1093/bioinformatics/btr627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, et al. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- Quince C, et al. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Gonzalez R, et al. PyroClean: denoising pyrosequences from protein-coding amplicons for the recovery of interspecific and intraspecific genetic variation. PLoS One. 2013;8:e57615. doi: 10.1371/journal.pone.0057615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat. Methods. 2010;7:668–669. doi: 10.1038/nmeth0910-668b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo AG, et al. Quantitation of target molecules from polymerase chain reaction-based limiting dilution assays. AIDS Res. Hum. Retroviruses. 1997;13:737–742. doi: 10.1089/aid.1997.13.737. [DOI] [PubMed] [Google Scholar]
- Rousseau C, et al. Large-scale amplification, cloning and sequencing of near full-length HIV-1 subtype C genomes. J. Virol. Methods. 2006;136:118–125. doi: 10.1016/j.jviromet.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Salmela L, Schröder J. Correcting errors in short reads by multiple alignments. Bioinformatics. 2011;27:1455–1461. doi: 10.1093/bioinformatics/btr170. [DOI] [PubMed] [Google Scholar]
- Schacker T, et al. Clinical and epidemiologic features of primary HIV infection. Ann. Intern. Med. 1996;125:257–264. doi: 10.7326/0003-4819-125-4-199608150-00001. [DOI] [PubMed] [Google Scholar]
- Shao W, et al. Analysis of 454 sequencing error rate, error sources, and artifact recombination for detection of Low-frequency drug resistance mutations in HIV-1 DNA. Retrovirology. 2013;10:18. doi: 10.1186/1742-4690-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simen BB, et al. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 2009;199:693–701. doi: 10.1086/596736. [DOI] [PubMed] [Google Scholar]
- Stekler JD, et al. Are there benefits to starting antiretroviral therapy during primary HIV infection? Conclusions from the Seattle Primary Infection Cohort vary by control group. Int. J. STD AIDS. 2012;23:201–206. doi: 10.1258/ijsa.2011.011178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsibris AM, et al. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One. 2009;4:e5683. doi: 10.1371/journal.pone.0005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbroucke I, et al. Minor variant detection in amplicons using 454 massive parallel pyrosequencing: experiences and considerations for successful applications. Biotechniques. 2011;51:167–177. doi: 10.2144/000113733. [DOI] [PubMed] [Google Scholar]
- Wang C, et al. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 2007;17:1195–1201. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagordi O, et al. ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics. 2011;12:119. doi: 10.1186/1471-2105-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagordi O, et al. Deep sequencing of a genetically heterogeneous sample: local haplotype reconstruction and read error correction. J. Comput. Biol. 2010a;17:417–428. doi: 10.1089/cmb.2009.0164. [DOI] [PubMed] [Google Scholar]
- Zagordi O, et al. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Res. 2010b;38:7400–7409. doi: 10.1093/nar/gkq655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.