

Abstract
Adenosine deaminase-deficient severe combined immunodeficiency was the first disease investigated for gene therapy because of a postulated production or survival advantage for gene-corrected T lymphocytes, which may overcome inefficient gene transfer. Four years after three newborns with this disease were given infusions of transduced autologous umbilical cord blood CD34+ cells, the frequency of gene-containing T lymphocytes has risen to 1–10%, whereas the frequencies of other hematopoietic and lymphoid cells containing the gene remain at 0.01–0.1%. Cessation of polyethylene glycol-conjugated adenosine deaminase enzyme replacement in one subject led to a decline in immune function, despite the persistence of gene-containing T lymphocytes. Thus, despite the long-term engraftment of transduced stem cells and selective accumulation of gene-containing T lymphocytes, improved gene transfer and expression will be needed to attain a therapeutic effect.
Adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID) became an early disease candidate for studies of gene therapy based on the results of allogeneic bone marrow transplant. Transplantation of bone marrow from a fully HLA-matched, unaffected sibling donor allows SCID to be cured completely1,2. Patients develop a protective immune system of donor-derived T lymphocytes, despite the absence of significant numbers of donor-derived cells in other hematopoietic lineages3. This observation has been interpreted as indicating that the normal T-lymphoid cells have a selective advantage in SCID patients and repopulate the immune system from a small number of engrafted, genetically-normal donor hematopoietic stem cells. The selective advantage for normal T and B cells in SCID patients has been recently re-confirmed in two SCID patients (one with ADA-deficient SCID and one with the X-linked form) who had spontaneous improvements in their immune function4-5. Gene-correction of a small number of autologous hematopoietic stem cells could similarly result in the development of a functional immune system in SCID gene therapy recipients.
In May 1993, we treated three newborns with ADA-deficient SCID with a single infusion for each of retroviral vector-transduced autologous umbilical cord blood CD34+ cells. Initial observations during the first 18 months after this intervention showed the persistent presence of leukocytes in their peripheral blood and bone marrow which contained and expressed the inserted normal human ADA cDNA (ref.6). This observation was important because it provided convincing evidence that human hematopoietic stem cells could be transduced with retroviral vectors and re-engrafted in their hosts without administration of marrow cytoreductive conditioning. The low percentages of blood cells containing the gene reflected the present severely limited ability to transduce pluripotent human hematopoietic stem cells with murine retroviral vectors, and the lack of cytoreduction.
It is difficult to determine whether any of the subjects in this or other trials of gene therapy for ADA-deficient SCID have had improvements to their immune systems as a direct result of gene transfer, because all subjects have simultaneously been given exogenous enzyme replacement therapy in addition to the gene transfer7-10. Polyethylene glycol-conjugated ADA (PEG-ADA), injected intramuscularly twice each week, has been used successfully as the sole therapy in more than 50 ADA-deficient SCID subjects, with moderate-to-complete normalization of immune function in most subjects11. Thus, the relative contributions to immunity and health seen in subjects receiving both gene-modified cells as well as PEG-ADA therapy is difficult to discern.
To determine whether our subjects have had any immune reconstitution as a result of the presence of genetically corrected cells, we have progressively decreased their dosages of PEG-ADA during the past two-and-a-half years, while observing clinical signs, parameters of purine metabolism, immune cell numbers and function, and the frequency of gene-containing leukocytes. In one subject, enzyme therapy was stopped completely to assess the degree of immune function resulting solely from the progeny of the transduced hematopoietic stem cells.
Clinical course
The collection of autologous umbilical cord blood from the three ADA-deficient neonates, isolation of CD34+ cells, transduction with the LASN retroviral vector carrying normal human ADA cDNA and transplantation of the cells has been described6. Within the first few days after birth, the newborns were also started on enzyme replacement with PEG-ADA. Treatments with ancillary measures such as prophylactic trimethoprim-sulfamethoxazole and intravenous gammaglobulin were administered as determined by the responsible primary pediatric immunologists; this varied over time among the three subjects.
During the five years of our study, the subjects have had normal growth and development, with no severe infections. They have led generally unrestricted lives, and have attended pre-school without isolation precautions. Subject 1 has had recurrent otitis media and also had clinically diagnosed varicella-zoster on two occasions, at six months of age and again at 3 years. The clinical course of the varicella-zoster was mild and was treated with oral acyclovir in both instances.
Biochemical studies related to ADA deficiency and therapy
Treatment was started on days one to four after birth, with 30 U/kg PEG-ADA administered twice each week (Fig. 1a). This dosage maintained trough plasma ADA activity at 62–102 μmol/hr per ml, based on multiple blood samples from each subject during the first year (Fig. 1b). These levels are >1000 times the activity of ADA in normal plasma and >10 times the activity in normal whole blood (where >98% of the ADA activity is in erythrocytes). In pre-treatment samples, erythrocyte deoxyadenosine nucleotides (dAXP) were significantly elevated at 0.21–0.32 μmol/ml packed red blood cells (RBC; normal, <0.002). By two months of treatment, dAXP had declined to <0.01 μmol/ml and remained barely detectable as long as PEG-ADA therapy was continued (data not shown). Erythrocyte S-adenosylhomocysteine hydrolase activity was decreased in pre-treatment samples from each patient, ranging from 0.2 to 0.6 nmol/h per mg protein (normal, 4.2 ± 1.9). With PEG-ADA therapy, these values increased to 3–6 nmol/h per mg protein.
Fig. 1.
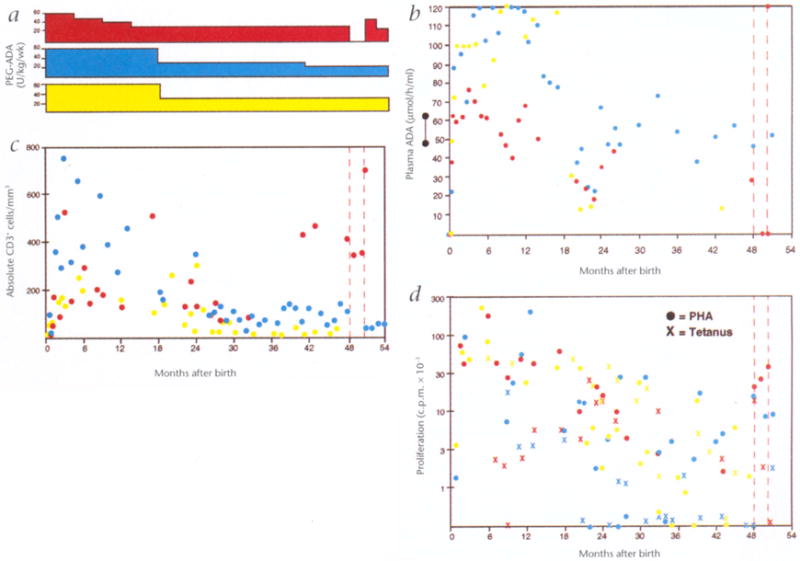
Purine metabolic parameters and immune function, a, PEG-ADA dosages. Dosages of PEG-ADA enzyme replacement therapy (U/kg per week) administered to subjects 1 (red), 2 (blue) and 3 (yellow), b, Plasma ADA levels. The levels of plasma adenosine deaminase enzymatic activity (●) measured at successive times are shown for subjects 1 (red), 2 (blue) and 3 (yellow), c, T-lymphocyte numbers. The absolute numbers of CD3+ T lymphocytes per mm3 of whole blood measured by FACS analysis are shown (●) for subjects 1 (red), 2 (blue) and 3 (yellow), d, T-lymphocyte blastogenesis. The proliferative responses of peripheral blood mononuclear cells to the T lymphocyte mitogen PHA (●) and the antigen tetanus toxoid (X) measured at successive times are shown for subjects 1 (red), 2 (blue) and 3 (yellow).
During the first 18 months of life, subject 1 was kept on a constant total amount of PEG-ADA so that, as he grew during infancy, the dose per kilogram he received declined gradually from 60 U/kg per week to 30 U/kg per week (Fig. 1a). Dosages for subjects 2 and 3 were modified every one to three months to adjust for weight gains, to maintain a dose of 60 U/kg per wk. At 18 months, with parental consent, the dosages of PEG-ADA for these patients were reduced to 30 U/kg per wk, based on stable clinical and immunologic status, and on evidence of sustained production of gene-containing leukocytes. When the PEG-ADA dosages were halved, plasma ADA levels fell accordingly by 50%, to 20–70 μmol/hr per ml (Fig. 1b). Despite these lower levels of plasma ADA activity, dAXP in RBC remained barely detectable (data not shown). ADA activity in hemolysates prepared from washed RBC of the three patients, measured many times during the first two years, remained in the deficient range, at 0.04–0.3 nmol/hr per mg protein (normal, 84.4 ± 40). These findings confirm that PEG-ADA does not enter erythrocytes, and indicate that there was minimal production of ADA from the transduced gene in erythrocytes. Because the RBC of the patients were deficient in ADA enzymatic activity, it seems that the reduced dosages of PEG-ADA provided adequate plasma ADA enzymatic activity to prevent detectable dAXP accumulation in RBC (consistent with experience in other patients11-13)
Lymphocyte numbers and function
The absolute numbers of T lymphocytes present in the umbilical cord blood and peripheral blood samples of the patients in the first few days of life were much less than those of normal neonates (Fig. 1c). The numbers of total (CD3+) T lymphocytes, as well as T-lymphocyte subsets, increased within the first few months on PEG-ADA treatment, although they remained below the lower limits of normal. The absolute numbers of T lymphocytes remained relatively stable in subject 1 during the four years, whereas they decreased by 50–80% in subjects 2 and 3 in years two to four as the dosages of PEG-ADA were reduced.
As with the numbers of T lymphocytes, T-lymphocyte function as measured by the proliferative responses to the T-lymphocyte mitogen phytohemagglutinin (PHA) was low in the cord blood and peripheral blood samples taken during the first few weeks after birth (Fig. 1d). After several months, PHA responses increased to the range of 30,000–100,000 cpm 3H-thymidine incorporation, which represents levels of blastogenesis at or slightly below the normal limits for our laboratory (>75,000 cpm) but significantly more than those seen in the neonatal period. PHA responses declined to the range of 3,000–30,000 cpm in all three subjects during years two to four (normal, >10,000 cpm). Similar results were seen after tetanus immunization at six months of age. Subject 1 showed low but stable levels of blastogenesis in response to tetanus toxoid, and subjects 2 and 3 had decreased but detectable blastogenesis in years two to four.
Frequency of gene-containing in granulocytes and PBMC
PCR was undertaken using DNA extracted from the subjects’ peripheral blood leukocytes, under conditions in which the frequency of cells containing the LASN vector sequences could be measured within half an order of magnitude over a range of positive cells from 1/30,000 (0.003%) to as high as 1/3 (33%). During the four-and-a-half years of evaluation, gene-containing leukocytes have been present in peripheral blood of all three subjects (Fig. 2). The levels of gene-containing cells in the granulocyte fraction increased slightly from the frequencies seen initially, ranging from 1/10,000–1/3,000 (0.01%–0.03%) to approximately 1/1,000 (0.1%). In contrast, the levels of gene-containing cells in the peripheral blood mononuclear cell (PBMC) fraction increased markedly (50- to 100-fold) in all three subjects, from the frequencies present in the first one to two years of 1/10,000 (0.01%) to approximately 1/100 (1%) in years two to four.
Fig. 2.
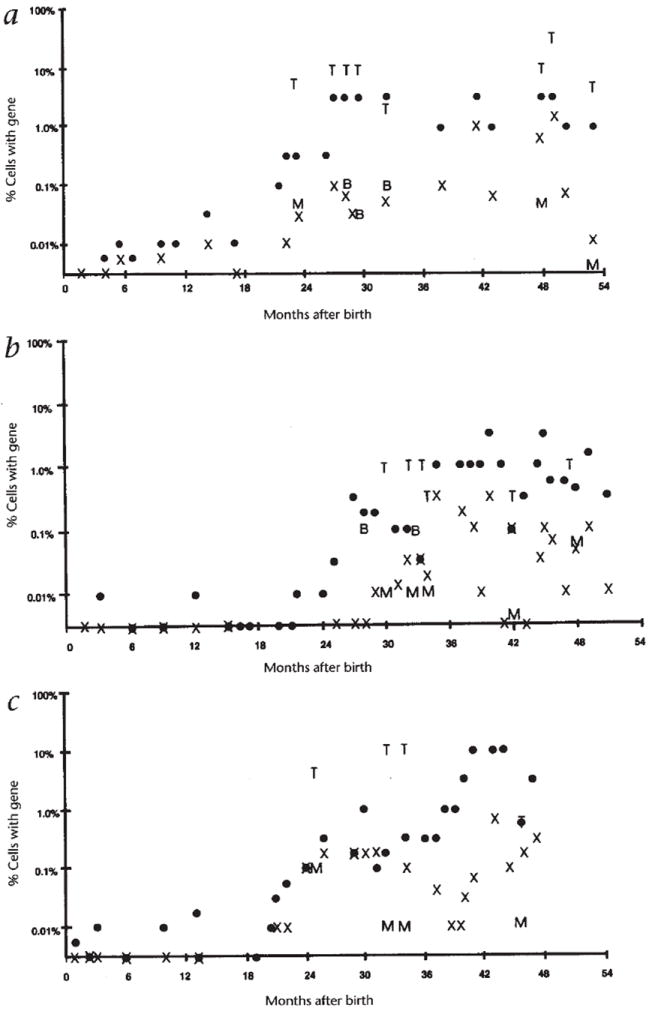
Frequency of gene-containing leukocytes. The frequency of cells containing the LASN vector sequences as measured by semi-quantitative PCR in PBMC fractions (●), granulocytic fractions (X), and FACS-sorted CD3+ T lymphocytes (T), CD13+/CD14+ monocytic cells (M) and CD19+ B lymphocytes (B) at successive times are shown for subjects 1 (a), 2 (b) and 3 (c).
Frequency of gene-containing cells in FACS-sorted fractions
When blood samples contained sufficient numbers of cells, peripheral blood leukocytes were sub-fractionated using a fluorescent activated cell sorter (FACS) for analysis of the frequencies of gene-containing cells in homogenous cell populations. Lineage-specific markers that were used for FACS sorting included: CD3 for mature T lymphocytes, a combination of CD13 and CD14 for myeloid cells (primarily monocytes from the PBMC), and CD19 for B lymphocytes. Typically, two million to four million cells were sorted, yielding 50,000–200,000 cells in each fraction. DNA was extracted from the sorted cells and semi-quantitative PCR was used to measure the frequency of gene-containing cells. In every sorted sample from all three subjects, the frequency of gene-containing cells in the CD3+ T-lymphocyte fraction (1–10%) was at least ten times as high as that in monocytes or B lymphocytes (0.01–0.1%)(Fig. 2).
Measurement of ADA expression
To determine the level of expression from the vector, reverse transcription-polymerase chain reaction (RT-PCR) was undertaken, using primers designed to measure specifically vector-derived transcripts (Fig. 3). Vector-derived transcripts were not detectable in freshly isolated PBMC (or granulocytes). Culturing the PBMC for five days to stimulate T-lymphocyte proliferation with PHA and IL-2 produced a large increase in the level of expression from the vector.
Fig. 3.
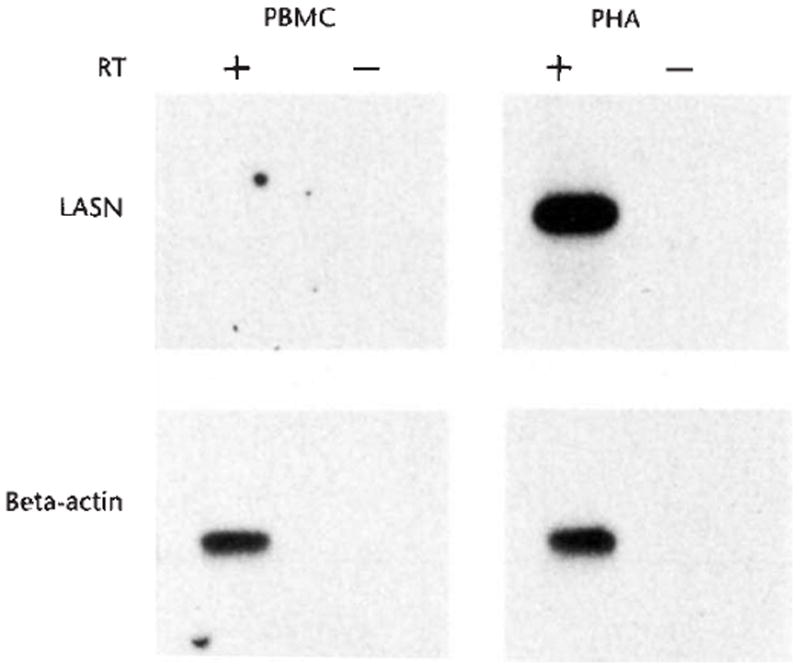
RT-PCR analysis of vector expression in leukocytes. The products were generated by reverse transcription of RNA using a primer which was antisense to the ADA cDNA, followed by PCR amplification of cDNA using primers from the LASN vector. Samples were from peripheral blood leukocytes obtained from subject 1 four-and-a-half years after gene transfer. Samples were analyzed in duplicate, with (+) or without (−) reverse transcriptase, to control for DNA contamination. Leukocyte samples include PBMC (PBMC) and PHA-stimulated PBMC (PHA). Portions of each RNA sample were also reverse transcribed and amplified with primers to β-actin (beta-actin), to verify that equivalent amounts of RNA were assayed.
Effects of complete cessation of PEG-ADA therapy in one subject
In May 1997, PEG-ADA was stopped completely in subject 1, who had the highest levels of gene marking and T-lymphocyte numbers, to assess his independent immune function. Two months after ceasing enzyme therapy, he developed an upper respiratory tract infection, with sinusitis diagnosed by X-ray. On physical examination, he was moderately ill-appearing. Notably, he had also developed white-colored oral lesions, consistent with oral monilia. Additionally, his serum alanine aminotransferase, which had been normal on all previous measurements, was increased at 244 and 71 U/ml (normal range, 3–46 U/ml) during clinic visits at one-and-a-half and two months, respectively, after stopping PEG-ADA. This observation is consistent with evidence that ADA deficiency may result in metabolic liver injury14.
Because of this evidence of the development of a possible opportunistic infection, the subject was re-started on PEG-ADA at a dose of 45 U/kg per week. He was also treated with oral amoxicillin and nystatin. At the next clinic visit, one month later, he had recovered his normal state of health, had gained 2 kg in weight, and was free of signs of upper respiratory infection or thrush. His PEG-ADA dosage was then returned to 40 U/kg per week, and he has remained well on this dosage for more than eight months.
During the two months without PEG-ADA therapy, he had a decline in his plasma ADA level, from >1000-fold to only 10-fold the level seen in the neonatal period (Fig. 1b, and Table 1). There was an associated 100-fold increase in the levels of erythrocyte deoxyadenosine nucleotides, to a range exceeding that seen in the neonatal period, before enzyme replacement began. At the same time, S-adenosylhomocysteine hydrolase activity in erythrocytes decreased to 0.1 nmol/h per mg protein (data not shown). The absolute number of circulating CD3, CD4 and CD8 T lymphocytes remained relatively stable (Fig. 1c, and Table 1). In sharp contrast, there were large decreases in the absolute numbers of B lymphocytes and NK cells. Within one month after reinstitution of enzyme replacement at 45 U/kg per week, the numbers of B lymphocytes and NK cells returned to the levels seen before PEG-ADA therapy was stopped.
Table 1.
Laboratory results before, during and after PEG-ADA cessation
| Month (Normal): | PEG-ADA (U/kg/wk) | ADA (μmol/hr/ml) | dAXP (μmol/ml) | PHA tetanus (c.p.m × 10-3) | Absolute cell numbers/mm3 | ALT (U/L) | Frequency of gene-containing cells | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD3 | CD4 | CD8 | C19 | CD16/56 | |||||||||||
| – | (6–12) | (<0.01) | (>100) | (>5) | (>820) | (>440) | (>180) | (>50) | (>80) | (3-46) | PBMC | Gran | T | Mono | |
| 30 | 30 | 35.0 | 0.004 | – | – | – | – | – | – | – | 36 | 0.3 | 0.03 | 10 | 0.5 |
| 41 | 20 | – | – | 6.2 | 9.3 | 738 | 198 | 342 | 162 | 918 | – | 1.0 | 0.07 | – | – |
| 48 | 15→0 | 28.6 | 0.006 | 20.7 | 13.4 | 405 | 211 | – | – | – | – | 3.0 | 0.8 | 10 | 0.05 |
| 49 | 0 | – | – | 25.5 | 1.7 | 297 | 158 | 139 | 56 | 557 | – | 3.0 | 2.0 | 30 | n.d. |
| 49.5 | 0 | 0.33 | 0.712 | 32.7 | 0 | 370 | 139 | 218 | 9 | 35 | 244 | 1.0 | 0.08 | – | – |
| 50 | 0→45 | 0.26 | 0.272 | – | – | – | 291 | 313 | 22 | 29 | 71 | – | – | – | – |
| 51 | 45→40 | 119.5 | – | – | – | – | 419 | 987 | 239 | 987 | 37 | – | – | – | – |
| 53 | 40 | 36.2 | 0.000 | – | – | – | – | – | – | – | – | 1.0 | 0.01 | 5.0 | 0.003 |
| 54 | 40 | – | – | 65.9 | 12.4 | 329 | 145 | – | – | – | – | – | – | – | – |
During the time without PEG-ADA therapy, the proliferative responses to PHA remained constant (Fig. 1d, and Table 1). However, the proliferative response to tetanus toxoid was lost. Upon re-institution of PEG-ADA, the proliferative response to tetanus toxoid again returned. Consistent with the dependence of tetanus-responsive blastogenesis on exogenous PEG-ADA, none of ten tetanus-responsive T-lymphocyte clones grown from subject 1 at three years of age contained the LASN vector sequences (data not shown).
The cessation and re-institution of PEG-ADA therapy had minimal effects on the frequencies of gene-containing leukocytes. A blood sample obtained two months after stopping PEG-ADA showed that the percentages of circulating T lymphocytes containing the introduced ADA gene had increased from 10% to 30%, whereas the absolute numbers of T lymphocytes remained stable (Fig. 2). Measurements after adenosine metabolic and immune function parameters had returned to their baseline after resumption of PEG-ADA therapy showed essentially unchanged frequencies of gene-containing cells.
Discussion
These studies extend our previous observation of successful gene transduction and autologous engraftment of umbilical cord blood CD34+ cells in ADA-deficient neonates. Observations for more than four years demonstrate the continuing presence of peripheral blood leukocytes containing the transferred normal ADA gene. The levels of gene-containing cells have been stable, suggesting that long-lived stem cells were transduced and engrafted to produce cells of multiple hematopoietic and lymphoid lineages. Life-long follow-up will be needed to determine the full proliferation capacity and ultimate longevity of the genetically-modified stem cells.
Additionally, during the past two to three years, we have found that the frequency of T lymphocytes containing the transferred ADA gene has greatly exceeded the frequency of transduced cells of other hematopoietic and lymphoid lineages. This finding is consistent with the well-known results with allogeneic transplantation of genetically normal stem cells into SCID patients in whom normal T-lymphocyte numbers develop from a few engrafted donor hematopoietic stem cells3.
Whether the higher steady-state level of T lymphocytes reflects a relative increase in the production or the survival of T lymphocytes containing the ADA gene, compared with other cell types, cannot be determined from the available information. The stage of T-lymphoid development in which the transduced ADA gene confers an advantage (pre-, intra- or post-thymic) is also not known. ADA levels are normally highest in thymic T lymphocytes, and it has been postulated that absence of ADA has its most critical impact at the production stage of T lymphopoiesis13,15. However, a study by Ferrari et al.16 demonstrated that ADA gene-corrected peripheral blood T lymphocytes had prolonged survival after injection into SCID-mice, compared with non-corrected, ADA-deficient T lymphocytes. Thus, ADA expression may also be essential for the survival of mature peripheral blood T lymphocytes.
When the dosages of PEG-ADA enzyme were decreased, the plasma levels of ADA enzymatic activity decreased accordingly. We intended to reduce the dosages of PEG-ADA sufficiently to create an environment in which T lymphocytes expressing endogenous ADA would have a greater selective advantage. However, erythrocyte deoxyadenosine nucleotide levels did not increase significantly, suggesting that even the decreased dosages of PEG-ADA were sufficient to effectively catabolize circulating deoxyadenosine metabolites. Nevertheless, there were decreases in both T-lymphocyte numbers and function after the PEG-ADA dosage decreases. These observations may reflect the greater sensitivity of lymphoid cells to ADA substrates, due to their ability to more efficiently activate deoxyadenosine to toxic nucleotides15.
While the PEG-ADA dosage was lowered, the percentage of T lymphocytes containing the inserted ADA gene increased by more than two orders of magnitude, whereas the absolute numbers of T lymphocytes decreased by only 50–75%. Thus, the increase in the frequency of transduced T lymphocytes was not secondary to the relative lymphopenia associated with reduction of PEG-ADA, but instead represented an increase in the absolute number of transduced T lymphocytes.
During the two-month period of PEG-ADA cessation in subject 1, there was no evidence that immune function was sustained solely by the gene-containing cells. From the rise in RBC dAXP and fall in S-adenosylhomocysteine hydrolase activity after cessation of PEG-ADA therapy, it is evident that the total amount of ADA gene-transduced cells must provide far less ADA activity than was being provided by PEG-ADA therapy.
There was a rapid loss of pre-existing, antigen-specific T lymphocytes reactive to tetanus toxoid during this period without enzyme therapy, indicating that PEG-ADA was required for their persistence. The absence of detectable transduced tetanus-specific T lymphocytes may be the result of administration of tetanus toxoid immunization in the first year of life, when he was receiving full dosages of PEG-ADA, obviating a selective advantage and resulting in a frequency of transduced antigen-specific T lymphocytes as low as for other leukocytes (1/3,000–1/10,000). However, the numbers of T lymphocytes and their proliferative responses to the mitogen PHA remained unchanged. Additionally, the frequency of transduced CD3+ T lymphocytes increased from 10% on PEG-ADA to 30% during the two months without PEG-ADA therapy. Thus, some T lymphocytes do seem to be supported by the presence of the inserted ADA gene.
The precipitous decreases in absolute numbers of B lymphocytes and NK cells after PEG-ADA cessation demonstrates that production or survival of these cell types are dependent on exogenous ADA enzyme, either directly or indirectly (by maintenance of a helper T-cell population which was lost with PEG-ADA withdrawal). B-lymphocyte counts often rise before T-cell counts when PEG-ADA therapy is begun in SCID patients11,13,14, which indicates a direct dependence.
Poor or inconsistent expression of the transferred ADA gene may limit the ability of T lymphocytes to metabolize deoxyadenosine and selectively survive. The Moloney murine leukemia virus (MoMuLV) long-terminal repeat (LTR), which drives expression of the ADA gene in the LASN vector, is most active during intra-thymic T-lymphocyte development17,18. It may be at that stage of T-lymphocyte differentiation that endogenous ADA expression can confer a selective advantage to transduced T lymphocytes, compared with non-transduced thymocytes, and allow their relative increase in frequency. Recent reports describe relatively low expression from the MoMuLV LTR in quiescent, mature human T lymphocytes19,20, similar to our results. Inadequate ADA expression in circulating antigen-specific T lymphocytes, which are largely quiescent, may be responsible for the loss of response to tetanus after PEG-ADA withdrawal.
The results described here indicate the capabilities and limits of current gene transfer techniques to treat disorders of hematopoietic and lymphoid cells. Although the results represent a modest degree of success, it is evident that significant advances are needed in gene transfer and expression technology to achieve clinical benefits for immune deficiencies, hemoglobinopathies and lysosomal storage diseases using gene therapy with hematopoietic stem cells.
Methods
Cells and vectors
The LASN retroviral vector was constructed and packaged in the PA317 amphotropic packaging cell line in the laboratory of A. D. Miller at the Fred Hutchinson Cancer Research Center21,22 (Seattle, Washington). LASN carries a normal human ADA cDNA under transcriptional control of the MoMuLV LTR and an internal neomycin resistance gene controlled by an SV40 promoter. Clinical grade supernatant was produced by Genetic Therapy (Gaithersberg, Maryland).
Peripheral blood samples were obtained during clinic visits every 1–3 months by the physicians providing primary care to the subjects. Samples for measurement of plasma ADA and RBC dAXP levels were collected with the addition of preservative-free heparin and kept at 4 °C during shipment to the Biochemistry Laboratory at Duke University Medical School. The collection of blood samples was synchronized to precede a scheduled administration of PEG-ADA, to represent a trough measurement. Peripheral blood samples to measure lymphocyte numbers, immunophenotype, and blastogenic responses and to extract of genomic DNA to measure the frequency of gene-containing cells were collected with the addition of preservative-free heparin and assayed at the primary care institutions or shipped at ambient temperature to the Clinical Immunology Laboratory at Children’s Hospital Los Angeles.
Analysis of peripheral blood leukocytes
Peripheral blood samples were separated into PBMC and granulocyte fractions by centrifugation on ficoll-hypaque gradients (Pharmacia). PBMC were fractionated further by labeling with fluorescent-labeled antibodies followed by sorting using a FACS Vantage (Becton-Dickinson Immunocytometry Systems; San Jose, California). Monoclonal antibodies used for sorting were conjugated to either fluorescein or phycoerythrin and included: Leu-4 (anti human CD3 for T lymphocytes), Leu-M7 and Leu-M3 (anti-human CD13 and CD14, respectively, for monocytic cells), Leu-12 (anti-human CD19 for B lymphocytes), and Leu-11c and Leu-19 (anti-human CD16 and CD56, respectively, for NK cells), all from Becton-Dickinson Immunocytometry Systems (San Jose, California). Cells isolated by ficoll-hypaque or FACS sorting were frozen as cell pellets for subsequent DNA or RNA analysis. Immunophenotypic analyses of PBMC were done using fluorescently-labeled monoclonal antibodies as described12. Blastogenic responses of PBMC to PHA and tetanus toxoid were measured by 3H-thymidine incorporation as described12. Plasma levels of ADA enzymatic activity and erythrocytic levels of deoxyadenosine nucleotides were measured as described12. Semi-quantitative PCR of leukocyte DNA and RT-PCR of RNA to determine the frequency of peripheral blood leukocytes containing and expressing the LASN vector were done as described6. In every set of PCR reactions, DNA or RNA was extracted in parallel from normal control leukocytes and assayed to detect false positive reactions and a standard curve was generated using mixtures of a human T-cell line containing a single copy of the LASN vector diluted with non-transduced cells.
Acknowledgments
This work was supported by a General Clinical Research Center award (3 MO1 RR0043-35S1), and a SCOR grant (1P50 HL54850) from the National Institutes of Health (NIH). M.S.H. was supported by a grant (DK20902) from the NIH and a grant from Enzon, Inc. D.B.K. is the recipient of an Elizabeth Glaser Scientist Award from the Pediatric AIDS Foundation.
References
- 1.Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet. 1968;2:1366–1369. doi: 10.1016/s0140-6736(68)92673-1. [DOI] [PubMed] [Google Scholar]
- 2.Parkman R, Gelfand EW, Rosen FS, Sanderson A, Hirschhorn R. Severe combined immunodeficiency and adenosine deaminase. N Engl J Med. 1975;292:714–719. doi: 10.1056/NEJM197504032921402. [DOI] [PubMed] [Google Scholar]
- 3.Tjonnfjord GE, Steen R, Veiby OP, Friedrich W, Egelan T. Evidence for engraftment of donor-type multipotent CD34+ cells in a patient with selective T-lymphocyte reconstitution after bone marrow transplantation for B- SCID. Blood. 1994;84:3584–3589. [PubMed] [Google Scholar]
- 4.Hirschhorn R, et al. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nature Genet. 1996;13:290–295. doi: 10.1038/ng0796-290. [DOI] [PubMed] [Google Scholar]
- 5.Stephan V, et al. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N Engl J Med. 1996;335:1563–1567. doi: 10.1056/NEJM199611213352104. [DOI] [PubMed] [Google Scholar]
- 6.Kohn DB, et al. Engraftment of gene-modified cells from umbilical cord blood in neonates with adenosine deaminase deficiency. Nature Med. 1995;1:1017–1026. doi: 10.1038/nm1095-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaese RM, et al. T lymphocyte-directed gene therapy for ADA- SCID: Initial trial results after 4 years. Science. 1995;270:475–480. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 8.Bordignon C, et al. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science. 1995;270:470–475. doi: 10.1126/science.270.5235.470. [DOI] [PubMed] [Google Scholar]
- 9.Hoogerbrugge PM, et al. Bone marrow gene transfer in three patients with adenosine deaminase deficiency. Gene Ther. 1996;3:179–183. [PubMed] [Google Scholar]
- 10.Hershfield MS. PEG-ADA: an alternative to haploidentical bone marrow transplantation and an adjunct to gene therapy for adenosine deaminase deficiency. Hum Mutat. 1995;5:107–112. doi: 10.1002/humu.1380050202. [DOI] [PubMed] [Google Scholar]
- 11.Hershfield MS. PEG-ADA replacement therapy for adenosine deaminase deficiency: an update after 8.5 years. Clin Immunol Immunopathol. 1995;76:S228–232. doi: 10.1016/s0090-1229(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 12.Weinberg K, et al. T lymphocyte ontogeny in adenosine deaminase-deficient severe combined immunodeficiency after treatment with polyethylene glycol-modified adenosine deaminase. J Clin Invest. 1993;92:596–602. doi: 10.1172/JCI116626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hershfield MS, Mitchell BS. In: The Metabolic and Molecular Bases of Inherited Disease. 7. Scriver CS, Beaudet AL, Sly WS, Valle D, editors. McGraw-Hill; New York: 1995. pp. 1725–1768. [Google Scholar]
- 14.Bollinger ME. Brief report: hepatic dysfunction as a complication of adenosine deaminase deficiency. N Engl J Med. 1996;334:1367–1371. doi: 10.1056/NEJM199605233342104. [DOI] [PubMed] [Google Scholar]
- 15.Hirschhorn R. Overview of biochemical abnormalities and molecular genetics of adenosine deaminase deficiency. Pediatr Res. 1993;33(Suppl):S35–S41. doi: 10.1203/00006450-199305001-00194. [DOI] [PubMed] [Google Scholar]
- 16.Ferrari G, et al. An in vivo model of somatic cell gene therapy for human severe combined immunodeficiency. Science. 1991;251:1363–1366. doi: 10.1126/science.1848369. [DOI] [PubMed] [Google Scholar]
- 17.Tsichlis PN. Oncogenesis by Moloney murine leukemia virus. Anticancer Res. 1987;7:171–180. [PubMed] [Google Scholar]
- 18.DesGroseillers L, Jolicoeur P. Mapping the viral sequences conferring leukemogenicity and disease specificity in Moloney and amphotropic murine leukemia viruses. J Virol. 1984;52:448–456. doi: 10.1128/jvi.52.2.448-456.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agarwal M, Austin TW, Morel F, Chen J, Böhnlein E, Plavec I. Scaffold attachment region-mediated enhancement of retroviral expression in primary T cells. J Virol. 1998;72:3720–3728. doi: 10.1128/jvi.72.5.3720-3728.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruhl E, Lum LG, Trevor KT. T cell activation status modulates retrovirus-mediated gene expression. Blood. 1997;90(Suppl 1):417b. doi: 10.1089/hum.1998.9.10-1457. [DOI] [PubMed] [Google Scholar]
- 21.Hock RA, Miller AD, Osborne WR. Expression of human adenosine deaminase from various strong promoters after gene transfer into human hematopoietic cell lines. Blood. 1989;74:876–881. [PubMed] [Google Scholar]
- 22.Miller AD, Buttimore C. Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production. Mol Cell Biol. 1986;6:2895–2902. doi: 10.1128/mcb.6.8.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]