

Abstract
Cellular processes that drive sterile inflammatory injury after hepatic ischemia/reperfusion (I/R) injury are not completely understood. Activation of the inflammasome plays a key role in response to invading intracellular pathogens, but mounting evidence suggests it also plays a role in inflammation driven by endogenous danger-associate molecular pattern (DAMP) molecules released after ischemic injury. The nucleotide-binding domain, leucine-rich repeat containing protein 3 (NLRP3) inflammasome is one such process, and the mechanism by which its activation results in damage and inflammatory responses following liver I/R is unknown. Here we report that both NLRP3 and its downstream target Caspase-1 are activated I/R and are essential for hepatic I/R injury as both NLRP3 and Caspase-1 KO mice are protected from injury. Furthermore, inflammasome-mediated injury is dependent on Caspase-1 expression in liver non-parenchymal cells. While upstream signals that activate the inflammasome during ischemic injury are not well characterized, we show that endogenous extracellular histones activate the NLRP3 inflammasome during liver I/R through Toll-like Receptor-9 (TLR9). This occurs through TLR9-dependent generation of reactive oxygen species. This mechanism is operant in resident liver Kupffer cells, which drive innate immune responses after I/R injury by recruiting additional cell types, including neutrophils and inflammatory monocytes. These novel findings illustrate a new mechanism by which extracellular histones and activation of NLRP3 inflammasome contribute to liver damage and activation of innate immunity during sterile inflammation.
Background
Ischemia reperfusion (I/R) injury is a dynamic process that involves the deprivation of blood flow and oxygen followed by their restoration, which leads to ischemic organ damage and inflammation-mediated reperfusion injury (1). Liver I/R injury inevitably occurs after liver resection, organ transplantation, massive trauma and hemorrhagic shock. There are two distinct stages of liver I/R. First, the ischemic insult causes sub-lethal cellular damage through oxidative stress and reactive oxygen species (ROS) production. Reperfusion then augments the injury by propagating the sterile inflammatory, innate and adaptive immune responses (2,3). Liver resident Kupffer cells (KCs), the major population of non-parenchymal cells (NPCs), have crucial roles in the initial inflammatory stages by phagocytosing necrotic cells, secreting cytokines, and recruiting other inflammatory cells such as neutrophils and circulating monocytes (1). Because of their immune triggering capability, responses driven by KCs have been recognized as a key mechanisms in liver I/R injury (3).
Nucleotide-binding domain, leucine-rich repeat containing protein 3 (NLRP3), also known as NALP3 or cryopyrin, is an intracellular nucleotide-binding oligomerization domain (NOD)-like receptor that functions as a danger signal sensor that becomes activated in response to a diverse range of microbial and non-microbial cellular stressors (4). The activation of NLRP3 leads to the assembly of NLRP3 inflammasome, which includes pro-caspase-1 and the adaptor apoptosis-associated speck-like protein containing a CARD (ASC), resulting in the production of pro-inflammatory cytokines IL-1β and IL-18. This multi-protein complex plays an important role for host responses to microbial pathogens and several multifaceted diseases (4). There are numerous exogenous agonists that can activate the NLRP3 inflammasome, including several pathogen-associated molecular pattern (PAMP) molecules. Additionally, a number of endogenous agonists have also been recently described. These molecules are danger-associated molecular pattern (DAMP) molecules, and include ATP, amyloid β, monosodium urate, and cholesterol crystals (5). While PAMP molecules are generally recognized in response to invading pathogens, DAMPs are the major mediators of sterile inflammatory injury, such as hepatic I/R. Recently, it has been shown that the activation of the inflammasome plays a crucial role in both cardiac and hepatic I/R injury (6,7), as gene silencing of NLRP3 results in protection from inflammation and hepatocyte injury after liver I/R. This protective effect is associated with reduced production of pro-inflammatory cytokines including IL-1β, IL-18, TNF-α, and IL-6 (6). While activation of the inflammasome has been shown to play a key role in these processes, upstream ligands responsible for initiating these responses are unknown.
The functions of extracellular histones have been intensely studied in several inflammatory models. Although low levels of extracellular histones have shown to be present in normal human circulation (8); their levels are greatly increased during sepsis (9), and systemic lupus erythematosus (10). In a mouse model of sepsis, extracellular histones have been demonstrated to be major mediators of endothelial dysfunction, organ failure and death (11). Additionally, histones contribute to death in inflammatory injury and chemical-induced liver injury (12). We have recently shown that extracellular histones act as a new class of DAMPs and augment inflammation and organ damage through TLR9 after liver I/R (13). We hypothesized that extracellular histones might also play a role in inflammasome activation after I/R. In this study, we demonstrate that extracellular histones released after I/R activate the NLRP3 inflammasome in KCs through TLR9-dependent generation of ROS. This mechanism propagates organ damage through neutrophil and inflammatory monocytes recruitment. These findings reveal a novel pathway in KCs by which endogenous DAMP molecules - histone proteins - drive innate immune responses in hepatic I/R through ROS generation, NLRP3 inflammasome activation, and further recruitment of pro-inflammatory cells.
Materials and Methods
Animals
Eight- to 12-week-old male wild-type (WT) C57BL/6 mice, IL-1R knockout (KO), IL-18 were purchased from The Jackson Laboratory (Bar Harbor, Me), NLRP3−/−, caspase-1−/−, TLR9CpG/CpG mutant mice were provided by Dr. Timothy Billiar from the University of Pittsburgh Medical Center (Pittsburgh, PA). Animal protocol approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh and the experiments were performed in adherence to National Institutes of Health guidelines for the use of laboratory animals.
Chimeric mice
Chimeric mice were produced by adoptive transfer of donor bone marrow cells into irradiated recipient animals using combinations of caspase-1 WT and caspase-1 KO mice in the following recipient/donor combinations: WT/WT, WT/KO, KO/KO, KO/WT. Recipient mice were exposed to an otherwise lethal 1000 cGy from a Cesium source (Nordion International) 6 h before receiving 2.5 × 106 bone marrow cells by tail vein injection. The bone marrow cells were prepared in a sterile manner from the tibia and femur bones of the donor mice. All animals were monitored two to three times weekly for the first 2 weeks to ensure successful bone marrow engraftment. The chimeric mice underwent hepatic I/R after another 8–10 weeks to ensure stable engraftment.
Liver I/R
A nonlethal model of segmental (70%) hepatic warm ischemia and reperfusion was used (14). Under sodium ketamine (100 mg/kg body weight, i.p.) and xylazine (10 mg/kg) anesthesia, a midline laparotomy was performed. The liver hilum was dissected free of surrounding tissue. All structures in the portal triad (hepatic artery, portal vein, bile duct) to the left and median liver lobes were occluded with a microvascular clamp (Fine Science Tools) for 60 min, and reperfusion was initiated by removal of the clamp. Throughout the ischemic interval, evidence of ischemia was confirmed by visualizing the pale blanching of the ischemic lobes. After the clamp was removed, gross evidence of reperfusion that was based on immediate color change was assured before closing the abdomen with continuous 4–0 polypropelene suture. Temperature during the ischemia was maintained at 31°C by a warming incubator chamber (Fine Science Tool). Sham animals underwent anesthesia, laparotomy, and exposure of the portal triad without hepatic ischemia. At the end of the observation period following reperfusion, the mice were anesthetized with inhaled isoflurane and sacrificed by exsanguination.
Experimental Design
Mice received anti-histone H3 or H4 antibodies (20 mg/kg) (12), control IgG (I5006, Sigma-Aldrich), TLR9 agonist (100 μg per mouse; ODN1668, Invivogen), caspase-1 inhibitor (100 μg per mouse; Z-YVAD-FMK, R&D Systems) (15) or DMSO intravenously 30 minutes before ischemia, as previously described (13). Calf thymus histones (25 mg/kg; H9250, Sigma-Aldrich), control CpG (Invivogen), TLR9 antagonist (100 μg per mouse; ODN2088, Invivogen), or phosphate-buffered saline (PBS) were injected intraperitoneally immediately after ischemia.
Liver Damage Assessment
Serum alanine aminotransferase (sALT) levels were measured using the DRI-CHEM 4000 Chemistry Analyzer System (Heska). The extent of parenchymal necrosis in the ischemic lobes was evaluated using H&E stained histological sections. Images covering all necrotic areas were captured at a magnification of 40× (16). The necrotic area was quantitatively assessed by using Image J (NIH). Results were presented as the mean of percentage of necrotic area (mm2) with respect to the entire area of one capture (mm2). Histological sections were assessed in a blinded manner by two individual examiners, who were unaware of the treatment group assignment of the animals, and quantified using a semi-quantitative scoring system to assess liver damage (17).
ELISA
Serum IL-1β and IL-18 levels in the mouse were detected by ELISA (eBioscience or MBL international) according to the manufacturer’s instructions.
Isolation and Culture of Hepatocytes, Nonparenchymal Cells and Kupffer cells
Hepatocytes and NPCs were isolated from normal WT C57BL/6 mice. Briefly, the portal vein was cannulated and the liver was perfused for 3 min with 1× HBSS (Invitrogen Life Technologies) supplemented with 0.96 g of sodium bicarbonate/500 ml (Perfusate I) at a flow rate of 10 ml/min. Then, the liver was perfused with a 0.2% protease (Sigma-Aldrich) in Perfusate I for 3 min. The liver was dissected out and placed in a petri dish with Perfusate II and diced into 2- to 3-mm pieces. The supernatant was then filtered. NPCs were separated from the hepatocytes by one cycle of differential centrifugation (400 rpm for 5 min). The supernatant was further centrifuged (400 rpm for 5 min and two cycles of 1500 rpm for 5 min) to obtain NPCs. The NPCs did not contain hepatocytes as detected by light microscopy. Hepatocytes were further purified over a 90% Percoll gradient. Hepatocyte purity exceeded 98% as assessed by light microscopy, and viability typically was 95% as determined by trypan blue exclusion assay. NPCs were resuspended and added to 3.0 ml of 40% (w/v) Optiprep (Sigma-Aldrich) to remove debris and enrich the NPC. The NPCs-enriched fraction was collected, washed in PBS, and positively selected using PE-conjugated anti-F4/80 (Cedarlane), and magnetic MicroBeads following the manufacturer’s protocol (Miltenyi Biotec). NPCs (50 × 106) were plated as described (18,19). KCs (50 × 106) were plated.
In Vitro Coculture Assays
The in vitro experimental procedure was modified as described by Gowda et. al (20). WT KCs were cultured under different time of hypoxia (0 to 24 hour), or stimulated with different doses of histones (0 μg/mL to 50μg/mL) or TLR9 agonist (ODN 1668, 0 μg/mL to 15 μg/mL), or TLR9 antagonist (ODN2088 15μg/mL) or histone + TLR9 antagonist, or anti-oxidant N-acetylcysteine (NAC) (25 mM or 50 mM) for 12 hours. Additionally, WT KC’s were stimulated with either histones (50 μg/mL), DNase I (100 U/mL), or Trypsin (100 μg/mL). For combination experiments, histones were pre-treated with trypsin or DNase I for 30 minutes prior to addition to KC cells.
Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis and Western Blotting
Western blot analysis for Caspase-1 (1:1000; Cell Signaling Technology), IL-1β (1:1,000; Abcam), IL-18 (1:800; MBL International), functional TLR9 (1:1,000; eBioscience), were performed. Protein extraction and Western blot analysis were performed following a standard protocol as described previously (21).
SYBR Green Real-Time Reverse-Transcription PCR
Total RNA was extracted from the liver using the RNeasy Mini Kit (Qiagen). mRNA for TNF-α, IL-6, and β-actin was quantified in duplicates by SYBR Green Reverse-Transcription PCR. PCR reaction mixture was prepared using SYBR Green PCR Master Mix (PE Applied Biosystems) using described primers (21).
Caspase Activity Assay
Caspase-1 activity was determined in freshly prepared whole liver lysates with a colorimetric assay as described previously (22). The caspase-1 activity analysis was based on the cleavage of the WEHD-pNA (Trp-Glu-His-Asp-pnitroanilide) substrate (R&D Systems).
Immunofluorescent Staining for Activated Caspase-1
Kupffer cells were stimulated with hypoxia, 15 μg/mL TLR9 agonist (ODN 1668 Invivogen) or 25 μg/mL exogenous histones for 12 h, then were incubated with cell permeable caspase-1 carboxyfluorescein-labeled fluorochrome inhibitor of caspase-1 (FLICA®660 caspase-1 kit, ImmunoChemistry Technologies), which binds to activated caspase-1, in serum-free DMEM for 1 h, followed by 3 washes, fixed with 4% paraformaldehyde in PBS for 15 min at room temperature, and incubated with 1 mg/ml Hoechst for 15 min at room temperature in the dark. Cells were mounted with Vecta-Shield Mounting media. Slides were viewed with Olympus confocal microscopes.
Quantitation of Confocal Immunofluorescent
All images were quantitated as previously described (23) using MetaMorph™ software (Molecular Devices, Downingtown, PA). In brief, all confocal immunofluorescent images were uniformly gated for inclusive threshold to measure cleaved caspase-1 content. The total area of the fluorescent signal specific to cleaved caspase-1 was measured by using the Show Region Statistics application from the measure menu. The total number of nuclei was determined, for the purpose of normalizing amount of cleaved caspase-1 by sample size within the sample field, by applying a threshold excluding local background fluorescence and utilizing the Count Cells application from the Apps menu.
Detection of Cellular Reactive Oxygen Species Production
Cellular ROS production was detected by using an Image-it™ live green reactive oxygen species detection kit (Molecular Probes/Invitrogen) and analyzed using a high content analysis platform on an Arrayscan VTi (Cellomics) following the manufacture’s protocol. Or Cells were incubated with 10 μM DCF diacetate (DCF-DA) (Invitrogen). After stimulation, cells lysate supernatants were read in a fluorescence spectrophotometer (SpectraMAX Gemini XS; MDS Analytical Technologies) with 485-nm excitation and 530-nm emission wavelengths, as described previously (24).
Flow Cytometry Analysis
Ischemic liver lobes were aseptically harvested from NLRP3 KO and WT mice at 6 h reperfusion after 1 h ischemia and prepared as a single cell suspension. RBC were lysed, and the cells were analyzed by flow cytometry for innate immune cell populations using the following antibodies as described (25): dendritic cells [MHC class II (I-Ab)+CD11c+], neutrophils (CD11b+Ly6G+), inflammatory monocytes (CD11b+Ly6Chi), and Kupffer cells (CD11bloF4/80+). Antibodies were purchased from eBioscience: PE anti-NK1.1 PK136 and PE-Cy7 anti-CD11b M1/70, BD Bioscience: APC anti-CD11c HL3 and FITC anti-Ly6C AL-21, or Biolegend: FITC anti-I-Ab AF6-120.1, APC anti-CD11b M1/70, PE anti-F4/80 BM8, PE-Cy7 anti-Ly6G 1A8, or from Miltenyi Biotec: eflour450 ME-9F1. For measurement of mitochondria-associated ROS production, NPCs were stained with MitoSOX (Molecular Probes/Invitrogen) following the manufacture’s protocol for FACS analysis (26). Data were acquired with a BD FACS LSR Fortessa flow cytometer (BD Biosciences) and analyzed with FlowJo analytical software (Treestar). Each experiment was repeated a minimum of three times.
Statistical Analysis
Results are expressed as the mean ± SE. Statistical analysis was performed using the Student’s t-test or one-way analysis of variance (ANOVA). All statistical analyses were performed using Sigma Stat version 3.5 (Systat Software, Inc.). Graphs were generated using Sigma Plot version 10 (Systat Software, Inc.). P < 0.05 was considered statistically significant.
Results
Genetic Deletion of nlrp3 or caspase-1 is Protective in Liver I/R Injury
To determine whether the NLRP3 inflammasome and its downstream protein, caspase-1, contribute to hepatic organ damage after ischemic stress, we subjected NLRP3 KO and Caspase-1 KO mice to liver I/R. Both NLRP3 KO and Caspase-1 KO mice were significantly protected from hepatic I/R injury compared to WT mice (Fig. 1A). Based on a semi-quantitative scoring system to assess liver damage (17), histology was concordant with the serum ALT estimation of liver damage with the presence of severe sinusoidal dilatation and confluent pericentral hepatocellular necrosis in liver tissue from WT mice but not within NLRP3 or Caspase-1 KO mice (Fig. 1B). To further confirm the role of caspase-1 in liver I/R, we used the caspase-1 inhibitor Z-YVAD-FMK. WT mice treated with the Z-YVAD-FMK inhibitor were significantly protected following hepatic I/R compared to mice treated with control DMSO (Fig. 1A). Liver histology was also consistent with these results (Fig. 1B).
Figure 1.
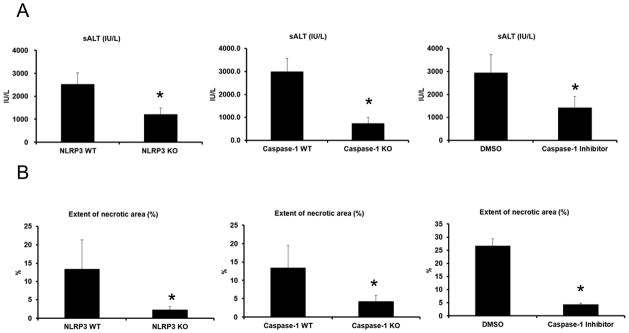
Genetic deletion of NLRP3 (NLRP3 KO), caspase-1 (caspase-1 KO) or inhibition of caspase-1 protects against hepatic I/R injury. (A) Serum ALT levels in NLRP3 KO, caspase-1 KO, WT mice treated with caspase-1 inhibitor and their corresponding control animals after liver I/R. WT mice were given caspase-1 inhibitor (Z-YVAD-FMK 100 mg/mouse) or 0.1% DMSO (control) intravenously 30 minutes before ischemia. Data represent the mean ± SE (n = 6 mice per group). Student’s t-test, *P < 0.05 vs. WT control. (B) Quantification of necrotic hepatocytes in hematoxylin and eosin-stained liver sections from NLRP3 KO, caspase-1 KO, caspase-1 inhibitor treated mice and WT animals 6 hours after reperfusion. The graph is representative of liver sections from six mice per group. Student’s t-test, *P < 0.05 vs. WT control. Histological sections were assessed in a blinded manner by two individual examiners, who were unaware of the treatment group assignment of the animals, and quantified using a semi-quantitative scoring system to assess liver damage (17).
The NLRP3 Inflammasome is Activated in Liver I/R Injury
Using NLRP3 KO and capase-1 KO mice in I/R, we first verified that caspase-1 activity was significantly reduced in NLRP3 KO mice and absent in caspase-1 KO mice after liver I/R (Fig. 2A). NLRP3 activation leads to caspase-1 activation, which causes the maturation and secretion of IL-1β and IL-18 among other substrates (5). After hepatic I/R, liver tissue expression of activated caspase-1 and downstream matured IL-1β and IL-18 were all up-regulated in the liver after I/R injury (Fig. 2B). Furthermore, these levels were reduced in NLRP3 KO mice, caspase-1 KO mice, and in mice treated with a caspase-1 inhibitor. We also assayed serum levels of IL-1β and IL-18 at the same time point and found reductions of these levels in both NLRP3 and caspase-1 KO mice (Figure 2C)
Figure 2.

Activation of NLRP3 inflammasome is involved in liver I/R. (A) Activation of caspase-1 in NLRP3 KO, caspase-1 KO and WT mice subjected to liver I/R compared to the WT sham group, assessed by colorimetric assay. Data represent the mean ± SE (n = 6 mice per group). ANOVA Tukey test, *P < 0.05, NLRP3 KO group versus WT group after I/R. (B) Western blot images showing the protein levels of activated (cleaved) caspase-1, IL-1β and IL-18 in liver of WT (C57BL/6) mice, NLRP3 KO, caspase-1 KO and caspase-1 inhibitor treated-mice at 6 hours after reperfusion. Each lane represents a separate animal. The blots shown are representatives of three experiments with similar results. (C) Serum levels of IL-1β and IL-18 obtained from NLRP3 KO, caspase-1 KO mice and their WT at 6 hours after reperfusion were measured by ELISA and compared to the sham group. Data represent the mean ± SE (n = 6 mice per group) ANOVA Tukey test, *P<0.05 vs. WT control. (D) IL-6 and TNF-α mRNA levels in NLRP3 KO or caspase-1 KO mice vs WT mice after 6 hours I/R, ANOVA Tukey test, *P < 0.05 WT vs. sham, NALP3 KO, caspase-1 KO or caspase-1 treated mice.
We hypothesized that release of IL-1β and IL-18 downstream of NLRP3 inflammasome activation would contribute to pro-inflammatory cytokine signaling. The cytokines, TNF-α and IL-6 can be readily assessed as global markers of inflammation and organ damage during hepatic I/R injury (21). NLRP3 KO, caspase-1 KO and caspase-1 inhibitor-treated mice exhibited significantly lower mRNA level of TNF-α and IL-6 compared with the WT mice after liver I/R (Fig. 2D). We also determined that the lack of IL-1β or IL-18 signaling protected mice from I/R injury using IL-18 KO and IL-1R1 KO mice. These mice were subjected to liver I/R and exhibited decreased hepatocellular damage compared to WT (data not shown), suggesting that IL-1β or IL-18 contribute to organ damage downstream of inflammasome activation. This data suggests that the NLRP3 inflammasome, caspase-1, and cytokines IL-1β and IL-18 play key roles in organ injury following hepatic I/R by driving pro-inflammatory signaling and promoting hepatocyte necrosis.
Functional caspase-1 on bone marrow-derived cells is required for liver I/R injury
The pathophysiology of liver I/R is dependent on both hepatocellular mechanisms as well as interactions with bone marrow-derived NPCs, including resident KCs, infiltrating neutrophils, and dendritic cells, among others (2). We hypothesized that bone-marrow derived cells would responsible for the effect of inflammasome-driven inflammation given that NLRP3 activity is robust in macrophages and other immune cells (27). Additionally, another recent study examined inflammasome activation in all cell types of the liver (28) and found strong activation in KCs, the liver’s resident macrophages. We generated caspase-1 chimeric mice by adoptive transfer of donor bone marrow cells into irradiated recipient animals using combinations of caspase-1 WT and caspase-1 KO mice. First, we confirmed functionality of this model by assaying caspase-1 activity (Fig. 3A). WT/WT mice had significantly higher levels of caspase-1 activity after I/R, while KO/KO mice had no increase in activity after I/R. In caspase-1 KO mice with caspase-1 WT bone-marrow cells (KO/WT), caspase-1 activity was restored to levels near WT/WT mice, and there was no significant difference between these groups. However, in caspase-1 WT mice with caspase-1 KO bone-marrow (WT/KO), caspase-1 activity was significant reduced after I/R to levels near KO/KO mice. These results suggested that the majority of caspase-1 signaling after I/R is dependent on bone-marrow derived cells. We also assayed liver damage and cytokine levels. KO/KO chimeric mice were protected from liver I/R compared with WT/WT mice as measured by serum ALT levels (Fig. 3B), histological damage (Fig. 3C), serum IL-1β and IL-18 (Fig. 3D), liver IL-1β and IL-18 (Fig. 3E), and by reductions in serum cytokines IL-6 and TNF-α (Fig. 3F). Caspase-1 WT mice with caspase-1 mutant bone marrow cells (WT/KO) were also protected from hepatic I/R in a similar fashion compared to WT/WT controls (Fig. 3B–F). In contrast, caspase-1 mutant mice with caspase-1 WT bone marrow cells (KO/WT) displayed a phenotype after I/R injury similar to WT/WT mice, with no significant differences in sALT levels, serum IL-1β, liver IL-1β and IL-18, and serum TNF-α (Fig. 3B–F). However, we did observe that KO/WT mice had significantly less histological damage (Fig. 3C), serum IL-18 (Fig 3D), and serum IL-6 (Fig 3F), suggesting that the observed effects of inflammasome activation after I/R may not be totally dependent on bone marrow derived cells, and that other systemic mechanisms may be contributing to this pattern. Nonetheless, these results strongly suggest that inflammatory damage driven by inflammasome activation after I/R is largely due to bone-marrow derived cells.
Figure 3.

Functional caspase-1 on bone marrow derived cells, not parenchymal cells, is required for liver I/R injury. (A) Activation of caspase-1 in WT/WT, WT/KO, KO/WT and WT/WT mice subjected to liver I/R compared to the WT sham group, assessed by colorimetric assay. Data represent the mean ± SE (n = 6 mice per group). ANOVA Holm-Sidak method, *P < 0.05 WT/KO vs. KO/WT, WT/WT vs. WT/KO; N.S., not significant WT/WT vs. KO/WT. (B) Serum ALT levels in caspase-1 chimeric mice after liver I/R. Data represent the mean ± SE (n = 4–6 mice per group). ANOVA Holm-Sidak method, *P < 0.05 WT/KO vs. KO/WT, WT/WT vs. WT/KO; N.S., not significant WT/WT vs. KO/WT. (C) Quantification of necrotic hepatocytes in hematoxylin and eosin-stained liver sections from caspase-1 chimeric mice 6 hours after reperfusion. The graph is representative of liver sections from 4–6 mice per group ANOVA Holm-Sidak method, *P < 0.05, WT/KO vs. KO/WT, WT/WT vs. WT/KO, WT/WT vs. KO/WT; N.S., not significant. (D) Serum levels of IL-1β and IL-18 obtained from caspase-1 chimeric mice at 6 hours after reperfusion were measured by ELISA. Data represent the mean ± SE (n = 4–6 mice per group). ANOVA Holm-Sidak method, *P < 0.05, WT/KO vs. KO/WT, WT/WT vs. WT/KO, WT/WT vs. KO/WT; N.S., not significant WT/WT vs. KO/WT. (E) Western blot images showing the protein levels of activated (cleaved) IL-1β and IL-18 in liver of caspase-1 chimeric mice at 6 hours after reperfusion. Each lane represents a separate animal. The blots shown are representatives of three experiments with similar results. (F) IL-6 and TNF-α mRNA levels in NLRP3 KO or caspase-1 KO mice vs WT mice after 6 hours I/R, ANOVA Holm-Sidak method, *P < 0.05, WT/KO vs. KO/WT, WT/WT vs. WT/KO, WT/WT vs. KO/WT; N.S., not significant WT/WT vs. KO/WT.
Extracellular Histones Activate the NLRP3 Inflammasome through the TLR9 Signaling
We have recently reported that extracellular histones contribute to hepatic I/R injury by functioning as DAMPs, leading to a pro-inflammatory immune responses and organ damage (13). At high systemic doses they cause direct endothelial damage and organ injury, and our previous results suggest that they also participate in innate immune responses by stimulating hepatic non-parenchymal cells (NPCs) to secrete cytokines through unknown mechanisms. Although the NLRP3 inflammasome has previously been shown to be activated by several endogenous agonists (5), the contribution of histone proteins has not been examined. We hypothesized that histones might play a role in inflammasome activation. Therefore, in order to determine whether NLRP3 is activated by extracellular histones after liver I/R, we treated mice with neutralizing antibodies to either histone H3 or H4 and found that mice treated with neutralizing anti-histone antibodies clearly exhibited less activated caspase-1 as well as matured IL-1β and IL-18 compared to IgG-treated mice (Fig. 4A). In contrast, we found that mice treated with exogenous histones immediately after ischemia had significantly greater protein level of activated caspase-1 as well as matured IL-1β and IL-18 compared to both PBS-treated and sham mice (Fig. 4A). Caspase-1 activity evaluated by colorimetric assay was also consistent with these results (Fig. 4B).
Figure 4.

Extracellular histones activate NLRP3 inflammasome during liver I/R. (A) Western blot images showing inflammasome protein level activated caspase-1, IL-1β and IL-18 in liver of WT (C57BL/6) mice at 6 hours after reperfusion. Sham or I/R-treated mice was given either a nonlethal dose of exogenous histone mixture (25 mg/kg body weight), vehicle PBS, anti-histone H3 antibody, anti-histone H4 antibody (20 mg/kg body weight) or control antibody intravenously 30 minutes prior to ischemia. Each lane represents a separate animal. (B) Activation of caspase-1 in WT mice treated with vehicle PBS, exogenous histone mixture, anti-histone H3 or anti-histone H4 antibody that were subjected to liver I/R compared to sham treated-mice, assessed by colorimetric assay. Data represent the mean ± SE (n = 6 mice per group). ANOVA Tukey test, *P < 0.05, PBS vs. Histones; **P < 0.05, anti-histone H3 or H4 vs. PBS. Extracellular histones activate NRLP3 inflammasome through TLR9 signaling pathway during liver I/R. (C) Protein levels of activated caspase-1, IL-1β and IL-18 in liver of TLR9 mutant or WT mice treated with PBS or exogenous histones (25 mg/kg body weight) and quantitative densitometry of the protein expressions of activated caspase-1. (D) Protein levels of activated caspase-1, IL-1β and IL-18 in liver of TLR9 inhibitor treated- or control CpG treated-mice administrated with PBS or exogenous histones and quantitative densitometry of the protein expressions of activated caspase-1. Each lane represents a separate animal and each animal was harvested after 6 hours of reperfusion. The blots shown are representatives of three experiments with similar results Student’s t-test, *P < 0.05, PBS vs. Histones.
We recently established that TLR9 and its downstream signaling molecule MyD88 are involved in histone-mediated damage during sterile inflammatory injury induced by I/R (13). Since we found that histones stimulate inflammasome activation after I/R, we sought to determine whether TLR9 activation is an intermediary between extracellular histones and activation of the NLRP3 inflammasome. TLR9 mutant (TLR9CpG/CpG) and their WT counterparts were administered either exogenous histones or PBS after ischemia. Protein levels of activated caspase-1 in TLR9 mutant mice were significantly less than in WT mice after ischemia (Fig. 4C). Activated caspase-1 was increased in TLR9 WT mice given exogenous histones, whereas they failed to enhance the activation of caspase-1 in TLR9 mutant mice (Fig 4C). Similarly, lower levels of activated caspase-1 were also observed in WT mice treated with a TLR9 inhibitor (ODN2088) compared to PBS injection (Fig. 4D), corroborating these findins. These results demonstrate that inflammasome activation after I/R injury is a result of upstream TLR9 activation stimulated by extracellular histones.
Extracellular Histone Mediated Liver I/R Injury is Dependent on the NLRP3 Inflammasome
Our previous study demonstrated a role for TLR9 signaling in mediating extracellular histone-mediated damage (13), although the precise mechanism accounting for this phenomenon remains unanswered. Since our data showed blocking the effects of histones or TLR9 activation after I/R reduced NLRP3 inflammasome activation, we sought to investigate the role of NLRP3 in histone-mediated damage after I/R injury. When exogenous histones were administered to NLRP3 KO mice they failed to exacerbate liver damage compared to KO mice treated with PBS, whereas damage as assessed by sALT and liver histology was significantly increased in the corresponding WT controls (Fig. 5A and B).
Figure 5.

Extracellular histones mediated hepatic I/R injury depends on NLRP3 inflammasome. (A) Serum ALT levels in NLRP3 KO or WT mice after 6 hours of reperfusion that were treated with PBS or exogenous histones (25 mg/kg body weight). Data represent the mean ± SE (n = 7 mice per group). Student’s t-test Mann-Whitney Rank Sum Test, N.S., not significant PBS vs. Histones. (B) Quantification of necrotic hepatocytes in hematoxylin and eosin-stained liver sections from histone treated- or PBS treated-NLRP3 KO mice 6 hours after reperfusion. The graph is representative of liver sections from six mice per group. ANOVA Tukey test, N.S., not significant PBS vs. Histones (C) Protein levels of activated caspase-1, IL-1β and IL-18 in liver of TLR9 mutant or WT mice treated with PBS or exogenous histones. Each lane represents a separate animal. The blots shown are representatives of three experiments with similar results. (D) Serum levels of IL-1β and IL-18 obtained from NLRP3 KO mice and WT counterparts at 6 hours after reperfusion that were treated with PBS or exogenous histones (25 mg/kg body weight) were measured by ELISA and compared to the sham group. Data represent the mean ± SE (n = 6 mice per group). NLRP3 KO mice vs WT mice after I/R, Student’s t-test, N.S., not significant PBS vs. Histones.
The reduction in injury in NLRP3 KO mice after exogenous histones treatment was further validated by protein analysis of downstream pro-inflammatory cytokines. Exogenous histones failed to enhance the activation of caspase-1, mature IL-1β and IL-18 in NLRP3 KO mice. In contrast, those protein levels were dramatically increased in WT mice treated with histones (Fig. 5C, 5D). The decrease in mature IL-1β and IL-18 in NLRP3 KO mice likely contributed to the attenuated damage after liver I/R.
The NLRP3 Inflammasome in KCs is Activated by Extracellular Histones through TLR9
Next, we sought to determine potential in vitro mechanisms through which extracellular histones, TLR9, and the NLRP3 inflammasome mediate organ damage after I/R. Since our chimeric mouse data suggested a strong role for bone-marrow derived cells, we examined the cell non-parenchymal cell types of the liver that might be responsible for mediating the effect of inflammasome activation after I/R. While we found that the NLRP3 inflammasome was weakly activated in liver sinusoidal epithelial cells (data not shown), we also considered that these cell types are not re-populated in the liver after adoptive bone marrow transfer and thus were not responsible for the observed effects of inflammasome activation in NPCs. Strong activation of the NLRP3 inflammasome has been demonstrated in KCs (28), and we also found robust activation of the inflammasome in these cells in vitro. We used hypoxia as a stimulus as it simulates the ischemic microenvironment during I/R. We observed an increase in the activation of caspase-1 as well as IL-1β and IL-18 in a time-dependent-manner in KCs exposed to hypoxic conditions compared to normoxia (Fig. 6A). Treatment of KCs with H2O2 showed similar results occurring in a dose-dependent manner (data not shown). Confocal microscopy confirmed that both hypoxia- and H2O2- treatment induced an increase in caspase-1 activity in KCs (Fig. 6B).
Figure 6.

NLRP3 inflammasome in KCs is activated by extracellular histones through TLR9 pathway. Whole cell lysate from KCs after stimulation was subjected to western blot analysis of activated caspase-1, IL-1β and IL-18. For Western blot images: The blots shown are representatives of three experiments with similar results. (A) Cultured mouse KCs were exposed to hypoxia (1% O2) from 0 to 12 hours. (B) The activated caspase-1 in cultured KCs obtained WT mice stimulated with normoxia or hypoxia (1% O2) overnight was visualized with caspase-1 fluorochrome inhibitor of caspase-1 reagent and observed under confocal microscope. Green, actin; blue, nuclei; red, activated caspase-1. (C) Cultured KCs from WT or TLR9 mutant mice were stimulated with exogenous histones from dosage of 0 to 50 μg/mL for 12 hours. (D) The activated caspase-1 in cultured KCs that were stimulated with exogenous histones (25 μg/mL) or TLR9 agonist (15μg/mL) or PBS for 12 hours, was visualized with caspase-1 fluorochrome inhibitor of caspase-1 reagent and observed under confocal microscope. Green, actin; blue, nuclei; red, activated caspase-1. (E) Cultured mouse KCs were stimulated with TLR9 agonist from dosage of 0 to 15μg/mL for 12 hours. (F) Quantitation of activated caspase-1 in cultured mouse KCs was measured using the analytical software MetaMorph™ and normalized to nuclei within the sample field. ANOVA Holm-Sidak method, *P < 0.05; Histones or TLR9 agonist vs. PBS treated-KCs, **P < 0.05; Hypoxia vs. Normoxia treated KCs. (G) Cultured mouse KCs were stimulated with TLR9 antagonist or/and exogenous histones for 12 hours.
We next determined whether exogenous histones could activate the NLRP3 inflammasome in KCs. KCs obtained from WT mice exposed to different concentrations of exogenous histones resulted in a dose-dependent increase in the activation of caspase-1 whereas the NNLRP3 inflammasome is not activated in KCs from TLR9 mutant mice (Fig. 6C). Consistent results were obtained when KCs were stimulated with the TLR9 agonist (ODN1668) exhibiting increased activation of caspase-1, IL-1β and IL-18 in a dose-dependent manner (Fig. 6D and 6E). Immunofluorescence quantification of activated caspase-1 confirmed the activation of inflammasome in mouse KCs after stimulation of histones, TLR9 agonist or hypoxia compared to normal controls (Fig. 6F). Furthermore, KCs were co-stimulated with histones and the TLR9 antagonist ODN2088. A reduction in activation of caspase-1, IL-1β and IL-18 was observed with ODN2088 treatment (Fig. 6G). While our results suggested a strong role for TLR9 in mediating extracellular histone-mediated inflammasome activation in hypoxic KCs, we also considered that other mechanisms may be contributing to our observed effects. In hepatic I/R, other members of the TLR family - mainly TLR4 and TLR2 - have been shown to contribute to I/R damage through propagation of DAMP signaling and downstream inflammatory pathways (29). Our own results showed a strong but non-significant trend in protection from histone-mediated damage after I/R in TLR4 KO mice (13). We stimulated TLR2 and TLR4 KO KCs with histones and assayed for inflammasome activation (Supplemental Figure 1). We found a dose-dependent increase in inflammasome activation in both TLR4 and TLR2 KO cells, suggesting that histone-mediated inflammasome activation is largely independent of these receptors. Collectively, these results suggest that extracellular histones are capable of mediating NLRP3 inflammasome activation in vitro through TLR9.
Extracellular Histone-Activated TLR9 Leads to Mitochondrial ROS Production in KCs and Subsequently Activates the NLRP3 Inflammasome
Our data show that histones are capable of activating the inflammasome in KCs through TLR9, but the intermediate signaling pathways that link TLR9 to inflammasome activation are unknown. Kupffer cells are well-known to mediate damage after I/R through reactive oxygen species (ROS) (30) and recent data suggests that mitochondrial-associated ROS production activates the NLRP3 inflammasome (31,32). Therefore, we sought to discern whether ROS linked upstream histone/TLR9 engagement to downstream NLRP3 inflammsome activation. Using high content analysis, we studied ROS production WT and TLR9 mutant KCs in response to a TLR9 agonist or histones. Levels of ROS significantly increased in both WT and TLR9 mutant KCs after stimulation with tert-butyl hydroperoxide, a known stimulus of ROS production in hepatocytes (33). Stimulation with either extracellular histones or a TLR9 agonist resulted in a statistically significant increase in total cellular ROS production in KCs from TLR9 WT mice. However, both these treatments failed to enhance the production of ROS in KCs from TLR9 mutant mice (Fig. 7A). Additionally, we found that mitochondrial ROS were also elevated in a similar fashion in WT cells stimulated with either histones or TLR9 agonist (Fig. 7B). Given that ROS production has been extensively shown to activate the NLRP3 inflammasome (34), we hypothesized that ROS production downstream of TLR9/histone engagement might provide the stimulus for NLRP3 inflammasome activation. Indeed, we found that the increased ROS production in WT KCs stimulated with either histones or TLR9 agonist was significantly neutralized by anti-oxidant N-acetylcysteine (NAC) (Fig. 7C). Neutralization of ROS by treatment with the NAC completely abolished inflammasome activation in KCs stimulated with histones or a TLR9 agonist (Fig. 7D), suggesting that production of ROS is the mechanism by which extracellular histones - through TLR9 signaling - activate the NLRP3 inflammasome in KCs.
Figure 7.

TLR9 signaling pathway is involved in the activation of NLRP3 inflammasome through the increase of mitochondrial and total cellular ROS production. (A) Cellular ROS production detected by high content analysis in cultured whole NPCs, which were obtained from WT or TLR9 mutant mice, were stimulated with exogenous histones or TLR9 agonist and then compared with negative and positive control stimulation. ANOVA Dunn’s method, *P < 0.05 compared to negative control. **P < 0.05 compared to negative control. N.S., not significant compared to negative control. (B) Flow cytometry of cultured KCs stimulated with exogenous histones, TLR9 agonist or PBS were stained with MitoSOX and were compared with negative and positive controls. The bar graph represents pooled data from three experiments. ANOVA Tukey test, *P < 0.05 compared to PBS treatment. (C) KCs from WT mice stimulated with NAC (0, 25 or 50 mM) Cells were subjected to exogenous histones (50 μg/mL), TLR9 agonist (15 μg/mL) or PBS for 12 hours. ROS production measured by DCF assay shown is the percent increase relative to respective normoxic controls. ANOVA Holm-Sidak method, *, P < 0.05 versus NAC treated KCs. N.S., not significant. (D) KCs from WT mice stimulated with NAC (50mM) were subjected to exogenous histones (50 μg/mL), TLR9 agonist (15 μg/mL) or PBS for 12 hours. Whole cell lysate from KCs after stimulation was subjected to western blot analysis of activated caspase-1, IL-1β and IL-18. For Western blot images: The blots shown are representatives of three experiments with similar results.
NLRP3 Inflammasome Regulates Innate Immune Cells in Liver I/R
To determine the role of NLRP3 inflammasome in modulating other innate immune cells in the liver following I/R, flow cytometry analysis with quantitative evaluation of dendritic cells (DCs), neutrophils, and inflammatory monocytes in homogenized ischemia liver lobes was performed. Liver I/R injury in WT mice led to a significant increase in total cell numbers of neutrophils and inflammatory monocytes but a significant decrease in DCs compared with sham WT mice (Fig. 8). However, the ablation of NLRP3 conferred a stable innate immune environment that DCs, neutrophils and inflammatory monocyte numbers remained unchanged after liver I/R compared with sham KO mice. This data demonstrate that the elimination of the NLRP3 inflammasome down-regulates innate immune response by decreasing the influx of innate immune cells in the ischemic lobes after liver I/R.
Figure 8.

NLRP3 inflammasome regulates innate immune cells in liver I/R. Flow cytometry analysis with a quantitative evaluation of NPCs in homogenized ischemia liver lobes in NLRP3 KO and WT mice. Gated percentages of conventional dendritic cells (cDCs), neutrophils, and inflammatory monocytes were evaluated. (B) Absolute numbers (in million of cells) of cDCs, neutrophils, and inflammatory monocytes cells. Data represent the mean ± SE (n = 6 mice per group). ANOVA Tukey test, *P < 0.05 compared to sham mice N.S., not significant sham vs. liver I/R. Each experiment was repeated a minimum of three times.
Discussion
Ischemia reperfusion injury contributes to morbidity and mortality in a broad range of pathologies. It has been established that the sterile inflammatory response plays a key role in the pathogenesis of I/R injury. This involves DAMPs activating signaling through pattern-recognition receptors, such as TLRs, with the subsequent recruitment and activation of innate immune cells (35). In addition to TLRs, emerging evidence illustrates the importance of NOD-like receptors with their formation of the multi-protein complex inflammasome in I/R injury (36). Iyer et al. found that acute necrotic cell death due to renal I/R injury triggers an inflammatory response through the NLRP3 inflammasome and results in collateral tissue damage (37). Shigeoka et al. also found NLRP3-deficient mice showed protection against renal I/R injury (38), and results from Kawaguchi et al. suggest that the inflammasome is a potential novel therapeutic target for preventing myocardial I/R injury (7). The purpose of this study was to determine the mechanisms by which NLRP3 inflammasome activation results in damage and inflammatory response following liver I/R. The major and novel findings of this investigation are: (a) extracellular histones serve as novel activators of the NLRP3 inflammasome; (b) functional caspase-1 in liver bone-marrow derived cells, not parenchymal hepatocytes, is required for liver I/R injury; (c) the activation of NLRP3 inflammasome by extracellular histones is dependent on TLR9 suggesting interplay between TLR9 and NLRP3 signaling pathways; (d) KCs play an essential role in histone-mediated activation of the NLRP3 inflammasome; (e) activation of the NLRP3 inflammasome in KCs drives the innate immune response by mediating the infiltration of innate immune cells during liver I/R.
A recent experimental study in mice with NLRP3 silenced by small hairpin RNA revealed that NLRP3 signaling is involved in liver I/R and silencing of NLRP3 can protect the liver from I/R injury by reducing IL-1β, IL-18, TNF-α, IL-6, and HMGB1 release (6). Our present findings are consistent with the study in that the activation of the NLRP3 inflammasome was found to have detrimental effects in liver tissues after I/R injury. We further demonstrate that genetic elimination of NLRP3 or caspase-1, and pharmacologic inhibition of caspase-1 ameliorated the innate immune response and resultant organ injury associated with liver I/R. In contrast to our data, Menzel et al. suggests that caspase-1 is hepatoprotective through the regulation of cell death pathways in the liver after major trauma, and that caspase-1 activation does not depend on NLRP3 in the hemorrhagic shock/bilateral femur fracture mouse model (39). We believe that this discrepancy on the role of NLRP3 and caspase-1 in liver injury may be model-related. Our study uses a model of local injury to the liver where caspase-1 in KCs is dominant; whereas Menzel and colleagues utilized a systemic injury model where caspase-1 in hepatocytes is clearly involved. Additionally, they suggest that other inflammasomes instead of NLRP3 serve as activators of caspase-1 in hepatocytes after hemorrhagic shock. We, as well as others (36), demonstrated reduction of activated caspase-1 after eliminating NLRP3 during I/R injury, suggesting that activation of caspase-1 is dependent on the activation of NLRP3 during I/R in KCs. However, the contribution of NLRP3 signaling in hepatocytes remains to be seen in warm hepatic I/R injury.
We find that extracellular histones released following liver I/R serve as novel activators of NLRP3, which in turn stimulates the formation of NLRP3 inflammasome and activates downstream caspase-1, IL-1β and IL-18. Besides pathogen-associated stimuli, a number of endogenous danger signals such as DNA fragments and HMGB1 have also been reported to activate the NLRP3 inflammasome and contribute to the pro-inflammatory response in liver I/R (40–44). We recently reported that extracellular histones may also function as DAMPs, mediating liver I/R injury through the TLR9 signaling pathway (13). Thus, we hypothesized that extracellular histones can also activate NLRP3 inflammasome via a mechanism that is dependent on TLR9 signaling pathway during liver I/R. We provide evidence that the administration of exogenous histones can activate the NLRP3 inflammasome, whereas anti-histone antibody treatment, neutralizing endogenously release histones, reduced the activation of NLRP3 during liver I/R. Furthermore, NLRP3 activation was reduced when TLR9 antagonist was introduced to the system along with exogenous histones suggesting NLRP3 activation by extracellular histones is dependent on TLR9 signaling pathway. Interestingly, in two different models of inflammation (acetaminophen-induced hepatic injury and acute pancreatitis in mice), TLR9 alone was shown to promote pro-IL-1β through the ubiquitously activated NF-κB; and, activation of the NLRP3 inflammasome was suggested to be independent of TLR9 signaling pathway (45,46). On the contrary, our study shows a clear association between the TLR9 signaling pathway and the activation of NLRP3 during liver I/R.
Though these studies provide data to show that histones augment inflammasome activation through TLR9, one limitation of this study is that the role of nucleosomes and free DNA in inflammasome activation were not examined in our model. Indeed, several studies recent studies have examined the effects of histones, free DNA, and their combination in the form of nucleosomes in both TLR9 and inflammasome activation. In a model using parasitic p. falciparum, Gowda et al. showed that nucleosomes were the immunostimulatory component responsible for activating human dendritic cells (20), and that degradation of either histones or DNA resulted in abolished DC activation. They hypothesized that while negatively charged DNA is incapable of being taken up by DC’s, the combination of highly positively charged histones in the form of nucleosomes allowed for cell uptake and efficient delivery to intracellular TLR9-containing endosomes. In our in vivo model of hepatic I/R, the mechanism of uptake and delivery of histones and DNA to TLR9 remains an unanswered question. Furthermore, the individual roles of histones and free DNA have not been examined. However, our data in these studies along with our previously published data (13) clearly show that a systemically non-toxic dose of histones (25 mg/kg) exacerbates I/R injury, and that this histone-specific toxicity is dependent on both TLR9 and NLRP3 inflammasome activation in bone marrow-derived inflammatory cells to promote the full extent of inflammatory injury. Whether this phenomenon is due to direct histone-mediated toxicity or through enhanced delivery of TLR9 ligands to endosomes remains unknown. However, we did find in vitro in KCs that full activation of the inflammasome likely requires both histones and free DNA (Supplemental Figure 2). We found that co-treatment of KCs with histones and either DNase or trypsin reduced inflammasome activation. Furthermore, with both DNase and trypsin, inflammasome activation was nearly abolished completely. These data suggest that both histones and DNA work in combination to activate the inflammasome.
While our data shows that histones drive inflammasome-driven organ inflammation and damage downstream of TLR9 activation, other TLRs have been shown to play a role in histone-mediated cell toxicity. Specifically, TLR4 and TLR2 have been shown to play prominent roles in mediating the toxic effects of histones in ConA-induced acute liver injury (12), acute kidney injury (47), and thrombosis (48). In hepatic I/R, our own data shows a strong but non-significant trend in which TLR4 KO mice are afforded protection from histone toxicity after I/R (13). Several studies have examine the role of TLRs in hepatic I/R (43), and while TLR4, TLR2, and TLR9 mice are globally protected from I/R injury, our data suggest that TLR9 is most important for histone mediated toxicity in hepatic I/R. While I/R injury is exacerbated with histone injections in TLR2 and TLR4 KO mice, this response is blunted most dramatically in TLR9 KO mice. Similar to other DAMP molecules, histones appear to have both direct and indirect effects on cell signaling pathways. Directly, they disrupt cell membranes causing electrolyte imbalances (11) and may directly bind to TLR2, TLR4, and TLR9. Indirectly, they may function to enhance the cytokine activity of other DAMP molecules whose effects are primarily mediated through TLRs, including TLR2, TLR4, and TLR9.
Finally, we found that activation of the NLRP3 inflammasome mediates the innate immune response by inducing the infiltration of innate immune cells during liver I/R. We further confirmed the reduction of DCs in ischemic liver lobes after liver I/R as Bamboat et al. observed (25). However, in our study we observed a much lower percentage of cDCs compared to theirs. This may possibly be a result of differences in the cell collection method and time point of NPC harvest. We used an earlier time point of 6h of reperfusion while Bamboat and colleagues harvested cells 12h after ischemia. Interestingly, total cell numbers of cDCs were significantly lowered in NLRP3 KO sham mice compare with WT sham mice, the mechanism of this requires further investigation. Reduction of neutrophils and inflammatory monocytes in NLRP3 KO mice after liver I/R suggests the importance of the NLRP3 inflammasome in recruiting innate immune cells after liver damage. This is consistent the findings of McDonald et al. that NLRP3 deficiency significantly reduced the quantity of recruited neutrophils after local liver necrosis (49). Collectively, these studies in addition to our data suggest that inflammasome activation may regulate innate immune cell numbers in sterile inflammation. However, the mechanism by which NLRP3 inflammasome regulates innate immune cells remains to be established.
In conclusion, the NLRP3 inflammasome is activated by endogenous histones during liver I/R, which contributes to organ damage through activation of caspase-1 and increased production of the pro-inflammatory cytokines IL-1β and IL-18. Activation of NLRP3 inflammasome in KCs by histones is through a TLR9-dependent pathway, which mediates ROS production. Activation of KCs ultimately leads to alterations in the innate immune cell composition of the liver after I/R (Figure 9). Our findings illustrate a novel mechanism by which I/R may lead to injury and potentially provide additional therapeutic targets to limit the sterile inflammatory response.
Figure 9.

Hypothetical model illustrating the role of extracellular histones in the activation of NLRP3 inflammasome through TLR9 in liver I/R injury. During liver I/R injury, extracellular histones are sensed by TLR9, which initiates the cellular and mitochondrial ROS production. ROS then activates the NLRP3 inflammasome. Activation of NLRP3 inflammasome further regulates innate immune cells in liver I/R.
Supplementary Material
Acknowledgments
We thank Xinghua Liao and Nicole Hays for technical assistance in preparing the manuscript. We also thank Marc Monestier at Temple University for providing hybridoma cells for producing LG2-1 and BWA-3.
This work was supported by Howard Hughes Medical Institute Physician-Scientist Award, Society of University Surgeons Junior Faculty Award, and National Institutes of Health Grant R01 GM95566 (to A.T.)
Reference List
- 1.Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. 2011;11:1563–1569. doi: 10.1111/j.1600-6143.2011.03579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vardanian AJ, Busuttil RW, Kupiec-Weglinski JW. Molecular mediators of liver ischemia and reperfusion injury: a brief review. Mol Med. 2008;14:337–345. doi: 10.2119/2007-00134.Vardanian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klune JR, Tsung A. Molecular biology of liver ischemia/reperfusion injury: established mechanisms and recent advancements. Surg Clin North Am. 2010;90:665–677. doi: 10.1016/j.suc.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 5.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu P, Duan L, Chen J, Xiong A, Xu Q, Zhang H, Zheng F, Tan Z, Gong F, Fang M. Gene Silencing of NALP3 Protects Against Liver Ischemia-Reperfusion Injury in Mice. Hum Gene Ther. 2011;22:853–864. doi: 10.1089/hum.2010.145. [DOI] [PubMed] [Google Scholar]
- 7.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 8.Pemberton AD, Brown JK, Inglis NF. Proteomic identification of interactions between histones and plasma proteins: implications for cytoprotection. Proteomics. 2010;10:1484–1493. doi: 10.1002/pmic.200900818. [DOI] [PubMed] [Google Scholar]
- 9.Zeerleder S, Zwart B, Wuillemin WA, Aarden LA, Groeneveld AB, Caliezi C, van Nieuwenhuijze AE, van Mierlo GJ, Eerenberg AJ, Lammle B, Hack CE. Elevated nucleosome levels in systemic inflammation and sepsis. Crit Care Med. 2003;31:1947–1951. doi: 10.1097/01.CCM.0000074719.40109.95. [DOI] [PubMed] [Google Scholar]
- 10.Muller S, Dieker J, Tincani A, Meroni PL. Pathogenic anti-nucleosome antibodies. Lupus. 2008;17:431–436. doi: 10.1177/0961203308090030. [DOI] [PubMed] [Google Scholar]
- 11.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187:2626–2631. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, Liao X, Billiar T, Xu J, Esmon CT, Tsung A. Endogenous histones function as alarmins in sterile inflammatory liver injury through toll-like receptor 9. Hepatology. 2011 doi: 10.1002/hep.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsung A, Stang MT, Ikeda A, Critchlow ND, Izuishi K, Nakao A, Chan MH, Jeyabalan G, Yim JH, Geller DA. The transcription factor interferon regulatory factor-1 mediates liver damage during ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1261–G1268. doi: 10.1152/ajpgi.00460.2005. [DOI] [PubMed] [Google Scholar]
- 15.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang H, Deng M, Jin H, Liu A, Dirsch O, Dahmen U. Hepatic arterial perfusion is essential for the spontaneous recovery from focal hepatic venous outflow obstruction in rats. Am J Transplant. 2011;11:2342–2352. doi: 10.1111/j.1600-6143.2011.03682.x. [DOI] [PubMed] [Google Scholar]
- 17.Gu Y, Dirsch O, Dahmen U, Ji Y, He Q, Chi H, Broelsch CE. Impact of donor gender on male rat recipients of small-for-size liver grafts. Liver Transpl. 2005;11:669–678. doi: 10.1002/lt.20408. [DOI] [PubMed] [Google Scholar]
- 18.Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, Zlotnicki J, Billiar T, Tsung A. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 2010;285:39888–39897. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, Lotze MT, Geller DA, Billiar TR. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661–7668. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 20.Gowda NM, Wu X, Gowda DC. The nucleosome (histone-DNA complex) is the TLR9-specific immunostimulatory component of Plasmodium falciparum that activates DCs. PLoS One. 2011;6:e20398. doi: 10.1371/journal.pone.0020398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, Monga SP, Geller DA, Lotze MT, Tsung A. High mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012 doi: 10.1002/hep.25572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loughran PA, Stolz DB, Barrick SR, Wheeler DS, Friedman PA, Rachubinski RA, Watkins SC, Billiar TR. PEX7 and EBP50 target iNOS to the peroxisome in hepatocytes. Nitric Oxide. 2013 doi: 10.1016/j.niox.2013.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, Dematteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest. 2010 doi: 10.1172/JCI40008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boaru SG, Borkham-Kamphorst E, Tihaa L, Haas U, Weiskirchen R. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease. J Inflamm (Lond) 2012;9:49. doi: 10.1186/1476-9255-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arumugam TV, Okun E, Tang SC, Thundyil J, Taylor SM, Woodruff TM. Toll-like receptors in ischemia-reperfusion injury. Shock. 2009;32:4–16. doi: 10.1097/SHK.0b013e318193e333. [DOI] [PubMed] [Google Scholar]
- 30.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol. 1991;260:G355–G362. doi: 10.1152/ajpgi.1991.260.3.G355. [DOI] [PubMed] [Google Scholar]
- 31.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 33.Martin C, Martinez R, Navarro R, Ruiz-Sanz JI, Lacort M, Ruiz-Larrea MB. tert-Butyl hydroperoxide-induced lipid signaling in hepatocytes: involvement of glutathione and free radicals. Biochem Pharmacol. 2001;62:705–712. doi: 10.1016/s0006-2952(01)00704-3. [DOI] [PubMed] [Google Scholar]
- 34.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 35.Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leemans JC, Cassel SL, Sutterwala FS. Sensing damage by the NLRP3 inflammasome. Immunol Rev. 2011;243:152–162. doi: 10.1111/j.1600-065X.2011.01043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, Sutterwala FS. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shigeoka AA, Mueller JL, Kambo A, Mathison JC, King AJ, Hall WF, Correia JS, Ulevitch RJ, Hoffman HM, McKay DB. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol. 2010;185:6277–6285. doi: 10.4049/jimmunol.1002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menzel CL, Sun Q, Loughran PA, Pape HC, Billiar TR, Scott MJ. Caspase-1 is hepatoprotective during trauma and hemorrhagic shock by reducing liver injury and inflammation. Mol Med. 2011;17:1031–1038. doi: 10.2119/molmed.2011.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest. 2009;119:3502–3511. doi: 10.1172/JCI40599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 42.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 43.Evankovich J, Billiar T, Tsung A. Toll-like receptors in hepatic ischemia/reperfusion and transplantation. Gastroenterol Res Pract. 2010;2010 doi: 10.1155/2010/537263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, Scott MJ, Billiar TR, Wilson MA, Fan J. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol. 2011;187:4809–4817. doi: 10.4049/jimmunol.1102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119:305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, Mehal W. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. 2011;141:358–369. doi: 10.1053/j.gastro.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hagele H, Lichtnekert J, Hagemann JH, Rupanagudi KV, Ryu M, Schwarzenberger C, Hohenstein B, Hugo C, Uhl B, Reichel CA, Krombach F, Monestier M, Liapis H, Moreth K, Schaefer L, Anders HJ. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. 2012;23:1375–1388. doi: 10.1681/ASN.2011111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118:1952–1961. doi: 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.